
Abstract
Traumatic brain injury (TBI) was shown to impair pressure-induced myogenic response of cerebral arteries, which is associated with vascular and neural dysfunction and increased mortality of TBI patients. Hypertension was shown to enhance myogenic tone of cerebral arteries via increased vascular production of 20-hydroxyeicosatrienoic acid (HETE). This adaptive mechanism protects brain tissue from pressure/volume overload; however, it can also lead to increased susceptibility to cerebral ischemia. Although both effects may potentiate the detrimental vascular consequences of TBI, it is not known how hypertension modulates the effect of TBI on myogenic responses of cerebral vessels. We hypothesized that in hypertensive rats, the enhanced myogenic cerebrovascular response is preserved after TBI. Therefore, we investigated the myogenic responses of isolated middle cerebral arteries (MCA) of normotensive and spontaneously hypertensive rats (SHR) after severe impact acceleration diffuse brain injury. TBI diminished myogenic constriction of MCAs isolated from normotensive rats, whereas the 20-HETE-mediated enhanced myogenic response of MCAs isolated from SHRs was not affected by TBI. These results suggest that the optimal cerebral perfusion pressure values and vascular signaling pathways can be different and, therefore, should be targeted differently in normotensive and hypertensive patients following TBI.
Introduction
T
TBI was shown to impair the autoregulation of cerebral blood flow (CBF), which is associated with increased mortality and worse outcome of TBI patients. 4 –9 Autoregulation of CBF is an important homeostatic mechanism, which maintains stable cerebral perfusion and, therefore, cerebral volume when systemic blood pressure changes. 10,11 Pressure-induced myogenic response of cerebral arteries and arterioles plays a central role in autoregulatory function by coupling cerebrovascular resistance inversely to the changes in intraluminal pressure. 12 It has been demonstrated that TBI impairs myogenic mechanisms of cerebral arteries both in humans 9 and laboratory animals. 13 –15 Consequently, when blood pressure drops, the injured brain is exposed to hypoperfusion and ischemia as a result of inefficient vasodilation; on the contrary, when pressure increases, inefficient vasoconstriction leads to an increase in intracranial volume and pressure, thus exacerbating disruption of the blood–brain barrier (BBB) and formation/expansion of hemorrhages and cerebral edema. 16,17 Both hypotension and hypertension are associated with increased mortality in TBI patients. 18 –20
Pre-existing comorbid conditions lengthen hospital stay and increase mortality in TBI, most likely by exacerbating secondary injury of brain tissue. 21 Hypertension is the most common cardiovascular risk factor in TBI patients, 22,23 and has a substantial effect on cerebral myogenic autoregulation. 24,25 In chronic hypertension, myogenic constriction of cerebral resistance vessels is enhanced as a result of the hypertension-induced increased cerebrovascular production of the vasoconstrictor 20-hydroxyeicosatrienoic acid (20-HETE). 24 –26 The increased pressure-induced constrictor response of cerebral vessels shifts CBF autoregulation toward high pressure values. 24,25 These hypertension-induced changes of myogenic function of cerebral vessels have an important role in protecting the brain from pressure/volume overload, and at the same time they lead to increased susceptibility to hypotension-related cerebral ischemia; both of which may potentiate the consequences of TBI. Importantly, to our best knowledge, it is not known how brain trauma affects hypertension-induced 20-HETE-mediated increased myogenic constriction of cerebral arteries.
To test the hypothesis that in chronic hypertension, the enhanced myogenic cerebrovascular response is preserved after TBI, we examined the effect of severe brain trauma on myogenic properties of cerebral arteries in an established animal model of chronic hypertension, providing basis for further translational studies on cerebrovascular effects of TBI in hypertensive patients.
Methods
TBI in normotensive and hypertensive rats
All procedures were approved by the Institutional Animal Use and Care Committee of the University of Pecs Medical School and the National Scientific Ethical Committee on Animal Experimentation, Hungary (BAI/35/51-107/2016), and in accordance with the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines. Spontaneously hypertensive rats (SHR) (300–350 g, n = 12) and age-matched normotensive Wistar Kyoto rats (WKY) (300–350 g, n = 12) were purchased from Charles River Laboratories (Wilmington, MA). Severe impact acceleration diffuse brain injury was induced by Marmarou's weight drop model. 18,27 In brief, with the animal under isoflurane (2%) anesthesia, the skull was exposed by a midline incision between lambda and bregma, and a steel disc was fixed with cement on the skull. A 450 g cylindrical weight was dropped from 1.5 m onto the disc, causing severe diffuse TBI to the animals. Mortality rate was ∼15%. Blood pressure was measured before further experiments in all groups of animals, using the tail cuff method, as we previously described. 26 Middle cerebral arteries (MCA) were isolated from animals that survived 24 h after trauma, for further in vitro experiments.
