
Abstract
Traumatic brain injury (TBI) may be a significant risk factor for development of neurodegenerative disorders such as chronic traumatic encephalopathy (CTE), post-traumatic epilepsy (PTE), and Alzheimer's (AD) and Parkinson's (PD) diseases. Chronic TBI is associated with several pathological features that are also characteristic of neurodegenerative diseases, including tau pathologies, caspase-3-mediated apoptosis, neuroinflammation, and microvascular alterations. The goal of this study was to evaluate changes following TBI in cleaved-caspase-3 and caspase-3-cleaved tau truncated at Asp421, and their relationships to cellular markers potentially associated with inflammation and blood–brain (BBB) barrier damage. We studied astrocytes (glial fibrillary acidic protein [GFAP]), microglia (ionized calcium-binding adapter molecule 1 [Iba1]), BBB (endothelial barrier antigen [EBA]), and activated microglia/macrophages (cluster of differentiation 68 [CD68]). We employed immunohistochemistry at different time points from 24 h to 3 months after controlled cortical impact (CCI) injury in rats, with particular interest in white matter. The study demonstrated that CCI caused chronic upregulation of cleaved-caspase-3 in the white matter of the corpus callosum. Increases in cleaved-caspase-3 in the corpus callosum were accompanied by accumulation of caspase-3-cleaved tau, with increasing perivascular aggregation 3 months after CCI. Immunofluorescence experiments further showed cellular co-localization of cleaved-caspase-3 with GFAP and CD68 and its adjacent localization with EBA, suggesting involvement of apoptosis and neuroinflammation in mechanisms of delayed BBB and microvascular damage that could contribute to white matter changes. This study also provides the first evidence that evolving upregulation of cleaved-caspase-3 is associated with accumulation of caspase-3-cleaved tau following experimental TBI, thus providing new insights into potential common mechanisms mediated by caspase-3 and underlying chronic TBI pathologies and neurodegenerative diseases.
Introduction
T
A wealth of clinical and experimental data indicates that neurodegenerative disorders share many common features, including inflammatory and neurovascular pathologies associated with irregular accumulation of different isoforms of tau protein, 13 blood–brain barrier (BBB) extravasation, 14 abnormal angiogenesis, and apoptosis. 15 Although limited, current data suggest that these pathologies might be also involved in chronic sequelae of TBI. 16 TBI has been reported to cause significant neuronal degeneration and total brain atrophy, which are associated with progressive white matter degradation that continues for at least 1 year following the initial injury. 17,18 The molecular and cellular mechanisms that trigger the development of these pathologies and their progression following brain injuries are poorly understood.
Recent clinical and experimental data indicate that caspase-3-mediated apoptosis plays a key role in cleavage of tau, an initial process underlying formation of fibrillary tangles and amyloid plaques that are commonly observed in AD and other neurodegenerative disorders. 13,19,20 Numerous studies also report that caspase-3 upregulation in neuronal and glial cells also contributes to TBI pathology in human patients 21,22 and in animal models, including controlled cortical impact (CCI) 23,24 (see also a review by Zhang and colleagues 25 ). Moreover, pre-clinical in vivo and in vitro studies have shown that caspase-3 is involved in the proteolytic degradation of several brain proteins contributing to TBI pathology. 26 –28 Clinical data demonstrate increased levels of several αII-spectrin breakdown products (SBDPs) in the cerebrospinal fluid of TBI patients for up to 5 days following injury, including 120 kDa αII-spectrin breakdown product (SBDP120), a specific proteolytic product associated with caspase caspase-3 activity. 29 Another important biomarker of TBI is 150 kDa αII-spectrin breakdown product (SBDP150), 29 –31 which has been associated with activities of both calpain and caspase-3 proteases. 32 Serum levels of caspase-3 and another important caspase-3-specific protein cleavage product, caspase-3-cleaved cytokeratin-18 (CCCK-18), have been associated with outcome in patients with severe TBI. 21,33 Tau is also a well-recognized substrate of caspase-3 proteolytic activity, which yields two specific cleavage products. 34 Increased levels of tau and several of its isoforms are considered hallmarks of neurodegeneration. 35,36 Numerous publications have suggested that caspase-3-cleaved tau in neurofibrillary tangles might be one of the earliest biomarkers of AD. 13,20,37,38 Recent studies have also demonstrated increased levels of caspse-3-cleaved tau in brain extracts of patients with CTE 39 and in serum of both AD and TBI patients 40,41 (see also review by Glushakova and colleagues 42 ) Rohn and colleagues suggest that activation of the caspase-3 pathway and the consequent cleavage of tau are early events in the tangle pathology of AD, 34 leading to formation of neurofibrillary tangles and amyloid plaques 37,43 two of the most recognized hallmarks of AD. Further, data indicate that increased caspase-3 activity and neuronal apoptosis appear in perivascular regions, an observation suggestive of involvement of caspase-3 in cerebrovascular injury. 44 Finally, experimental data suggest that caspase-3 activity contributes to neuronal death and loss of brain tissue and that the inhibition of this pathway may be an effective treatment strategy for TBI. 45
A major chronic pathological finding appearing in later periods after TBI is formation of extensive white matter lesions and delayed diffuse axonal injury. 46,47 Similar observations have been reported in animal models of TBI. 48 Moreover, our recently published pre-clinical data demonstrated previously unrecognized pathologies of chronic TBI such as delayed punctate BBB concurrent with formation of microbleeds similar to those often observed in neurodegenerative disorders. 49 Importantly, these pathologies were associated with neuroinflammation and white matter damage, and although recognized in the clinic, such TBI pathological manifestations have been rarely examined in pre-clinical studies. 48 Moreover, these previous studies did not focus on more detailed characterizations of the mechanisms underlying these delayed and enduring neurodegenerative processes following TBI. Here, we expand our earlier investigations to examine the role of caspase-3-mediated activation and accumulation of proteolytic fragments of tau in perivascular events, including BBB damage that could ultimately contribute to evolving white matter damage and delayed axonal degeneration in white matter. We focus on the corpus callosum, a region vulnerable to chronic pathophysiological pathways initiated by TBI. 49 We hypothesized that the chronic caspase-3-mediated activation and consequent tau cleavage following TBI may represent an unrecognized component of cellular and molecular cascades that mediate evolving post-traumatic neurodegeneration, which could eventually result in chronic neurodegenerative diseases. As an initial test of this hypothesis, we determined spatiotemporal profiles of changes in selected biomarkers associated with neurodegenerative diseases, focusing on markers of caspase-3 activation (cleaved-caspase-3 and caspase-3-cleaved tau) to address associations of regional and cellular changes in these biomarkers with pathological microvascular alterations.
