
Abstract
Traumatic brain injury (TBI) is a devastating condition, often leading to life-long consequences for patients. Even though modern neurointensive care has improved functional and cognitive outcomes, efficient pharmacological therapies are still lacking. Targeting peripherally derived, or resident inflammatory, cells that are rapid responders to brain injury is promising, but complex, given that the contribution of inflammation to exacerbation versus improved recovery varies with time post-injury. The injury-induced inflammatory response is triggered by release of alarmins, and in the present study we asked whether interleukin-33 (IL-33), an injury-associated nuclear alarmin, is involved in TBI. Here, we used samples from human TBI microdialysate, tissue sections from human TBI, and mouse models of central nervous system injury and found that expression of IL-33 in the brain was elevated from nondetectable levels, reaching a maximum after 72 h in both human samples and mouse models. Astrocytes and oligodendrocytes were the main producers of IL-33. Post-TBI, brains of mice deficient in the IL-33 receptor, ST2, contained fewer microglia/macrophages in the injured region than wild-type mice and had an altered cytokine/chemokine profile in response to injury. These observations indicate that IL-33 plays a role in neuroinflammation with microglia/macrophages being cellular targets for this interleukin post-TBI.
Introduction
P
Neuroinflammation develops within hours post-TBI and can persist for months. The tissue damage leads to activation of microglia/macrophages, astrocytes, and precursors of oligodendrocytes as well as a catastrophic cascade of cytokines, chemokines, cytolytic enzymes, and reactive oxygen species. 2,7 Subsequent to infiltration of immune cells from the blood through damaged vessels, the affected tissue surrounding the injured regions of the brain also undergoes damage. 5 Additional release of cytokines and chemokines by these infiltrating cells activates glia cells and expands the injury, resulting in neuronal cell death, accumulation of cellular debris, and subsequent tissue cavitation. 3
Activated immune cells, including microglia, macrophages, neutrophils, mast cells, and others may play both beneficial or harmful roles in connection with TBI, 8 depending on the fragile equilibrium between the release of pro- and anti-inflammatory cytokines/chemokines that begins during the very first minutes post-injury. Like other tissues, the brain has a unique system for detection, containment, and repair of damaged tissue based on signaling pathways referred to as damage-associated molecular patterns (DAMPs), including alarmins. Alarmins are released rapidly after nonprogramed cell death and recruit immune cells expressing the appropriate receptor to promote reconstruction of the damaged tissue.
Interleukin 33 (IL-33), a nuclear alarmin, plays an important role in connection with inflammatory and autoimmune diseases, such as asthma, inflammatory bowel disease, or autoimmune hepatitis. 9,10 Experimental allergic encephalomyelitis and spinal cord injury (SCI) are conditions that lead to high levels of IL-33 messenger RNA (mRNA) followed by expression of the corresponding protein by a subpopulation of spinal cord astrocytes and microglia. 11 –13 Further, pathogens induce IL-33 expression in glial cultures. 14 We reported previously that glia cells, mainly astrocytes and oligodendrocytes, in the intact, noninflamed brain express IL-33 during early post-natal life. As the brain matures, IL-33 becomes downregulated, suggesting stringent regulation of IL-33 expression in the absence of a brain insult. 15
These observations led us to ask whether IL-33 is involved in TBI, and we found that IL-33 expression was indeed induced in vivo in a patient suffering from severe TBI, in tissue sections from 3 additional TBI patients versus 2 controls, as well as in mouse models of central nervous system (CNS) injury. To further elucidate the functional significance of IL-33, we determined the role of its receptor, ST2, in TBI-induced neuroinflammation. Post-TBI in ST2-deficient mice, the number of accumulated microglia was reduced and the chemokine profiles of their brains were different. We therefore propose that IL-33 signaling plays a part in microglia/macrophage infiltration post-TBI by altering the chemokines needed for their activation.
Methods
Preparation of traumatic brain injury micradialysis samples
Human serial microdialysis (MD) samples were obtained from a patient (male, 17 years of age) with TBI treated in the neurointensive care (NIC) unit at Uppsala University Hospital (Uppsala, Sweden). The post-resuscitation Glasgow Coma Scale (GCS) score was <9 and was graded as a severe TBI. Neuroimaging (computed tomography [CT], magnetic resonance imaging [MRI]) showed scattered petechial hemorrhages in the subcortical and periventricular white matter as well as the corpus callosum, but not in the brain stem. In addition, a hematoma (9 mm) was observed in the left thalamus with perifocal edema. The injury was classified as diffuse axonal injury (DAI), grade II. A tissue pressure transducer and an MD catheter were implanted in separate burr holes in the right frontal lobe according to the routine NIC protocol. 16 The MD catheter was a 71 high-molecular-weight cut-off MD catheter (M Dialysis AB, Stockholm, Sweden) with a 100-kDa nominal cutoff and a 10-mm membrane length. The perfusate was a sterile electrolyte solution (NaCl, 147 mmol/L; KCl, 2.7 mmol/L; CaCl2, 1.2 mmol/L; MgCl2, 0.85 mmol/L) with the addition of 1.5% human albumin solution, perfused at a rate of 0.3 μL/min. Samples were collected on an hourly basis and analyzed for routine low-molecular-weight biomarkers (glucose, lactate, pyruvate, glutamate, glycerol, and urea), using a point-of-care dedicated enzymatic MD analyzer (CMA 600; M Dialysis AB). Urea was used as an internal control for proper MD catheter performance. 17 The remaining sample material (approximately 12 μL/h) was stored at −70°C awaiting analysis.
The MD catheter was located in structurally normal appearing frontal lobe tissue as verified by CT+MRI (the golden tip of the catheter visible on CT+MRI). Given that this was a DAI, we consider the focal MD data to be highly representative of the global type of injury in this patient. MD sampling commenced 29 h post-injury following a 2-h equilibration period and continued for 5 days (120 h). Before cytokine analysis, the hourly MD samples were pooled into six 12-h and two 24-h fractions. During the 5-day MD monitoring period in the NIC unit, the patient was unconscious and mechanically ventilated. He then gradually regained consciousness, and after a 2-week stay in the NIC unit, he was transferred to his local hospital for neurorehabilitation. At 9 months post-injury, the outcome was graded as good recovery according to the Glasgow Outcome Scale, meaning that the patient was able to execute all activities of daily living independently. Ethical permission was granted by the Regional Ethics Review Board in Uppsala (Dnr: 2010/379), and the patient's relative gave written consent.