Assessment of pressure-induced responses in isolated MCA in normotensive and hypertensive rats after TBI
Previous studies demonstrated that TBI impairs myogenic constriction of isolated cerebral arteries (from both the ipsilateral and contralateral side of the brain) 24 h post-TBI, which may contribute to the development of secondary injury of brain tissue. 13,15 Therefore, pressure-induced diameter responses of isolated MCA of SHRs and WKYs were assessed using a pressure myograph 24 h after TBI.
Rats were anesthetized (IP pentobarbital sodium) and decapitated and the brains were removed and segments of MCAs were isolated using microsurgical technique under an operating microscope. 28,29 Then, the MCA segments were mounted onto two glass micropipettes in a custom-built organ chamber and pressurized to 80 mm Hg. The hydrodynamic resistance of the micropipettes was matched, and the inflow and outflow pressures were controlled and measured by a pressure servo-control system (Living Systems Instrumentation, Burlington, VT). Inner vascular diameter was assessed with a custom-built videomicroscopy system and continuously recorded using a computerized data acquisition system as reported. 15,16 All vessels stabilized their pressure-induced tone for 60 min in oxygenated (21% O2, 5% CO2, 75% N2) Krebs buffer (at 37°C).
Myogenic response of MCA was assessed by measuring changes in vascular diameter in response to stepwise increases (20 mm Hg steps, for 5 min each) in intraluminal pressure (from 0 to 140 mm Hg). To assess the role of 20-HETE in hypertension- and TBI-induced changes in the myogenic response, MCAs were incubated with the cytochrome P450 ω-hydroxylase inhibitor HET0016 (10−6 mol/L, for 30 min, purchased from Cayman Chemical Company, Ann Arbor, MI). Then, vascular responses to stepwise increases in intraluminal pressure were reassessed. HET0016 was previously reported to selectively inhibit the formation of 20-HETE by inhibiting CYP4A and CYP4F isoforms in renal microsomes isolated from spontaneously hypertensive rats (IC50: 35.2 nM) and in human kidney (IC50: 8.9 nM). 30
In additional experiments, to test the effect of hypertension and TBI on vascular reactivity to constrictor agonists, basilar arteries from the same animals were isolated and cut into rings, and isometric responses of the ring preparations to administration of the cerebrovascular constrictor U46616 (10−7 mol/L) using a DMT wire myograph, as previously described, were assessed. 31
At the end of each experiment, the passive diameter curves were obtained (0–180 mm Hg) in the presence of Ca2+-free Krebs buffer containing nifedipine (10−5 mol/L) to achieve maximal vasodilatation. Myogenic tone is defined as: [(DP−DA)/DP] × 100, where DP is the passive diameter and DA is the active diameter value of an isolated MCA at a given intraluminal pressure.
Quantitative real-time reverse transcription polymerase chain reaction (RT-PCR)
A quantitative real-time RT-PCR technique was used to analyze mRNA expression of the 20-HETE producing cytochrome isoforms Cyp4a1 and Cyp4a3 in MCAs of rats from each group. In brief, total RNA was isolated with the Pure Link™ RNA Mini Kit (Life Sciences, Carlsbad CA) according to the protocol suggested by the manufacturer. Samples were homogenized, and RNA was purified by ethanol treatment and eluted from the membrane. The total amount of RNA was determined by using NanoDrop (Thermo Scientific, Waltham MA). High Capacity cDNA kit was applied (Applied Biosystems, Foster City CA) to perform cDNA synthesis. For gene expression analysis, quantitative RT-PCR (qRT-PCR) was performed using SensiFast SYBR Green reagent (BioLine, Luckenwalde, Germany). Amplifications were run on ABI StepOnePlus system (Applied Biosystems, Foster City CA). StepOne software was used to analyze gene expressions, which was normalized to the 18S housekeeping gene. Based on the quality of the PCR reference curves, the 18S was chosen as a reference gene. The primer sequences are 18S forward: TTGCTGATCCACATCTG CTGG, reverse: ATTGCCGACAGGATGCAGAA; Cyp4a1 forward: AATGCTAACCCCAGCCTTCC, reverse: AGAGGAGTC TTGACCTGCCA; Cyp4a3 forward: CTGTAGCTTTTCCTCCAG ACTCCA, reverse: CAGTGGCTGGTCAGAGGTGAA. The amplification of PCR products was calculated according to the 2−ΔΔCt method.