Methods
Animals
All experimental procedures were approved by the University of Florida Institutional Animal Care and Use Committee (IACUC). Male Sprague–Dawley rats weighing 230–300 g (Harlan, Inc., Indianapolis, IN) were used. Rats were maintained in a specialized temperature- (20–24°C) and humidity- (30–70%) controlled facility on a 12 h light/dark cycle with access to a normal laboratory diet and potable water ad libitum, and were acclimated to these housing condition for at least 1 week before being used in the study. Animals used for brain immunohistochemistry were randomly assigned to control (sham) and CCI groups at varying time points following experimental TBI. Pre- and post-surgery pain management was maintained in all rats to ensure compliance with guidelines set forth by the University of Florida IACUC.
CCI
CCI brain injury was induced using the Benchmark™ Stereotaxic Impactor (MyNeurolab, USA) as we have previously described. 49 Briefly, all surgical procedures were performed with the animals under anesthesia induced with 4% isoflurane and maintained with 2.5% isoflurane in oxygen. Rats were placed in a stereotactic apparatus equipped with a temperature-controlled heating pad, and a 5 mm ipsilateral craniotomy was performed midway between bregma and lambda in the right parietal region, with the dura mater remaining intact. CCI was produced by a single impact using a cylindrical impactor with tip diameter of 4 mm. The CCI parameters were as follows: velocity 3.5 m/sec, compression distance 2.5 mm, and compression time 200 ms. The incision was closed using the Reflex™ Skin Closure System. Sham animals underwent anesthesia and all surgical procedures including craniotomy but not cortical impact. After surgery, rats were allowed to completely recover from anesthesia in a temperature-controlled recovery chamber and then were transferred to a housing facility.
Immunohistochemical analysis
Immunohistochemistry on paraffin-embedded coronal sections was performed as described previously 49,50 with minor modifications. Briefly, the rats were euthanized with a lethal dose of Beuthanasia-D solution and transcardially perfused with 4% paraformaldehyde. Immunohistochemical analyses were performed on paraffin-embedded 4 μm coronal brain sections attached to slides. The sections used for all immunohistochemical analyses were cut from a brain segment positioned within the midline plane of the impact site (between −3.8 mm and -2.8 mm from bregma). Two adjacent brain sections were placed on each slide. The brain sections from stereotactically corresponding regions were used in all animals. After deparaffinization, antigen retrieval with Trilogy solution (Cell Marque, Hot Springs, AR) and blocking of endogenous peroxide activity with 3% hydrogen peroxide, the sections were incubated overnight at 4° C with one or sequentially with several antibodies for single or multiple immunostaining experiments, respectively, followed by incubation with anti-species-specific corresponding secondary antibodies. The immunostaining was visualized with 3, 3'-diaminobenzidine (DAB) for brown color development, and sections were counterstained with hematoxylin (Dako, Carpinteria, CA). In this study, the following primary antibodies were used: (1) rabbit polyclonal anti-cleaved caspase-3 (Asp175) (Cat# 9661; Cell Signaling Technology, Danvers, MA); (2) mouse monoclonal anti-tau, caspase cleaved (truncated at Asp421) (Cat# MAB5430; EMD Millipore, Billerica, MA); (3) mouse monoclonal anti-rat BBB (endothelial barrier antigen [EBA]) (Cat #SMI-71R, BioLegend, San Diego CA); (4) goat polyclonal anti-Iba1 (Cat# ab5076; Abcam, Cambridge, MA); (5) mouse monoclonal anti-GFAP (Cat#3670; Cell Signaling Technology); (6) mouse monoclonal anti-macrophages/monocytes (cluster of differentiation 68 [CD68]) (Cat# MAB1435; EMD Millipore); (7) mouse monoclonal anti-NeuN (Cat# MAB377; EMD Millipore); and (8) mouse monoclonal anti-CNPase (Cat# ab6319; Abcam). The primary antibody dilutions were 1:500 or 1:1000, depending on applications.
For the quantitative immunohistochemical experiments, biotinylated secondary antibodies and avidin-HRP conjugate (LSAB+ kit, Cat #K0679, Dako, Carpinteria, CA) were used. For the immunofluorescence triple labeling experiments, the following species-specific secondary antibodies conjugated with Alexa Fluor dyes (all from Thermo Fisher Scientific, Waltham, MA) were used: (1) donkey anti-rabbit IgG/Alexa Fluor® 555 (Cat #A-31572) and (2) chicken anti-mouse IgG/Alexa Fluor® 488 (Cat# A-21200) and chicken anti-goat IgG/Alexa Fluor® 488 (Cat# A-21467). In all immunofluorescence experiments, 4',6-diamidino-2-phenylindole (DAPI) was used for visualization of nuclei. Following immunohistochemical and immunofluorescent staining procedures, the sections were washed with phosphate-buffered saline solution and deionized water and then cover-slipped with Faramount Mounting Medium (Dako, Carpinteria, CA) or SlowFade® Gold Antifade Mountant with DAPI (Thermo Fisher Scientific), respectively.