Luminex analysis
Human microdialysis samples were analyzed with a 14plex human cytokine panel (R&D Systems, Stockholm, Sweden) and quantified with the Luminex system (Luminex; R&D Systems). This panel detects: fibroblast growth factor 2, IL-1α, IL-1β, IL-6, IL-9, IL-33, leukemia inhibitory factor, platelet-derived growth factor (PDGF)-AA, PDGF-AB/BB, regulated on activation, normal T-cell expressed and secreted (RANTES), stem cell factor (SCF), stromal cell-derived factor (SDF) 1α/β, tumor necrosis factor alpha (TNF-α), and vascular endothelial growth factor. All values were obtained by comparison to standard curves generated with the corresponding recombinant human proteins provided with the commercial kit.
Sampling and preparation of brain tissues from patients
Human tissue was obtained in accord with an ethically reviewed and approved protocol (Uppsala Dnr 2008/303). Three patients (2 females and 1 male) with severe TBI, defined as post-resuscitation GCS scores ≤8, were included. Demographics and clinical characteristics are shown in Table 1. None of the patients had any other known neurological disorder or Down's syndrome. Patients were mechanically ventilated and sedated and continuous measurements of intracranial pressure (ICP) and cerebral perfusion pressure were performed (for details, see a previous work 16 ). TBI patients, from which brain tissue was included in this study, were subjected to focal surgical decompression attributed to life-threatening elevations of ICP and/or the presence of a space-occupying brain swelling or hemorrhage.
Initial surgery for a subdural hematoma, Ccx at resurgery.
TBI, traumatic brain injury; MVA, motor-vehicle accident; SBO, struck by object; R, right; F, frontal; PL, parietal lobe; Ccx, removal of cortical contusion; mGCS, motor component of the Glasgow Coma Scale.
Surgically removed brain tissue was placed in 4% buffered formalin (catalog no.: 02176; HistoLab Products AB, Gothenburg, Sweden). Samples were fixed for 24–72 h and then paraffin-embedded and processed by Tissue Tek VIP (Sakura, Torance, CA). Sections (6 μm thick) were cut using Thermo Scientific MicromHM355 S (Cellab Nordica AB, Sollentuna, Sweden) and placed on SuperFrost® plus slides (Menzel-Gläser, Vienna, Austria) for immunohistochemical IHC analysis.
Post-mortem brain tissue from the parietal cortex of 2 neurologically intact subjects, who had died from unrelated causes and did not have any known neurodegenerative disease, was included. The age of these neurologically intact subjects was 74 and 75 years (1 male and 1 female), and the post-mortem time from death to tissue fixation was <24 h. The tissue preparation of neurologically intact subjects followed the same protocol as described above for TBI patients.
Immunohistochemical staining of human brain
Slides were deparaffinized in xylene, rehydrated, and boiled in epitope-retrieval buffer (DAKO, Stockholm, Sweden). IHC staining with antibody against IL-33 (diluted 1:200, # AF3625; R&D Systems) was performed overnight at 4°C with 3,3’-diaminobenzidine as substrate. Slides were counterstained with hematoxylin and mounted before further analysis.
Animals and ethical approval
Adult C57BL/6 mice (6- to 7-week-old of both sexes; 20–25 g) were used in controlled cortical impact (CCI) and sciatic nerve ligation studies. Adult Balb/c mice (6- to 7-week-old, both sexes; 20–25 g), both wild type (WT) and ST2 deficient (ST2-KO [knockout]), were used in the stab wound experiments. In accord with the Swedish legislation, the local Ethics Committee for Laboratory Animal Experimentation pre-approved the animal protocol.
Traumatic brain injury in mice
Controlled cortical impact
Mice (5 per group) were restrained in a stereotaxic frame; anesthetized with 1.3% isoflurane in 70% nitrogen and 30% oxygen through a nose cone; and their core body temperature kept at 37 ± 0.3°C by a heating pad connected to a rectal probe (CMA150; CMA Microdialysis AB, Kista, Sweden). After injecting bupivacaine (Marcain®; AstraZeneca, Södertälje, Sweden), a midline incision was made on the scalp and a rectangular craniotomy (4 × 4 mm) was then created 1 mm posterior to bregma over the right parietal cortex. The injury was subsequently produced using a CCI device (2.5-mm-diameter piston, set at a compression depth of 0.5 mm and a speed of 3 m/s; VCU Biomedical Engineering Facility, Richmond, VA), bone flap replaced, wound closed with interrupted sutures, the animal moved to a heated cage, and, when fully awake, returned to its home cage.
Stab wound injury
The stab wound method was applied to ST2-deficient mice and their WT Balb/c controls. After isoflurane-induced anesthesia and local analgesia as described above, a midline incision of the scalp circular craniotomy (1-mm diameter) of each mouse (5 per group) was made 1 mm posterior to bregma over the right parietal cortex. The 0.5-mm stab wound was produced with a syringe needle (23G, No. 16, 0.6 × 25 mm; BD Microlance; Becton Dickinson, Franklin Lakes, NJ), after which the wound was closed with interrupted sutures and the animal moved to a heated cage for recovery. Animals were sacrificed 6 h, 3 and 7 days, and 1 and 2 months post-injury with an overdose of sodium pentobarbital (600 mg/kg). For histology, mice were perfused with sterile 0.9% NaCl followed by ice-cold 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS; 0.15 M, pH 7.4, 4°C). Brains were dissected out, the post-fixed in 4% PFA in PBS for 24 h. After fixation, brains were cryoprotected overnight in 10% sucrose in PBS at 4°C, frozen, and stored at −20°C.
For preparation of protein homogenate, hemispheres were dissected out and immediately frozen in liquid nitrogen, and stored at −20°C before chemokine/cytokine profiling.
Sciatic nerve ligation
Under isoflurane anesthesia, the right sciatic nerve of C57BL/6 mice was exposed, and approximately one third to one half the diameter of the nerve was tightly ligated with silk suture for 30 sec. In sham-operated mice, the nerve was exposed but left intact. After ligation, muscles were closed with sutures, and the skin was closed with clips. Animals were killed 3 and 7 days after sciatic nerve ligation. A 10-mm segment of the sciatic nerve, including ligation region, was removed and processed for IHC.
Immunohistochemical analysis of mouse tissue
Coronal cryostat sections (10 μm) were made from the region containing stab wound injury. Collected sections included the brain injury as well as the margins of injury. The needle tract and related tissue injury were examined in parallel sections, by staining with cresyl violet to keep track of the region of interest. Slides that were identified to contain the sections from the center of the cavity were marked and processed for IHC and further analysis.