Statistical analysis
Data were analyzed by t test and two-way analysis of variance (ANOVA) followed by Tukey post-hoc tests, as appropriate. A p value <0.05 was considered statistically significant. Data are expressed as mean ± SEM.
Results
Hypertension-induced increased myogenic constriction of cerebral arteries is intact after TBI
SHR with and without TBI had significantly (p < 0.05) higher systolic blood pressure values than sham-operated WKY and TBI rats (Fig 1A). We found that (in accordance with literature data and our previous studies 25,32 ) myogenic constrictions of MCAs of hypertensive rats were enhanced (p < 0.05) compared with normotensive WKY, and extended to high pressure values (Fig 1B). TBI impaired the myogenic response 13 –15 of MCAs of normotensive WKY rats as shown by significantly (p < 0.05) decreased pressure-induced constriction of vessels to stepwise increases in perfusion pressure compared with sham-operated normotensive controls (Fig 1B). In contrast, we found that myogenic responses of MCAs of hypertensive rats after TBI remained enhanced compared with arteries of both normotensive WKY and TBI rats, and that they did not differ from myogenic constrictions of MCAs from SHR (Fig 1B). Constrictor responses of basilar arteries to the thromboxane A2 agonist U46619 (10−7 mol/L) were not significantly different between the groups (Fig 1B inlet).
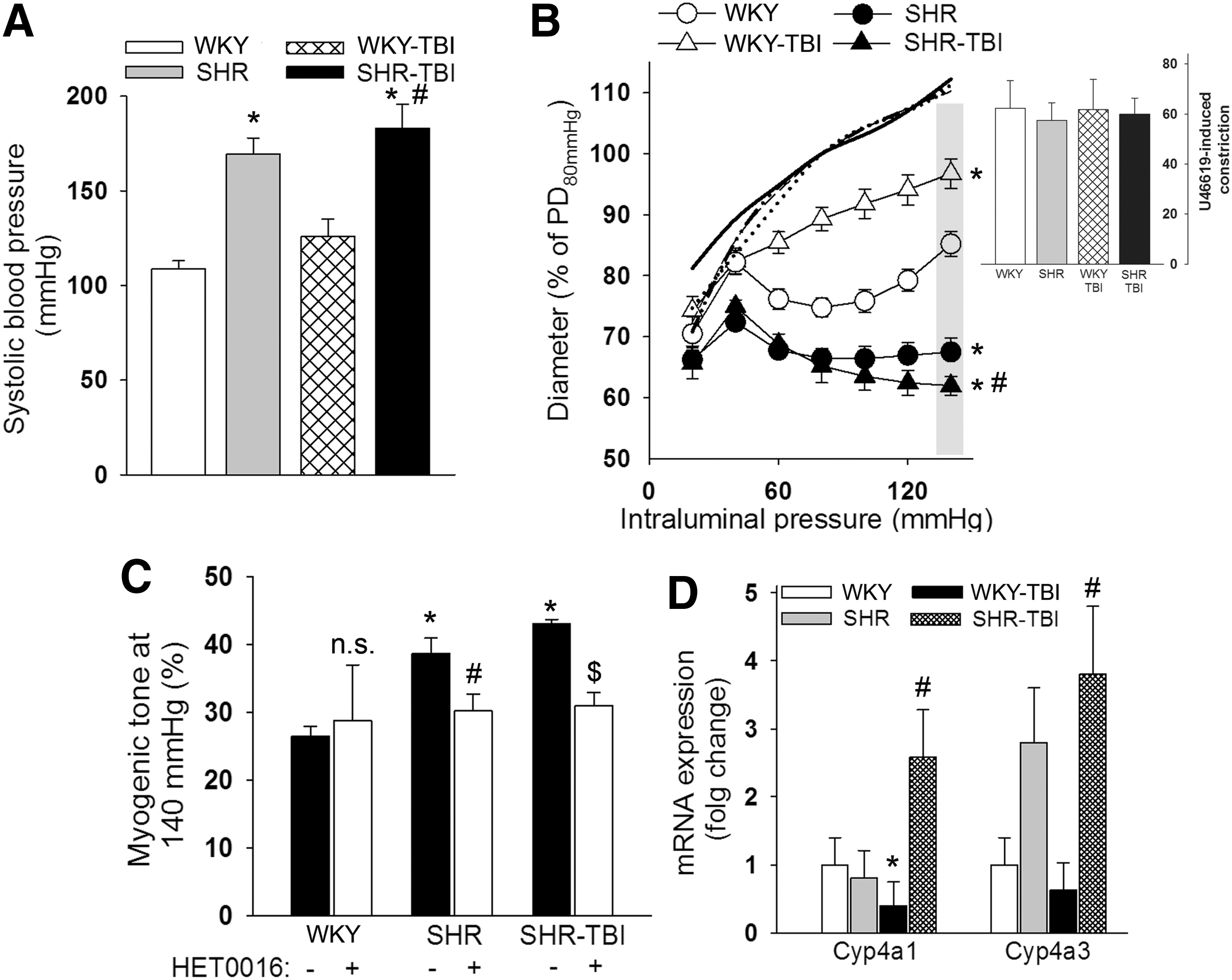
Hypertension-induced increased myogenic tone of cerebral arteries is not affected by traumatic brain injury: role of 20-hydroxyeicosatrienoic acid (20-HETE).