The slides with DAB-stained sections were scanned using the Aperio ScanScope GL (Aperio Technologies). Quantitative immunohistochemical analyses in a scanned slide image were performed using the ImageJ (National Institutes of Health, Bethesda, MD) software and were expressed as the numbers of immunopositive (or immunonegative) cells and other immunopositive structures (e.g., extracellular aggregates, puncta) per mm2. Morphological characteristics of cleaved-caspase-3 and cleaved caspase-3-cleaved tau were defined on the basis of counterstaining of specific DAB immunostaining for these antigens and nuclear stain hematoxylin. The intensity and visual appearance of the hematoxylin-stained cellular structures might be indicative of viable and dead or dying cells. In the viable cells, light hematoxylin staining represents round-shaped nuclear structure, whereas dead or dying cells can be distinguished clearly by condensed or fragmented nuclei, intensely hematoxylin-stained cytoplasm, and various degrees of cell body shrinkage. 51 Cleaved-caspase-3- and caspase-3-cleaved tau-immunopositive cells were identified by the presence of condensed DAB-stained material surrounding hematoxylin-stained nuclei or by co-localization of DAB with hematoxylin-stained structures, likely representing cells in different stages of the dying process. Immunonegative cells were identified by the absence of evident DAB staining within the cell body. Extracellular immunopositive material with punctate or diffuse appearance was distinguished by the absence of hematoxylin staining. In the corpus callosum, the same areas (600 × 250 μm) located within the interhemispheric fissure were used for quantification. In all experiments, quantitative analyses of the DAB-stained immunohistochemical sections were performed independently by two investigators blinded to animal groups (i.e., sham, surgery and time points). The analyses were performed in duplicate and mean values were used for final calculations and statistics. Triple immunofluorescence-labeled sections were examined using the Olympus IX70 Inverted Fluorescent Microscope equipped with a × 60 objective and QImaging Retiga 4000R Monochrome Camera with an RGB-HM-5 Color Filter, and analyzed using QImaging QCapture Pro 6.0 image analysis software.
Statistical analysis
For statistical analyses, a one way ANOVA followed by Newman–Keuls multiple comparison test or, where applicable, nonparametric one way ANOVA (Kruskal–Wallis statistic with Dunn's multiple comparison test) were used. Data were reported as mean ± standard error. P values <0.05 were considered significant. 52 Statistical analyses were performed using JMP (SAS Institute Inc., Cary, NC) and GraphPad (GraphPad Software Inc., La Jolla, CA) software.
Results
Cleaved caspase-3 and its cellular localization in the corpus callosum
Spatiotemporal profiles of caspase activation following TBI in the white matter of the corpus callosum showed persistent increases in cleaved-caspase-3 immunoreactivity in the corpus callosum for at least 3 months after CCI (Fig. 1A). Time-dependent changes in immunoreactivity, such as increases in number of cleaved-caspase-3-immunopositive cells and accumulation of cleaved-caspase-3 diffuse matter and puncta, were observed starting at 24 h after CCI and progressed over 3 months. Twenty-four hours following CCI and to a lesser extent in sham-injured rats, cleaved-caspase-3 was primarily co-localized with hematoxylin, suggesting its intracellular localization. In contrast, cleaved-caspase-3 puncta were observed only in CCI-injured animals and were prevalent at subacute and chronic time points. Cleaved-caspase-3 puncta were detectable as early as 24 h after CCI, and their numbers were increased at subsequent time points. Although cleaved-caspase-3-immunopositive cells were present to some degree in control rats, no extracellular aggregates and only negligible amounts of cleaved-caspase-3-immunopositive puncta were observed in these animals. In controls and predominantly at 24 h after CCI, cleaved-caspase-3-immunopositive cells were characterized by the presence of lightly stained cleaved-caspase-3 immunoreactivity around hematoxylin-stained cellular structures, suggesting the presence of cleaved-caspase-3 in the cytoplasm. In contrast, at subacute and chronic time points following TBI, cleaved-caspase-3 staining was more intense and was co-localized with hematoxylin, suggesting possible association of increased cleaved-caspase-3 upregulation with cell death processes. The latter observation is consistent with certain morphological features of cleaved-caspase-3-immunopositive cellular structures, such as an intense hematoxylin-stained cytoplasm containing shrunken cells, indicative of dead or dying cells seen in injured but not in control rats. In addition, we have observed cleaved-caspase-3-immunopositive material confined to small areas with no obvious co-localization with hematoxylin, suggesting the presence of cleaved-caspase-3-immunopositive “shadows” of dead cells or its extracellular aggregation.
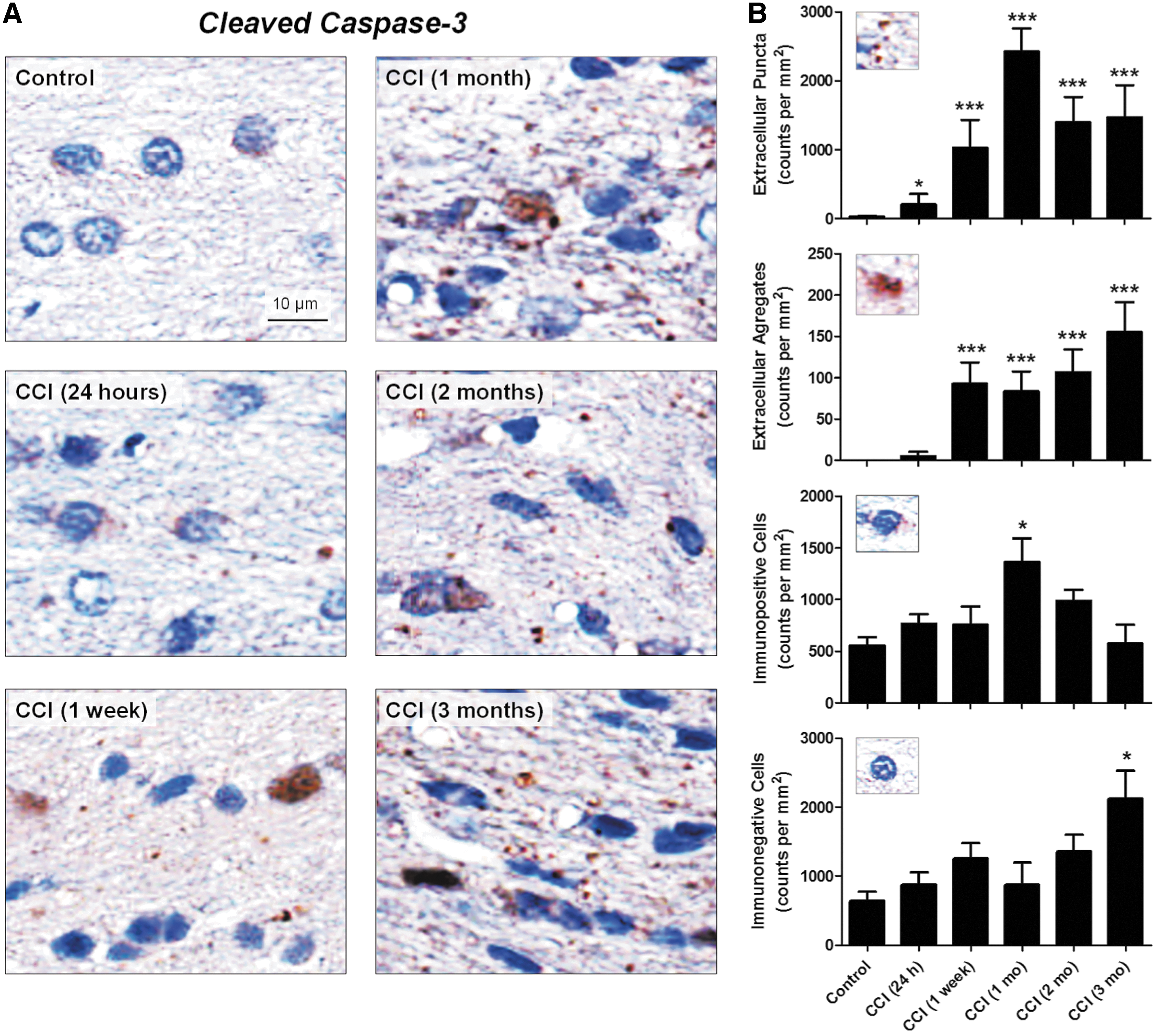
Morphology and temporal profile of cleaved-caspase-3 in the corpus callosum after experimental traumatic brain injury (TBI). (