Thawed tissue sections were first pre-incubated with blocking solution (1% bovine serum albumin, 0.3% Triton X-100, and 0.1% NaN3 in PBS) for 1 h at room temperature (RT), and then incubated overnight at 4°C with primary antibody (see below). After washing in 0.25% Triton X-100 in PBS, secondary antibodies (see below) were applied and the sample incubated for 2 more hours at RT. Finally, the samples were washed twice and mounted with DTG mounting media (2.5% DABCO [Sigma-Aldrich, St. Louis, MO], 90% glycerol, and 50 mM of Tris-HCl [pH 8.0]) with or without 0.375 ng/mL of DAPI (4’,6-diamidino-2-phenylindole; Sigma-Aldrich). The following primary antibodies were used to label tissues: goat anti-IL-33 (diluted 1:500, #AF3626; R&D Systems); mouse anti-GFAP (glial fibrillary acidic protein; diluted 1:200, #G3893; Sigma-Aldrich); rabbit anti-Olig-2 (oligodendrocyte lineage transcription factor 2; diluted 1:500, #AB9610; Millipore, Billerica, MA); mouse anti-Map2 (microtubule-associated protein 2; diluted 1:200, #M4403; Sigma-Aldrich) and rat anti-Mac2 (diluted 1:500, #CL8942AP; Cadarlane, Burlington, Ontario, Canada); NeuN (neuronal nuclei; diluted 1:200, #N5264; Sigma-Aldrich); nestin (diluted, 1:400, #MAB5326; Millipore); CNPase (2’,3’-cyclic nucleotide 3’-phosphodiesterase; diluted 1:500, #C5922; Sigma-Aldrich); and S100 (diluted 1:200; Abcam, Cambridge, MA). Secondary antibodies included cyanine 3–conjugated donkey anti-goat (diluted 1:200, #705-165-003; Jackson ImmunoResearch, Suffolk, UK), fluorescein isothiocyanate (FITC)-conjugated donkey anti-mouse (diluted 1:200, #715-095-150; Jackson ImmunoResearch), FITC-conjugated donkey anti-rat (diluted 1:200, #712-095-150; Jackson ImmunoResearch), and FITC-conjugated donkey anti-rabbit (diluted 1:200, #711-095-152; Jackson ImmunoResearch) immunoglobulin G.
Microscopy, image acquisition, and quantification
Stained slides were examined, in a blinded manner, with a Zeiss AxioVision fluorescence microscope (Carl Zeiss, Jena, Germany), and the raw images obtained in fixed exposition time as well as contrast/brightness settings were transferred to ImageJ (NIH, Bethesda, MD) and automated counts of cells were performed. Pre-processed, 8-bit, grayscale, single-channel images were processed through the Batch method using the customized “Analyze particles” function, or a customized macro that was prepared for each staining, and run on the raw files (multi-channel RGB, TIFF files). The parameters, which were adjusted in the macros before batch analysis includes: threshold (method: Huang); size (noise elimination); and Watershed function (nuclei counting). Each quantitative analysis was based on a minimum 3 mice, five sections, and 10 photographs per section containing a minimum of 4000 cells from the brain region. The percentage of cells positive for a specific marker was based on the total number of cells in the photographs from the region of interest (ROI). ROIs from TBI sections were selected to avoid the injury-induced cavity, obvious necrotic regions, as well as tissue handling-related artifacts.
Flow cytometry
Brain hemispheres from 5 C57BL/6 mice were mechanically and enzymatically dissociated. The crude cell suspension was passed through a 70-μm nylon cell strainer (BD Falcon; BD Biosciences, San Jose, CA) to remove all large fragments. Dissociated cells were washed twice in cold PBS and centrifuged for 10 min at 1000 rpm to remove debris. Suspensions of freshly isolated cells were fixed with 4% phosphate-buffered formaldehyde (Histolab AB) and 500,000 cells then stained for CD11b (conjugated with phycoerythrin [PE], #101208; BioLegend, San Diego, CA) and ST2 (conjugated with FITC, #101001F; MD Bioproducts, Zurich Switzerland). Unstained cells and cells stained with appropriate isotype controls for each of the antibodies used served as controls. Both stained cells and controls were analyzed in a BD LSR II flow cytometer (BD Biosciences, Oxford, UK), with 10,000 events in a window gated for the intersection of CD11b and ST2 staining being counted. For the analysis, we used scatter gating to remove the debris as well as small, pyknotic cells that were forward-scattered light (FSC) small and side-scattered light complex. Cell clumps were also eliminated using pulse geometry gating (FSC-HxFSC-A).
Cytokine/chemokine assay
To measure, in parallel, the relative levels of 36 selected cytokines and chemokines, the Proteome Profiler Mouse Chemokine Array Kit (ARY005; R&D Systems) was utilized in accord with the manufacturer's instructions. In brief, relative levels of mouse chemokines/cytokines after subjecting ST2-deficient mice and their WT Balb/c controls to stab wound injury were determined simultaneously from tissue lysates using a membrane-based sandwich immunoassay. Protein concentrations were established with a NanoDrop spectrophotometer (Thermo Scientific, Stockholm, Sweden) and equalized between lysates. Lysate samples were mixed with a cocktail of biotinylated antibodies and then incubated with the array membrane, on which were spotted capture antibodies to specific target proteins, in duplicate. Next, captured proteins were visualized with chemiluminescent detection reagents. Finally, positive signals were scanned, identified, and the pixel intensity of each spot was quantified with image analysis software.
Statistical analysis and image presentation
For statistical analysis, a post-hoc Tukey's multiple comparisons test and one-way analysis of variance were applied. Microsoft Excel (Microsoft Corporation, Redmond, WA) was used to generate graphs. Images were processed and arranged in Adobe Photoshop CS5 (Adobe Systems, San Jose, CA).
Results
Expression of interleukin-33 in human brain after traumatic brain injury
In vivo MD data from the patient with DAI was used to characterize the neurochemical status of the focal brain tissue in which the IL-33 was analyzed. 19,20 MD glucose was above the low critical level of 0.8 mmol/L and below 6 mmol/L during the entire MD monitoring period, suggesting an optimal glucose supply for the injured brain. The energy-metabolic state on day 1 was characterized by increasing levels of MD lactate and pyruvate with a virtually normal MD-lactate/pyruvate ratio (LPR), suggesting hyperglycolysis. Day 2 was characterized as a nonischemic energy crisis with LPR markedly above the critical level (>40), slightly elevated MD lactate (>4 mmol/L), slightly lowered pyruvate (<120 μmol/L), and normal glucose (vide supra). Days 3–5 showed a normal energy metabolic state. MD glutamate and glycerol, used as routine biomarkers of cellular distress, that is, excitotoxicity and cell membrane degradation/oxidative stress, respectively, were also evaluated. Glutamate showed dramatically elevated levels (>>15 μmol/L) during days 1 and 2, strongly suggesting excitotoxicity, challenging the energy-metabolic situation and mitochondrial function (i.e., nonischemic energy crisis). Glycerol showed fluctuating levels above the critical level (100 μmol/L) during days 2–5, suggesting ongoing cell membrane degradation/oxidative stress. 19,21 MD urea showed a stable and slowly increasing level, typically observed in NIC patients owing to protein catabolism, suggesting proper MD catheter performance throughout the MD monitoring period. 17
Serial samples of brain microdialysate collected at 12-h intervals during the first 5 days post-TBI contained markedly increasing levels of IL-33 with a peak at 72 h (Fig. 1A). The initially high levels of the proinflammatory cytokines IL-6 and RANTES declined during 5 days post-injury (Fig. 1B,C). The level of TNF-α was detectable and stable (Fig. 1D), whereas levels of SCF and SDF increased during the first days post-TBI, reaching peaks at 60 and 48 h, respectively (Fig. 1E,F).