Role of 20-HETE in hypertension-induced increased myogenic tone of MCAs post-TBI
Enhanced myogenic constriction of cerebral arteries in chronic hypertension has been demonstrated to be mediated by increased production of the vasoconstrictor 20-HETE by hypertension-induced increased vascular expression of the 20-HETE producing cytochrome P450 isoforms. 24,25,33 Accordingly, we demonstrate here that the significantly increased myogenic response of MCAs from SHR at 140 mm Hg was inhibited and reversed to the level of myogenic tone in MCAs of normotensive rats in the presence of the specific 20-HETE synthesis inhibitor HET0016 (Fig 1C). Similarly to hypertensive animals without TBI, in SHR, after TBI myogenic tone of MCAs was significantly (p < 0.05) increased at 140 mm Hg compared with vessels from WKY and WKY-TBI rats (Fig 1C), and the enhanced myogenic tone was blocked by HET0016 (Fig 1C). The expression of the 20-HETE synthetizing cyp450 isoforms Cyp4a1 and Cyp4a3 tended to be or was significantly increased in the cerebral arteries of SHR and SHR after TBI compared with normotensive WKY and WKY-TBI animals, respectively. TBI significantly decreased the cerebrovascular expression of Cyp4a1 in normotensive rats (Fig 1D).
Discussion
TBI is a major health problem worldwide, because of its high mortality and the significant remaining disabilities in survivors. 1 –3 After the impact and consequent primary injury, secondary damage of brain tissue, which develops in days after trauma, is a significant determinant of the survival and outcome of patients. 34 TBI-induced cerebrovascular dysfunction plays a central role in the development of secondary brain injury, and is associated with both increased mortality and worse outcome of TBI patients. 4 –9 Previous studies suggest that TBI impairs myogenic mechanisms of autoregulation of CBF. 9,13 –15 Myogenic autoregulatory responses contribute to the central homeostatic mechanism maintaining relatively constant blood flow despite changes in systemic blood pressure. 10 –12 This is achieved by the pressure-induced myogenic responses of cerebral vessels: cerebrovascular resistance is inversely adjusted to changes in perfusion pressure by constriction or dilation of cerebral arteries and arterioles. In accordance with the literature, 15 we show here that TBI impairs myogenic responses of MCAs isolated from normotensive rats 24 h after trauma (Fig. 1B). The pathophysiological importance of this finding can be envisioned as followed: the lack of appropriate myogenic response following brain trauma could expose the brain to hypoperfusion when blood pressure drops, which could lead to cerebral ischemia. Also, in cases in which systemic blood pressure is elevated, the distal part of cerebral microcirculation is unprotected from high blood pressure and volume. 16,17 The latter could lead to or exacerbate BBB disruption, TBI-induced edema formation, and consequent development of intracranial hypertension. 25,35 Both hypotension and hypertension are associated with worse outcome of TBI patients. 18,19,23
Comorbid conditions increase the mortality of TBI and lengthen hospital stay, most likely by exacerbating secondary injury of brain tissue. 21 Pre-existing hypertension is the most common cardiovascular risk factor in TBI patients. 23 Chronic hypertension has a significant effect on myogenic autoregulation of CBF. As we demonstrate here (Fig. 1B) and in previous studies, 24,25,33 in chronic hypertension, myogenic curves are shifted to the right, resulting in decreased myogenic dilation at low pressure but enhanced myogenic constriction at high pressure values. On the one hand, the decreased myogenic dilation makes hypertensive animals and patients prone to cerebral ischemia when blood pressure is low; however, enhanced myogenic constriction protects the cerebral microcirculation from pressure and volume overload. 25 Here we demonstrate for the first time that – unlike in arteries of normotensive animals – TBI does not affect the enhanced myogenic responses of cerebral arteries of rats with chronic hypertension (Fig. 1B). In theory, the decreased capacity of cerebral arteries of hypertensive animals and patients would augment hypotension-related brain damage following TBI, but the brains of hypertensive subjects would be protected from severely elevated blood pressure (at least during the first days after TBI). This hypothesis and its therapeutic consequences and the possibility of maintaining enhanced myogenic response in the later phase following TBI have yet to be tested by future studies.