Quantitative analyses (Fig. 1B) demonstrated that the number of cleaved-caspase-3-immunopositive cells peaked at 1 month after CCI, whereas extracellular cleaved-caspase-3-immunopositive puncta and extracellular aggregates were significantly increased starting at 24 h and 1 week after injury, respectively. Morphological characteristics of cleaved-caspase-3-immunopositive cells differed by intensity of DAB staining, with minimal staining around apparently viable nuclei in control animals and strong dense DAB staining of altered cellular structures overlapping with hematoxylin staining at chronic time points. A small but significant, increase in the number of immunoreactive puncta was observed as early as at 24 h after CCI, and the appearance of puncta preceded formation of cleaved-caspase-3-immunopositive extracellular aggregates. Interestingly, the increase in the punctate cleaved-caspase-3 immunoreactivity has a temporal profile similar to that observed for the numbers of immunopositive cells, a peak at 1 month after CCI. Although the cleaved-caspase-3-immunopositive cells at 3 months after experimental injury have an altered morphology, their number was not significantly different from the control value. However, the numbers of puncta remains significantly elevated up to the 3 month time point. In contrast, statistically significant increases in the numbers of cleaved-caspase-3-immunopositive extracellular aggregates were observed 1 week after CCI, and their maximal increase was observed at 3 months after experimental injury.
In addition, we analyzed the numbers of immunonegative cells and performed total cell counts, including both immunonegative and immunopositive cells (Fig. 1B). The results revealed that the numbers of immunopositive and immunonegative cells significantly increased at all time points, suggesting cellular proliferation of inflammatory cells such as astrocytes and microglia/microphages. 49 Further analyses have shown that at the 3 month time point, although the number of cleaved-caspase-3-immunopositive cells was not significantly different from the control, the increase in total cell number was the result of a significant increase in the number of cleaved-caspase-3-immunonegative cells (p < 0.05, n = 5–6 animals per time point) suggesting possible cellular reorganization of corpus callosum tissue caused by cell death and cellular proliferation processes.
Caspase-3 cleaved tau
To further investigate the physiological role of cleaved-caspase-3 upregulation after TBI, we performed immunostaining for one of the specific products of its proteolytic cleavage; that is, caspase-3-cleaved tau (truncated at Asp421) (Fig. 2). Increases in caspase-3-cleaved tau immunoreactivity in the corpus callosum of CCI-injured animals were observed starting at 24 h, and gradually increased during the 3 months following TBI (Fig. 2A). Only marginal caspase-3-cleaved tau immunoreactivity was observed in control animals. Caspase-3-cleaved tau presented as immunopositive material or aggregates with a diffuse appearance at early time points that became further condensed at chronic time points. Immunohistochemistry for caspase-3-cleaved tau counterstained with hematoxylin suggested both extracellular and cellular accumulations of caspase-3-cleaved tau around cell nuclei or dead cells, with prominent perivascular accumulation 3 months following CCI (Fig. 2B).

Temporal profile and morphological features of caspase-3 cleaved tau accumulation in the corpus callosum after experimental traumatic brain injury (TBI).
Cellular localization of cleaved-caspase-3 and caspase-3-cleaved tau processing in corpus callosum after TBI
To determine different cell types associated with caspase-3 activation, we performed a series of immunofluorescent co-localization studies using antibodies against cleaved-caspase-3 and cell-specific markers. In preliminary experiments, we performed immunostaining using neuronal (i.e., NeuN) and different glial cell-specific markers (i.e., GFAP [astrocytes], Iba1 [microglia], myelin basic protein [MBP, oligodendrocytes])
53
in naïve animals, to determine cell types present in the corpus callosum. These experiments confirmed that microglia and astrocytes are the prevailing cell types in the corpus callosum (Fig. S1) (see online supplementary material at
In addition, our preliminary experiments using immunohistochemistry for MBP and 2',3'-cyclic-nucleotide 3'-phosphodiesterase (CNPase), which labels the cell bodies of oligodendrocytes and the myelinated fibers,
53
in control and CCI-injured rats, suggest progressive loss of myelinated neuronal fibers and oligodendrocytes at chronic time points (Fig. S2), which is consistent with our previously published data demonstrating chronic white matter degeneration in the corpus callosum (see online supplementary material at

Co-localization of cleaved-caspase-3 with astrocytic and microglial markers after experimental traumatic brain injury (TBI). (
To further study possible involvement of glial cells in caspase-3-cleaved tau processing, we performed co-localization experiments using specific astrocytic and microglial markers. Immunofluorescent staining for caspase-3-cleaved tau revealed diffuse accumulation of immunopositive material as early as 24 h after CCI. Immunofluorescent co-localization experiments with caspase-3-cleaved tau and Iba1 revealed the presence of microglial cells in the areas showing caspase-3-cleaved tau accumulation. Counterstaining with DAPI further suggested that the perivascular accumulation of caspase-3-cleaved tau aggregates occurred around impaired microvessels, which was also especially evident at chronic time points (Fig. 4). Iba1-immunopositive microglia were present within both diffuse interstitial and perivascular caspase-3-cleaved tau accumulations. Some Iba1-positive structures, especially evident at the 24 h time point, overlapped with diffuse caspase-3-cleaved tau-positive material that somewhat resembled the shape of microglia (see cell examples in the excerpts marked with number [1] in Fig. 4), suggesting either intracellular or extracellular co-localization of these markers. Some Iba1-immunopositive cells at 24 h and most of the Iba1-immunopositive cells at 3 months after CCI were topologically congruent with condensed caspase-3-cleaved tau-positive material, suggesting possible intracellular co-localization of these markers (see cell examples in the excerpts marked with number [2] in Fig. 4). However, a significant amount of caspase-3-cleaved tau-positive material was located outside of Iba1-positive cellular structures, either surrounding DAPI-stained nuclei or located interstitially with a more diffuse appearance.