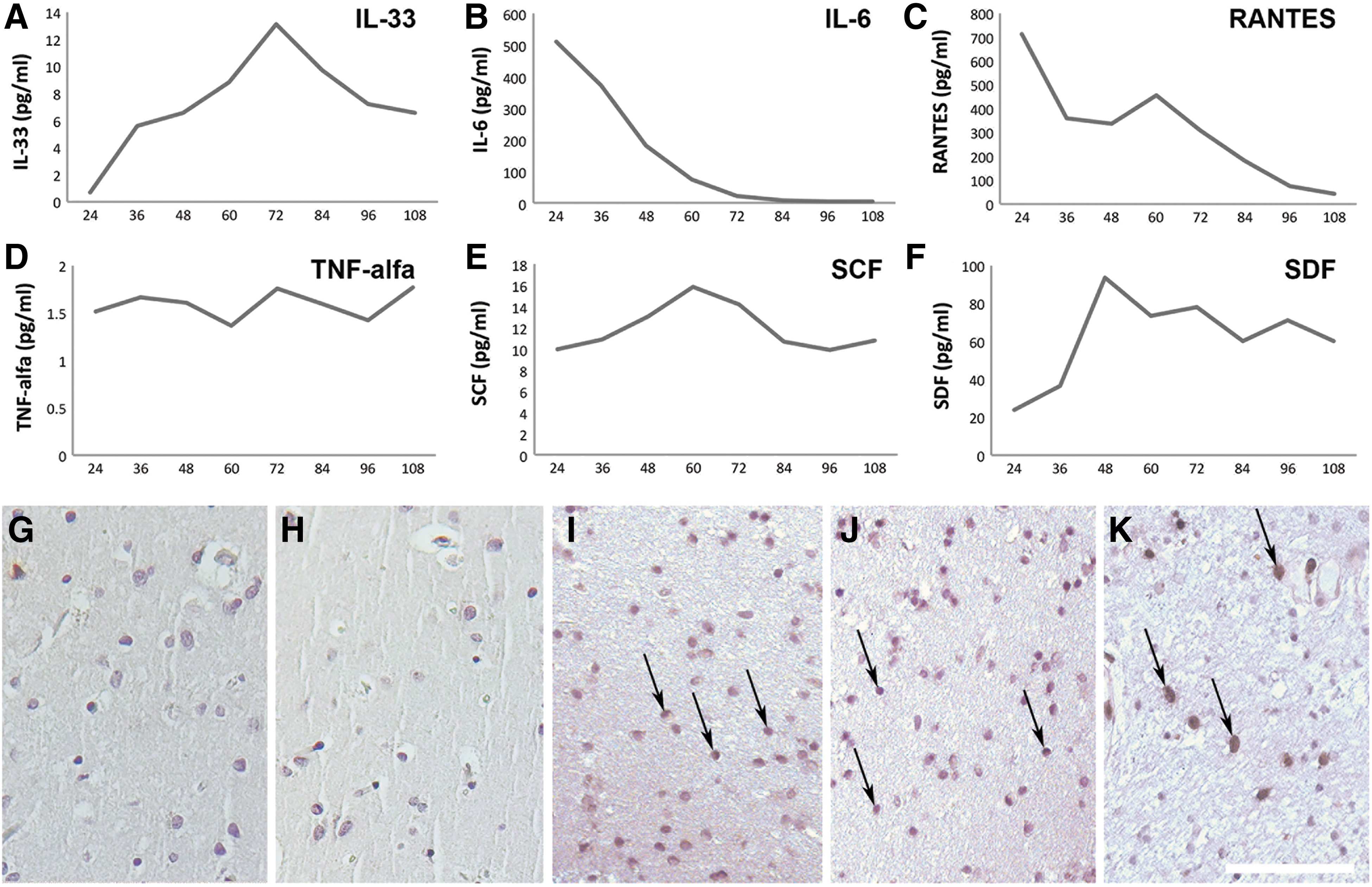
IL-33 is up-regulated in the human brain following TBI. The levels of (
To verify the expression of IL-33 upon TBI, we used IHC staining of sections of the frontal cortex from patients with three different causes of severe TBI (a motor vehicle accident, a fall, and an individual who was struck by an object). As expected, noninjured human brain revealed no or only weak immunoreactivity to IL-33 (Fig. 1G,H). Post-TBI, a large and intensely stained population of IL-33-expressing cells was observed in a section from the frontal lobe (Fig. 1I–K). IL-33 staining was localized primarily to the nucleus with some additional staining observed in the cytoplasm as well.
Expression of interleukin-33 following traumatic brain injury in mice
Having demonstrated that IL-33 is expressed in patients with TBI, we investigated IL-33 expression in mice post-CCI. CCI produced massive primary damage (Fig. 2A and inset) that was augmented during the week post-CCI, as indicated by extensive disruption of blood vessels followed by accumulation of cells from the blood in the affected hemisphere (Fig. 2A). Within a few weeks, the region of direct injury was characterized by cavitation and glia scar formation. No staining for IL-33 was detected in brains not subjected to injury (Fig. 2B). As in the human subjects, mouse brain subjected to TBI exhibited high expression of IL-33 by cells in the entire affected hemisphere with distinctive staining in dorsomedial cortex (Fig. 2C) as well as lateral cortex (Fig. 2E). Nuclear IL-33 appeared already at the 6-h time point post-injury and which remained for more than 1 week. Quantification revealed that IL-33 expression was highest at 3 days post-TBI (Fig. 2D).