We demonstrated earlier that increased myogenic tone of hypertensive mice and rats is mediated by the hypertension-induced increased cerebrovascular production of the vasoconstrictor 20-HETE. 24 –26 Accordingly, hypertension induces the cerebrovascular hydfroxylase isoforms expression of the 20-HETE producing cytochrome P450 ω-hydroxylase. 24 –26 We show here that, similar to sham operated hypertensive rats, specific inhibition of production of 20-HETE reverses increased myogenic tone of MCAs in SHR after TBI to the level of normotensive control animals (Fig. 1C). This suggests that TBI does not affect hypertension-induced cerebrovascular production of 20-HETE, which underlies the sustained myogenic constrictions. Supporting this, we found that the 20-HETE producing cytochrome isoforms are upregulated in the MCAs of SHR and SHR after brain trauma, as well (Fig. 1B).
The mechanisms through which 20-HETE mediates cerebral myogenic constriction in hypertension and TBI are likely multifaceted. The vasodilator calcium activated potassium (BKCa) channels are negative regulators of myogenic constriction activated by high pressure, 36 and have been demonstrated to contribute to the hypertension-induced increase in the myogenic tone of cerebral arteries. 24 Because 20-HETE inhibits the activation of BK channels, 37 this pathway is a plausible candidate to mediate the downstream vascular effects of 20-HETE in TBI and hypertension. This hypothesis is further substantiated by the findings that activation of BKCa channels contributes to the decreased myogenic constriction of cerebral arteries after TBI. 15 20-HETE-dependent activation of transient receptor potential channels (such as transient receptor potential cation channel, subfamily C [TRPC6]), which has also been demonstrated to increase cerebral myogenic tone in hypertension, represents another possible mechanism of increased cerebrovascular tone in hypertension and TBI. 24 These possibilities should be examined by future studies.
Limitations and perspectives of the study
In the present article, we did not compare survival and outcome of normotensive and hypertensive animals following TBI, which should be done in future studies, with special focus on the effect of controlled hypotension and hypertension. We did not measure the effects of TBI on autoregulation in vivo, and findings on isolated vessels can only be carefully extrapolated to in vivo conditions. However, myogenic response of cerebral vessels is a central mechanism of CBF autoregulation, 25 and can be studied in vitro in a well-controlled manner without the confounding effects of neural, glial, and other (i.e., innervation) mechanisms. Gender differences may substantially affect hypertension-related changes of vasomotor mechanisms; therefore, future studies should establish the effect of TBI on myogenic responses of cerebral vessels in female animals as well. It has been demonstrated that mild, repetitive trauma impairs myogenic autoregulation of CBF as well, and contributes to TBI-induced chronic cognitive problems. 38 Therefore, future studies should examine the effect of mild repetitive brain trauma on the myogenic autoregulatory function of cerebral resistance vessels in hypertensive animals and patients as well. Also, our results should be verified in other animal models of TBI.
Conclusion
In conclusion, we show here for the first time that 20-HETE-mediated enhanced myogenic constriction of cerebral arteries caused by the presence of chronic hypertension is preserved after TBI. Extrapolating these findings to in vivo conditions, we propose that in hypertension following TBI, animals and patients are likely to be prone to ischemic damage when blood pressure drops to a greater extent than in normotension. In the same time, they are more protected from the damaging effects of severely elevated blood pressure. Accordingly, the optimal cerebral perfusion pressure values and vascular signaling pathways/mechanisms can be different and, therefore, should be targeted differently in normotensive and hypertensive patients following TBI.
Footnotes
Acknowledgments
This work was supported by grants from the Marie Curie Actions SMARTER 7th Framework Program of the European Union 606998 (to N.S. and A.K.), the Hungarian Academy of Sciences (the Bolyai Research Scholarship BO/00634/15 to P.T.), the PTE AOK-KA 3/2016 04.01/F (to P.T.), the Hungarian Brain Research Program (Grant No. KTIA_13_NAP-A-II/8 to P.T., E.C. and A.B.) and the Hungarian Scientific Research Fund (K108444 to A.K.).
Author Disclosure Statement
No competing financial interests exist.