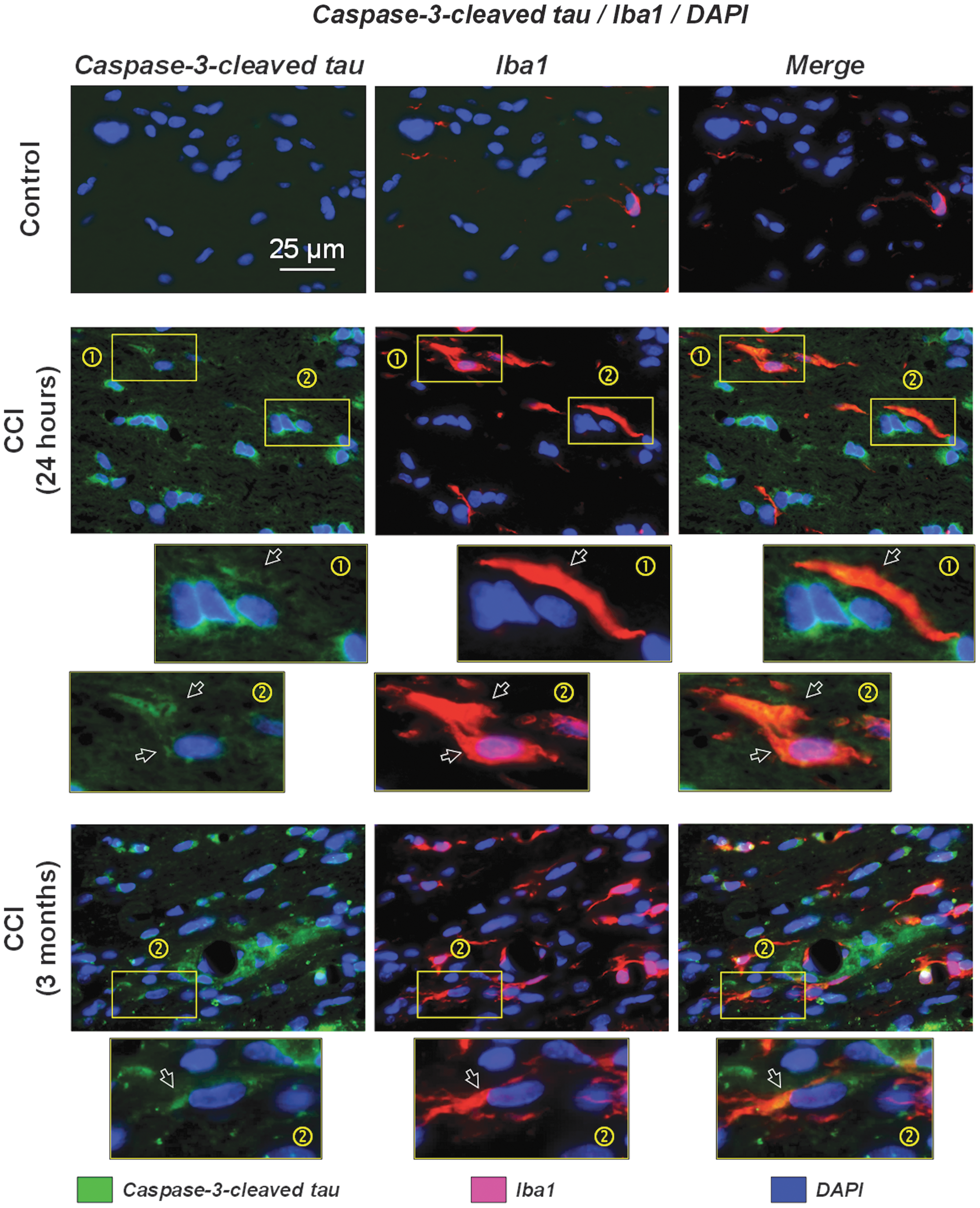
Co-localization of caspase-3-cleaved tau with ionized calcium-binding adapter molecule 1 (Iba1) (microglia) after experimental TBI. Representative photomicrographs of triple-labeled fluorescent immunostaining of brain sections from control and controlled cortical impact (CCI)-injured rats at 24 h and 3 months after experimental injury. Images show co-localization or close proximal association of caspase-3-cleaved tau (green) with microglial marker Iba1 (red) counterstained with nuclear marker 4',6-diamidino-2-phenylindole (DAPI) (blue). The immunofluorescent images suggest intracellular and extracellular proximal localization of caspase-3-cleaved tau with microglia. This co-localization was present in caspase-3-cleaved tau-positive aggregates with notable perivascular localization at 3 months after CCI. Images shown at the bottom of this panel represent zoomed excerpts ( × 6) from the corresponding images (shown with rectangles) demonstrating different morphological features of co-localization of caspase-3-cleaved tau-positive aggregates with Iba1 immunostained cells. Panels marked (1) demonstrate localization of the Iba1 immunopositive cells in the areas of diffuse accumulation of caspase-3-cleaved tau without the evident cellular co-localization most often observed at acute time points (e.g., 24 h after CCI). Panels marked (2) demonstrate the presence of cellular caspase-3-cleaved tau-positive aggregates surrounding DAPI-stained nuclei with shapes resembling Iba1-stained microglia and overlapping in the merged image suggesting possible cellular co-localization of these markers. The images also demonstrate a substantial number of extracellular caspase-3-cleaved tau-positive aggregates and immunopositive cells that are not co-localized with Iba1.