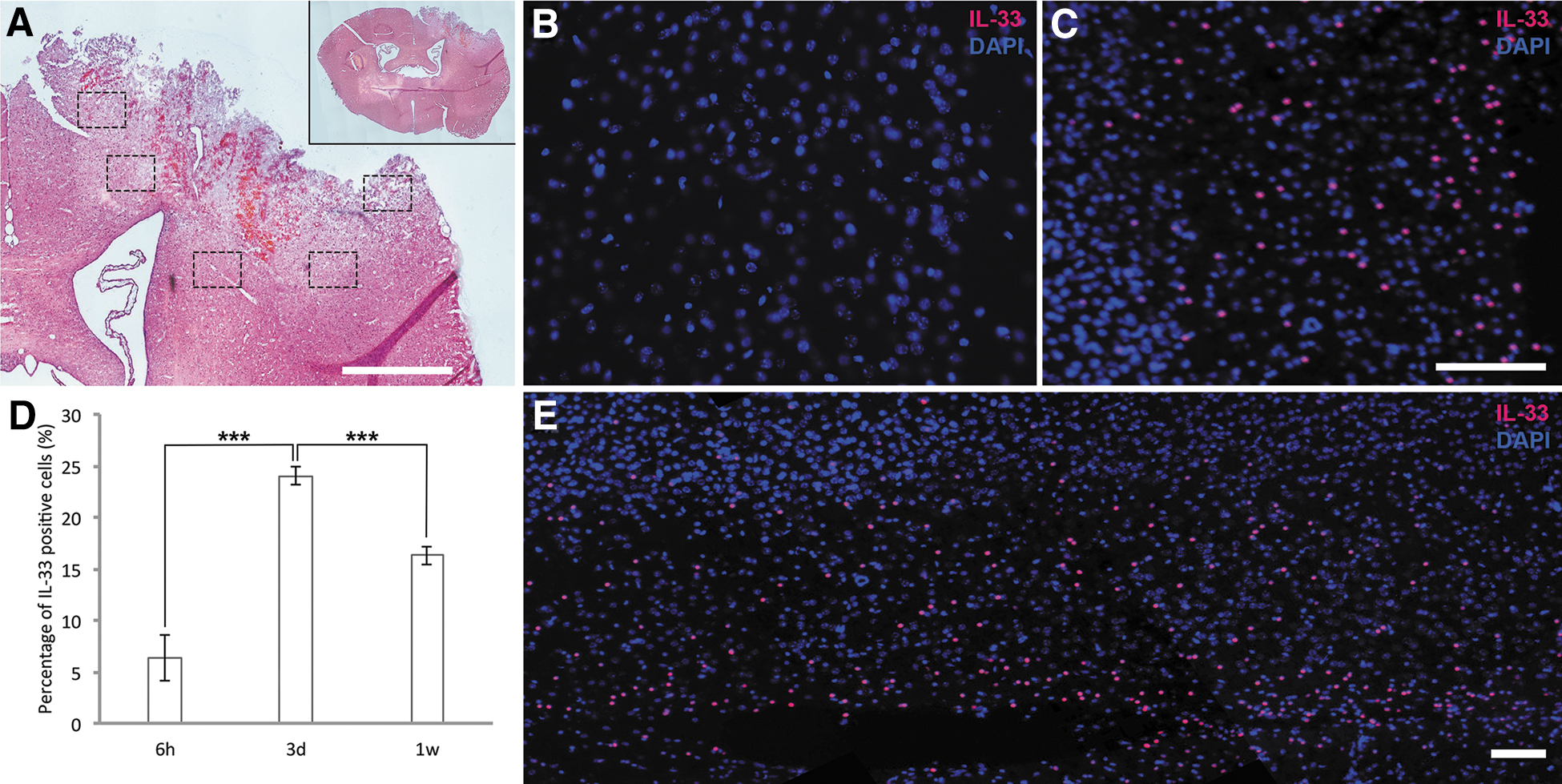
Expression of IL-33 after controlled cortical impact (CCI) in mice. (
Expression of interleukin-33 after sciatic nerve ligation
No expression of IL-33 was observed in the normal sciatic nerve included as a control for injury in the peripheral nervous system (Supplementary Fig. 1A) (see online supplementary material at
Identification of the interleukin-33-positive cells in the brain after traumatic brain injury
We next asked what type of cells produced IL-33 post-TBI in the mouse and therefore investigated the major CNS cell types. Double labeling with cell-type–specific markers was analyzed and verified with confocal imaging. Labeling with antibodies against the astrocytic marker, GFAP, or oligodendroglial markers, CNPase and Olig2, revealed double staining of IL-33-positive cells in the injured cerebral cortex with these markers (Fig. 3A–C, arrows). Moreover, IL-33-reactive cells did not stain positively for any of the following cell lineages: neuronal markers NeuN or Map2 (Fig. 3 D,E); the microglial/macrophage marker, Mac2 (Fig. 3F); the progenitor marker, nestin (Fig. 3G); or the blood vessel marker, CD31 (Fig. 3H). We therefore conclude that IL-33-positive cells in the injured brain were of macroglial origin.
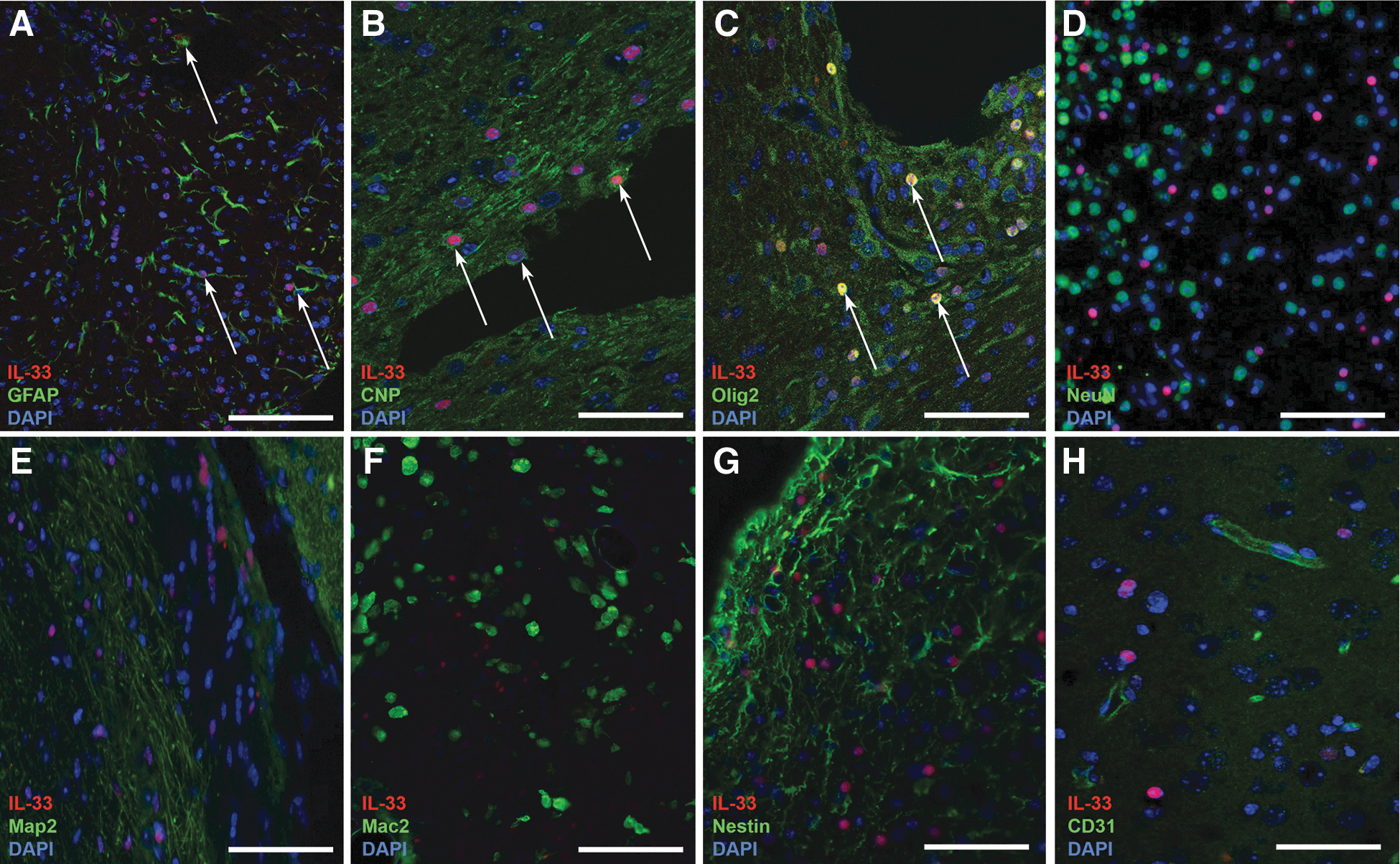
Identification of the IL-33-positive cells in the brain post-TBI. Immunostaining was performed to characterize IL-33-positive cells (red) in the cerebral cortex of mice subjected to TBI by CCI using cell lineage markers (green). DAPI nuclear stain (blue). (
ST2-deficiency influences the level of interleukin-33 expression in the brain of mice following stab wound injury
Next, we aimed to investigate whether ST2, the receptor to IL-33, was involved in the cellular response to brain trauma. Given that CCI resulted in massive tissue destruction with extensive necrotic regions, the stab wound injury model was used for further analysis and quantification of various parameters connected with the expression of IL-33 after a somewhat less-destructive version of TBI. Figure 4A and its inset display coronal sections through the mouse brain after stab wound of a WT mouse. We used both ST2-deficient