Possible involvement of cleaved-caspase-3 in BBB damage after TBI
To investigate the possible involvement of cleaved-caspase-3 in chronic microvascular damage and BBB breakdown, we performed immunofluorescent co-localization experiments in control and CCI-injured rats at 3 months after experimental TBI using EBA, a specific marker of the BBB, counterstained with cleaved-caspase-3 and DAPI. Figure 5A shows predominantly scattered immunopositivity in CCI-injured rats for cleaved-caspase-3 with prominent extracellular localization (suggested by DAPI nuclear counterstaining) in the corpus callosum. No cleaved-caspase-3 immunoreactivity was observed in control animals. Figure 5A also demonstrates the presence of cleaved-caspase-3 puncta overlying or touching the cellular structures immunopositive for the BBB marker EBA (shown with arrows). However, detailed morphological analysis suggested that there was no intracellular co-localization of cleaved-caspase-3 with EBA, and that the cleaved-caspase-3 immunopositive puncta could be localized extracellularly or were associated with other cell types.
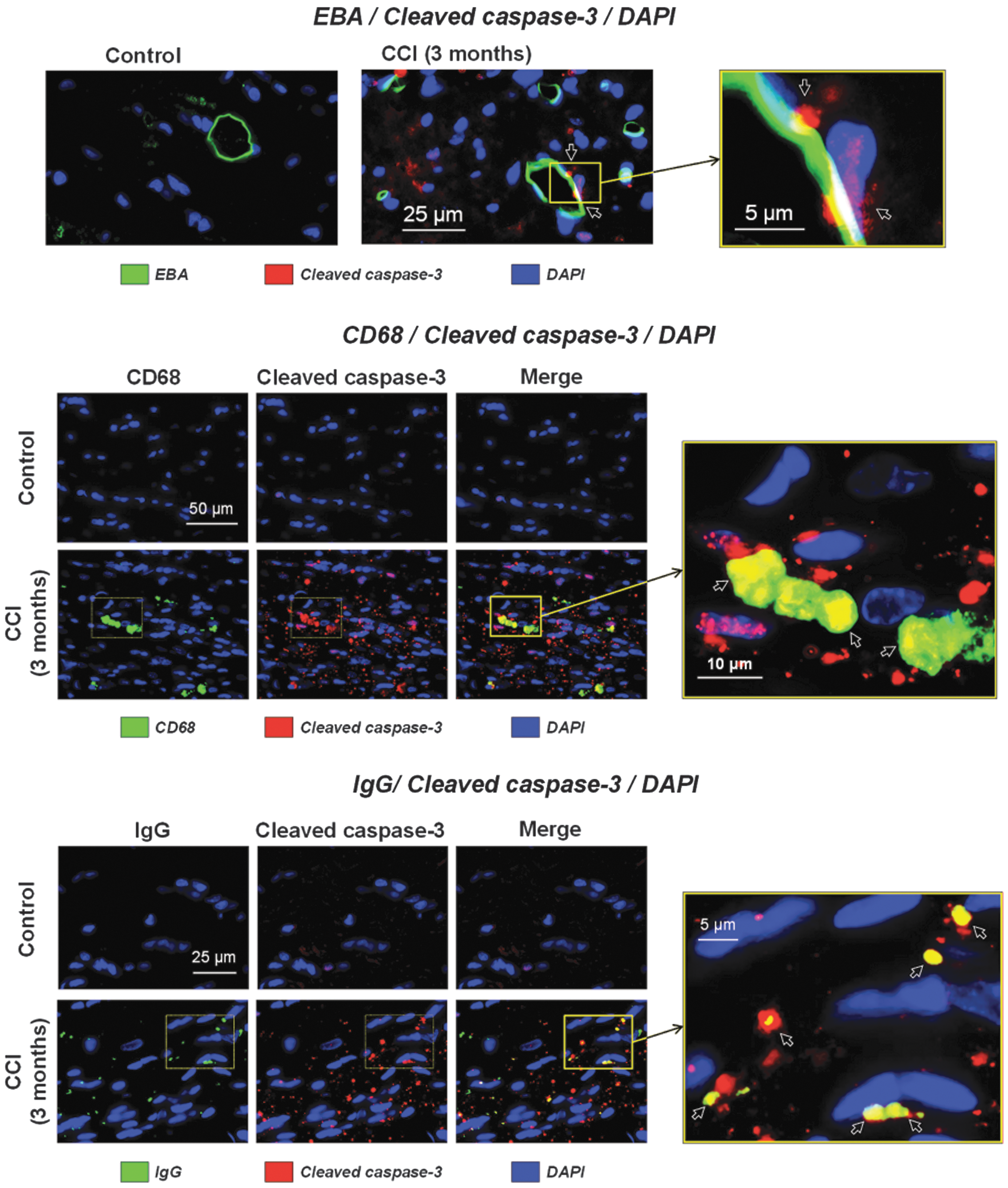
Co-localization of cleaved-caspase-3 with endothelial barrier antigen (EBA), a blood–brain barrier (BBB) marker, and markers of macrophage/monocytes and punctate BBB extravasation 3 months following controlled cortical impact (CCI). (
Based on our previous findings and published observations on the involvement of tissue-resident and/or infiltrating macrophages and monocytes in brain injuries, 49 we evaluated the possibility of cleaved-caspase-3 co-localization in macrophages using fluorescent counterstaining of cleaved-caspase-3 and CD68, a marker of macrophages. 54 Although CD68 is mainly expressed in macrophages, its expression is also characteristic of activated microglia and of infiltrating peripheral monocytes in brain tissue following injuries. 55 Immunofluorescent staining with CD68 revealed the presence of positive round-shaped cells in the corpus callosum (Fig. 5B), an observation consistent with our published study. 49 Although CD68 is also expressed in different cell types, including microglial cells, 56 the morphological features of the CD68-positive cells, including a rounded and/or kidney-shaped appearance, are characteristics of macrophages/monocytes rather than microglia, which generally have branched processes or an ameboid shape. The data further show that all CD68-immunopositive cells are also immunopositive for cleaved-caspase-3, suggesting possible involvement of macrophages in caspase-3-mediated apoptosis after TBI (Fig. 5B). Our previous data showed a co-localization of CD68-positive cells with chronic punctate BBB IgG extravasation in the corpus callosum, indicative of BBB damage. 49 Therefore, we performed immunofluorescent studies to determine potential co-localization of IgG puncta with cleaved-caspase-3. The results of these experiments revealed structural similarities and co-localization of cleaved-caspase-3 and IgG immunopositive puncta. The majority of IgG-immunopositive puncta were co-localized with cleaved-caspase-3, but a fraction of cleaved-caspase-3-immunopositive puncta was immunonegative to IgG (Fig. 5C).
Discussion
This study provides the first evidence of a significant prolonged upregulation of activated (cleaved) caspase-3 in the white matter of corpus callosum following TBI and of an association between upregulation of activated caspase-3 in the corpus callosum and an evolving accumulation of caspase-3-cleaved tau aggregates. This study also provides evidence for the involvement of astrocytes, endothelial cells, and, possibly, infiltrating macrophages/monocytes in perivascular caspase-3-mediated apoptosis. These results further suggest that caspase-3 activation is associated with chronic BBB disruption and with accumulation of perivascular caspase-cleaved tau aggregates that could potentially contribute to evolving and delayed white matter damage characteristic of TBI.
Caspase-3 activation after TBI
The major goal of this study was to investigate the role of caspase-3-mediated apoptosis in the corpus callosum as one of the possible mechanisms involved in chronic pathological processes associated with TBI, such as delayed development of white matter and microvascular pathologies that could result in development of neurodegenerative diseases. Clinical data indicate that chronic inflammation and white matter degeneration resulting from TBI can persist for years after even a single impact. 57 Although the specific pathological pathways contributing to progressive white matter degeneration after TBI have not been well defined, several possible mechanisms have been proposed, including apoptotic cell death 23,27,58,59 and inflammation. 60,61 In addition, pre-clinical and clinical studies have suggested that caspase-3 might be involved in brain injuries and neurodegenerative diseases via apoptosis-independent pathways such as proteolytic cleavage of structural proteins, including αII-spectrin. 29,62,63 However, a pre-clinical study using a fluid percussion model of TBI, which produces a markedly less severe magnitude of TBI than the CCI model used in this study, reported that caspase-3-mediated proteolysis of αII-spectrin showed only a small increase at acute time points in the cortex and was not detected in the corpus callosum. 63
Chronic duration of caspase-3 activation
Our study provides experimental evidence of involvement of caspase-3 in chronic sequelae of TBI as reflected in its differential upregulation in white matter for at least 3 months following TBI. Previous pre-clinical studies documented acute and subacute (up to 4 weeks) upregulation of activated caspase-3 in the traumatized brain in different pre-clinical models including CCI, 64,65 fluid-percussion injury, 66 and a model of surgical injury induced using a micro-knife. 67 These studies demonstrated acute activation of caspase-3 in injured gray matter regions only at acute time points, and do not provide direct evidence of involvement of caspase3 pathways in white matter. In contrast, our study demonstrates the progressive upregulation of cleaved-caspase-3 in the corpus callosum for at least 3 months after experimental injury, suggesting its involvement in chronic sequelae of TBI. A previous rat study using the CCI model of TBI demonstrated significant upregulation of activated caspase-3 mRNA in brain at 48 h after injury, and significant increases in activated caspase-3-immunopositive cells in the cortex and only a limited number of activated caspase-3-immunopsitive cells in the corpus callosum 68 at acute time points, which is consistent with the findings of our study. In contrast, in immature rats, cellular caspase-3 activation in the corpus callosum was only observed at 24 h after CCI. 69 Therefore, our study extends previous studies and demonstrates chronic activation of caspase-3 lasting for at least 3 months following experimental TBI, with time-dependent progression of distinctive morphological features of cleaved-caspease-3 immunohistochemistry including significant persistent increases in cleaved-caspase-3-positive puncta starting at 24 h and further progressing to formation of cleaved-caspase-3-positive aggregates starting at 1 week after CCI, and a transient increase in cleaved-caspase-3-positive cells with a significant change observed at 1 month after injury. These distinctive morphological features of cleaved-caspease-3 immunohistochemistry were associated with cellular and extracellular accumulation of caspase-3-cleaved tau starting as early as 24 h after CCI and progressing to distinctive perivascular accumulation of caspase-3-cleaved tau aggregates with significant increases starting at 1 week after CCI. The presence of these puncta may be the first morphological sign of caspase-3 activation. This possibility can be examined in future studies directly comparing enzymatic assays of caspase-3 activation and associated morphological changes.
Chronic evolving accumulation of caspase-3-cleaved tau
Numerous clinical and pre-clinical studies suggest that there is an involvement of tau proteins in the chronic sequelae of TBI. 70,71 Published pre-clinical data have demonstrated changes in the levels of tau proteins starting within the 1st week and lasting >1 month in different rodent TBI models. 72 –74 Data obtained using transgenic 3xTg-AD mice have shown that CCI independently affects amyloid β and tau abnormalities, supporting a causal role for TBI in acceleration of Alzheimer's-like neurodegenerative disease. 75
Activation of the caspase-3 pathway and the consequent cleavage of tau may be an early event in the tangle pathology of AD, 37 linking formation of neurofibrillary tangles and amyloid plaques, 43 two of the most recognized hallmarks of AD. A clinical study suggested that caspase-mediated cleavage of tau protein might be involved in the etiology of CTE. 39 However, the role of caspase-3 in tau pathologies after TBI is currently poorly understood. To the best of our knowledge, no published pre-clinical studies focus on the spatiotemporal distribution and association of tau deposition with cleaved-caspase-3 expression.