and WT mice, and first investigated whether they exhibit similar IL-33 expression after such injury (Fig. 4C,F). In both cases, the expression was limited to the region of injury (cerebral cortex) and no IL-33 expression was detected in the control brains not subjected to TBI (Fig. 4B,E). Both WT and ST2-deficient mice demonstrated high percentages of IL-33-positive cells in the injured cerebral cortex 6 h and 3 days later, but 1 week post-injury, this percentage was 2-fold higher in ST2-deficient mice (Fig. 4D).

Expression of IL-33 in cerebral cortex of wild-type and ST2-deficient mice subjected to TBI by stab wound injury. (
ST2 deficiency reduces the number of infiltrating microglia/macrophages in response to traumatic brain injury
To determine whether the lack of ST2 would change the amount of key cell lineages in the injured brain, we performed immune staining with markers for astrocytes, neurons, and microglia/macrophages. Quantification revealed no significant difference in the numbers of GFAP-positive cells (Fig. 5A) in brains of ST2-deficient and WT mice at any of the time points analyzed, suggesting that reactive astrocytes are not influenced by the IL-33/ST2 pathway. Furthermore, the numbers of NeuN-positive neurons were similar between the two genotypes 1 month post-injury (Fig. 5B), a time point when the acute inflammatory phase has mostly subsided. Interestingly, the number of infiltrating microglia/macrophages in the ST2-deficient brains were significantly reduced after 1 week compared to WT, becoming only half as many as the number in injured hemisphere of WT brains (Fig. 5C).
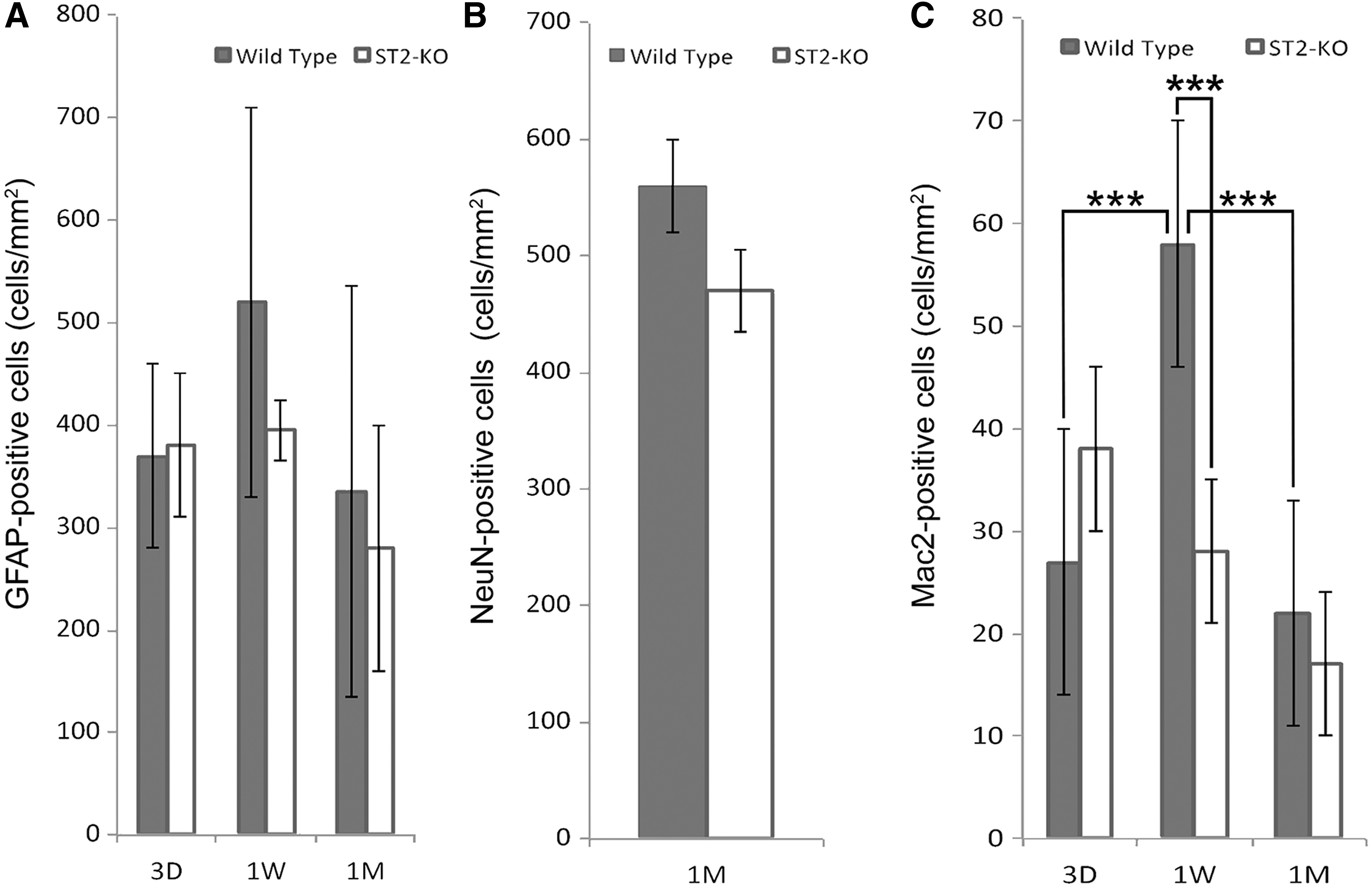
Alterations in numbers of glia and neurons in cerebral cortex of ST2-deficient and wild-type mice following stab wound injury. (
Microglia express the interleukin-33 receptor ST2
Because upregulation of IL-33 and the altered microglial/macrophage response in the brain post-TBI indicated that these cells might be the targets for IL-33, we analyzed expression of ST2 in naïve microglia utilizing flow cytometry. Indeed, 94% of the population of microglia isolated freshly from normal mouse brain was found to express this IL-33 receptor (Fig. 6).

Microglia express the IL-33 receptor, ST2. Flow cytometry analysis of microglia freshly isolated from normal mouse brain reveals that 94% of the CD11b-positive naïve microglia express the IL-33 receptor, ST2. FITC, fluorescein isothiocyanate; IL-33, interleukin-33; PE, phycoerythrin.
Differences in the cytokine/chemokine profiles in the brain of ST2-deficient and wild-type mice after stab wound injury
Differences between responses of ST2-deficient and WT mouse brain subjected to TBI were analyzed based on an array of 40 different cytokines and chemokines. Quantitative array analysis 3 days after stab wound injury revealed significant differences in several pro- and anti-inflammatory factors. Levels of chemokine (C-X-C motif) ligand (CXCL) 10/IP10 (interferon gamma-induced protein 10), M-CSF (macrophage-colony-stimulating factor), TNF-α, chemokine (C-C motif) ligand (CCL) 1/TCA-3 (T-cell activation gene 3), complement component 5 (C5)/C5a, and CXCL9/MIG (monokine induced by gamma interferon) were all lower in brain homogenates from ST2-deficient mice, whereas the level of chemokine CCL11/eotaxin-1 was higher in these homogenates (Fig. 7). No differences in expression of the other 33 cytokines or chemokines analyzed were observed.
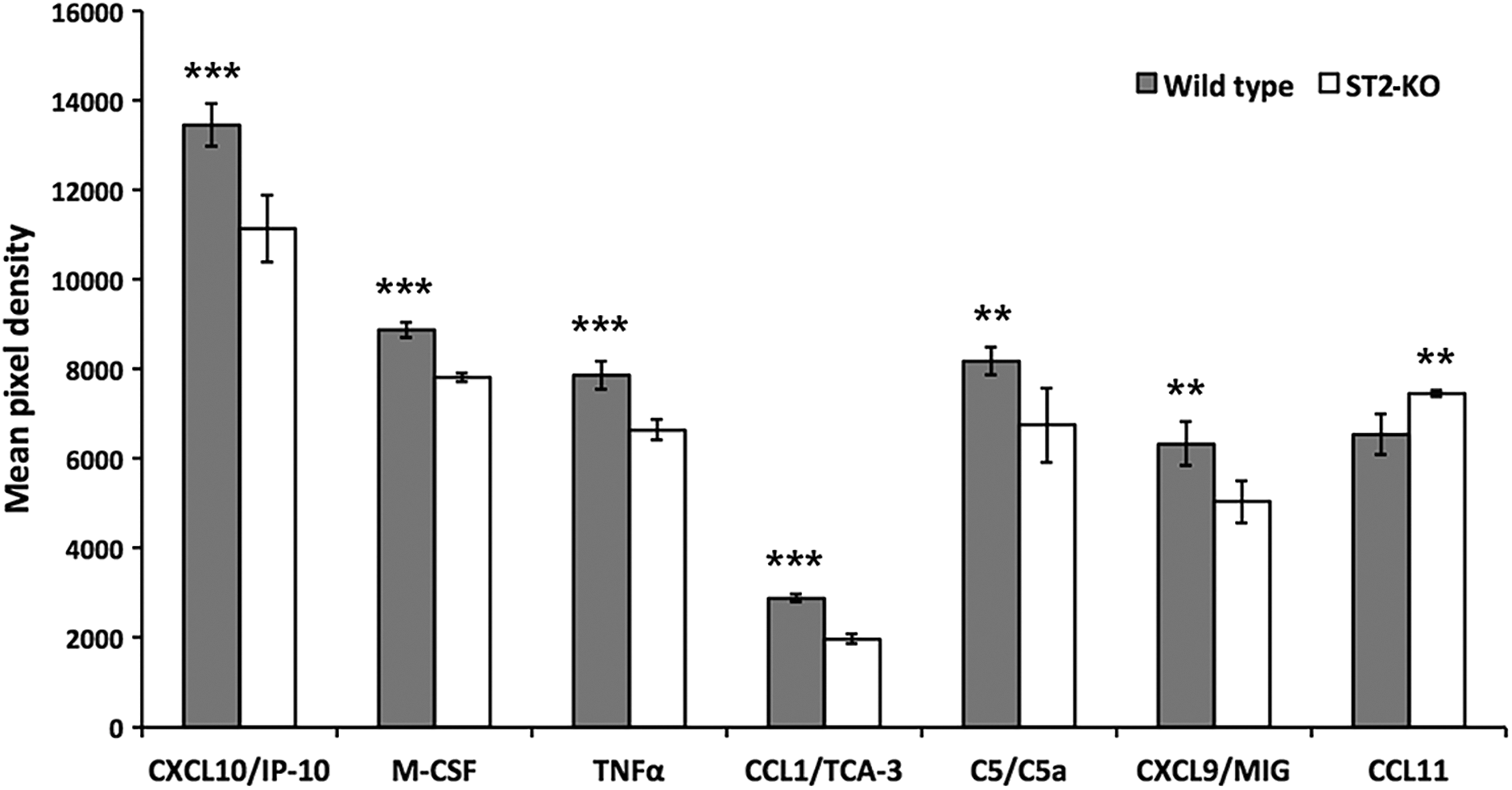
Cytokine/chemokine profiles in brain of ST2-deficient and wild-type mice after stab wound injury. Chemokine/cytokine profiles were analyzed by the Proteome Profiler Mouse Chemokine Array Kit (ARY005; R&D Systems, Stockholm, Sweden) in brain lysate from wild-type (gray bars) and ST2-deficient mice (open bars) 3 days after stab wound injury. ***p < 0.001 or **p < 0.01. C5, complement component 5; CCL, chemokine (C-C motif) ligand; CXCL, chemokine (C-X-C motif) ligand; IP-10, interferon gamma-induced protein 10; M-CSF, macrophage-colony-stimulating factor; MIG, monokine induced by gamma interferon; TCA-3, T cell activation gene 3; TNF-α, tumor necrosis factor.
Discussion
Although it is known that IL-33 expression is increased in response to various injurious stress or inflammatory stimuli, 11,12 the role played by this interleukin in trauma to the brain has not been previously investigated. Our present findings reveal that post-TBI, IL-33 is expressed at high levels in the brain with a pattern that corresponds to the neuroinflammatory response. Post-injury expression of both IL-33 and proinflammatory cytokines reaches a maximum after 3 days followed by a plateau or decline. Moreover, our observations here provide evidence that acting through its ST2 receptor, IL-33 influences the behavior of microglia/macrophages post-TBI.
In microdialysis samples from a patient suffering from severe diffuse TBI, we found that out of the proinflammatory chemokines/cytokines analyzed, IL-33 exhibit the most pronounced increase in expression at 3 days post-injury. By staining brain tissue samples from 3 additional TBI cases, we confirm that patients suffering from TBI strongly upregulate IL-33 immunoreactivity compared to control brain tissue. This is in line with a recent observation of expression of IL-33 in samples taken 26 h after murine SCI, 13 also indicating a role for IL-33 in the early phase of the immune response.
In the mouse, others have reported upregulation of IL-33 in glial cells after trauma to the spinal cord. 12 Moreover, Gadani and colleagues 12 found strongest expression of IL-33 in myelinated regions of the intact CNS, suggesting differences in the regulation and/or function of IL-33 in various regions of this system.
To explore the function of IL-33 post-TBI, we used two different mouse models (i.e., CCI and stab wound injury). The CCI causes severe damage to the ipsilateral hemisphere with a focal cortical contusion leading to a large cortical cavitation with widespread subcortical astrocyte activation and inflammatory cell infiltration. 2,23,24 In contrast, the injury caused by a stab is limited to a small portion of the ipsilateral cortex, but both injuries are accompanied by neuroinflammation. Remarkably, we observed a dynamic response of IL-33 expression in the traumatized mouse brain that was similar to that in human microdialysis samples, with an increased expression peaking at 3 days post-injury. In agreement with the data from cultured glia, 14 tissue from multiple sclerosis patients 25 and the injured rodent spinal cord, 11 we found that IL-33 expression is found in astrocytes where it is concentrated in the cell nucleus, but here we also detected oligodendrocytes displaying IL-33 immunoreactivity post-TBI. Our data agree with those observed for SCI, 13 where a subpopulation of oligodentrocytes in the traumatized area was also found to express IL-33. In contrast, we could not confirm a previous report on expression of IL-33 by spinal cord neurons, 26 suggesting possible differences between various parts of the CNS. Therefore, we propose that astrocytes and oligodendrocytes are the main sources of IL-33 in the injured brain that can act as immune response modulators to target cells that express the IL-33 receptor, ST2.
Signaling by IL-33 involves the ST2 receptor and interleukin-1 receptor accessory protein IL-1RAcP. Here, we found that the vast majority of microglia in normal mouse brain expresses ST2. In addition, brains of ST2-deficient mice exhibited fewer microglia, but more-potent upregulation of IL-33, than WT mice post-TBI, suggesting a deregulated feedback by abrogated IL-33/ST2 signaling. Our present findings indicate that microglia are the primary target cell in the brain for IL-33 post-TBI through ST2 receptor expression. This is seemingly in line with the observation that microglia activation peaks at day 7, beyond the peak of IL-33 expression in this CCI model. 2 It should be noted that differentiating between resident microglia and peripherally derived monocytes/macrophages post-TBI is difficult, and the infiltrating cell population positive for Mac2 post-injury, undoubtedly, consists of both microglia and macrophages. Given that macrophages have previously been shown to express ST2, 22 they add to the immune cells capable to respond to IL-33 post-TBI.
Onset of TBI, as mentioned above, involves an influx of cells from the periphery into the area of injury. 2 IL-33 has a multifunctional role in immune regulation, stimulating circulating immune cells such as macrophages, CD8+, CD4+ T cells, and others. 27 The lower expression of CXCL10/IP-10, CCL1/TCA-3, TNF-α, C5/C5a, M-CSF, and CXCL9/MIG and increase in CCL11/eotaxin-1 observed here in ST2-deficient compared to WT mice might explain the differences in microglia/macrophage number directly, but these molecules might also interact indirectly or synergistically with IL-33. It has been reported previously that CCL1, CXCL9, CXCL10, and TNF-α are potently expressed post-injury and play crucial roles in glial reactivity during the first days post-TBI. 28,29 Moreover, CCL1 and TNF-α enhance the chemotaxis, motility, and proliferation of microglia. 28 –30 Additionally, CXCL10/IP10 mRNA and protein expression are dramatically upregulated (>10-fold) early post-TBI in the present CCI model 31,32 and in microdialysis samples from severe diffuse TBI patients, 33 respectively, suggesting an important role in the neuroinflammatory respone and injury progression post-TBI and other neurodegenerative disorders. 32,34
Although it is believed that IL-33 mainly functions as a DAMP, that is, released from necrotic/injured cells to activate an inflammatory response, it can also be secreted in response to certain stimuli to act with a cytokine function. For example, astrocytes have been shown to secrete IL-33 in response to TNF. 35 Although IL-33, as a DAMP, has similar activities as secreted cytokine IL-33, the mechanism for its secretion is still not deciphered and it is unknown under which conditions IL-33 secretion (cytokine function) occurs.
Taken together, our present findings indicate that IL-33 plays a role in response to TBI, and that further exploration to alter the IL-33/ST2 pathway could suggest avenues to modulate neuroinflammation subsequent to these injuries.
Footnotes
Acknowledgments
This study was supported by grants from the Swedish Research Council.
The authors thank Mrs A. Hermansson for excellent technical assistance, and the personnel at the Animal Facility for animal care. Imaging was performed with support of the Science for Life Lab BioVis Platform, Uppsala. Prof. M. Ingelsson is acknowledged for his work with the human tissue biobank. Anonymized human brain tissue samples were supplied with support from Clinical Pathology and Cytology, Uppsala University Hospital.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.