Although caspase-3 activation associated with cleavage of several specific brain proteins including tau truncation at Asp421 is one of the major pathways involved in brain injuries and neurodegenerative disease, the etiopathology of these disorders also involves activation of different caspases and associated pathophysiological processes (see review by Glushakova and colleagues 42 ). Interestingly, recent data using transgenic mouse AD models expressing mutant human amyloid precursor protein or mutant human tau (i.e. J20 AAP and rTg4510 mice, respectively) suggest that caspase-2 activation involving specific cleavage of tau at Asp-314 can result in impairment of synaptic function and cognitive deficits in neurodegenerative disease. 76,77 Nevertheless, although there are similarities in caspase involvement in acute brain injuries and neurodegenerative diseases, activation of specific caspases might be different, depending on disease conditions. For example, experimental data suggest that caspase-2 is not involved in ischemic brain damage following experimental stroke. 78
On the other hand, the pathological sequelae of TBI involve complex pathways associated with tau processing that collectively could be associated with tauopathies and progression of neurodegenerative diseases such as CTE and AD following brain trauma. These pathologies could include tau dissociation from microtubules in axons, its cleavage by calpains and caspases, and abnormal hyperphosphorylation resulting from kinase activation. 6,42,79 A recent study by Kondo and colleagues using human CTE brain samples and murine blast and closed head injury models reported accumulation and perivascular localization of cis-phosphorylated tau isomers in human CTE compared with controls and in animal models following TBI associated with tau tangle pathology. 80 Further, animal experiments have shown that that cis-phosphorylated tau is an early marker of TBI associated with TBI severity. The presence of this form was restricted to the injured cortex between 24 h and 2 months following TBI, and was detected in other brain regions including hippocampus only at 6 months after experimental TBI. 80 In contrast, our study demonstrated the activation of caspase-3 in different inflammatory and endothelial cell types, and accumulation of caspase-3-cleaved tau in brain regions not directly impacted by primary injury including the white matter of the corpus callosum at both acute and chronic times. These data suggest that caspase-3-mediated tau cleavage represents a distinct complemented pathway involved in development of tau pathologies associated with neuroinflammation and BBB dysfunction in white matter.
In this study, we have hypothesized that caspase-3-mediated tau cleavage might be involved in the progression of chronic neuropathology after TBI, and that this may underlie neurodegeneration. Our study demonstrated a noticeable accumulation of caspase-3-cleaved tau aggregates in the corpus callosum, where increases in activated caspase-3 were seen predominantly as puncta staining. This phenomenon can be explained by the distribution of tau protein primarily in axons comprising the white matter of the corpus callosum and by tau proteolysis occurring within activated caspase-3 localization that results in the buildup of caspase-3-cleaved tau peptide and its further aggregation caused by altered physicochemical properties. These findings also provide evidence of delayed diffuse axonal injury in the white matter of the corpus callosum, and possible mechanisms involving capase-3-mediated apoptosis. Further, the possible involvement of activated caspase-3 via tau proteolysis that may potentially result in tau protein misfolding and delayed axonal injury is also suggested from the similarities between the temporal profile of the buildup of caspase-3-cleaved tau aggregates and that of white matter degeneration detected by Luxol Fast Blue stain in our previous study. 49 The diffuse accumulation of caspase-3-cleaved tau is evident at 24 h after CCI, and this diffuse caspase-3-cleaved tau accumulation further forms aggregates at chronic time points, noticeably with perivascular localization, suggesting development of chronic tau pathologies characteristic of neurodegenerative disorders.
Progression of chronic immunohistopathological changes after experimental TBI
TBI has been linked to chronic neurodegeneration and progressive brain atrophy lasting from months to 1 year after the initial injury, and these pathological alterations are associated with neuronal death and white matter degradation. 17,18,57 Our previous study using specific myelin staining with Luxol Fast Blue demonstrated chronic progressive white matter degeneration associated with neuroinflammation and microvascular alterations for at least 3 months after CCI. 49 Published data also suggest that TBI-induced white matter degeneration and myelin loss in the corpus callosum may result from oligodendrocyte apoptosis. An increased number of activated caspase-3-immunopositive oligodendrocytes in the corpus callosum was observed starting at 48 h after injury, remaining elevated for up to 3 weeks following fluid percussion injury in rats. This activated caspase-3 upregulation was associated with a decrease in numbers of healthy oligodendrocytes, suggesting their apoptotic death. 81 –83 Our initial data showed chronic progressive decrease in MBP and CNPase immunoreactivity for at least 3 months after CCI (Fig. S2), which is consistent with loss of myelinated nerve fibers and oligodendrocytes. Further, our study showed increases in cellular density, including both cleaved-caspase-3 immunopositive and immunonegative cells in the corpus callosum at 3 months after CCI, suggesting cellular reorganization of the corpus callosum, likely caused by chronic proliferation of microglia and astrocytes and infiltration of macrophages and other cell types after TBI. 49 Cellular co-localization of cleaved-caspase-3 was observed with GFAP and CD68, but not with Iba1, suggested that at chronic time points, apoptosis primarily affects astrocytes and macrophages/monocytes. These chronic changes in cellular organization of the corpus callosum were accompanied by chronic extracellular accumulation of cleaved-caspase-3-immunopositive structures (i.e., extracellular cleaved-caspase-3 aggregates and puncta), suggesting chronic progression of caspase-3-mediated apoptosis in selected cell types following TBI.
Glial, inflammatory, and microvascular changes associated with caspase-3 activation
To address possible pathological mechanisms and involvement of specific cell types in caspase-3-mediated proteolysis following TBI, we performed immunofluorescent staining to study co-localization of the cleaved-caspase-3 and different glial, microvascular, and peripheral inflammatory cell markers. It is well recognized that CCI results in acute and chronic gliosis, resulting in proliferation and activation of microglia and astrocytes in different gray and white matter regions, indirectly affected by CCI, including the corpus callosum. 49,84 –86 Published data suggest that following TBI, caspase-3 is upregulated primarily in neurons and, to a lesser extent, in glial cells, including astrocytes. 64,65 The results of our study extend the aforementioned published studies and suggest the presence of caspase-3 activation in certain cell types at chronic time points after experimental TBI, including populations of astrocytes and infiltrating CD68-immunopositive macrophages/monocytes.
Experimental studies have suggested that the delayed axonal damage induced by primary injury of cortical neurons results in a disruption of their projecting axons (Wallerian degeneration). This degeneration is accompanied by proliferation and prolonged activation of reactive microglia, which may also contribute to progression of secondary damage. 87 However, studies suggest a dual role of microglia in brain injuries; they may also have, under certain circumstances, a protective function. 88 Our study demonstrates that microglial cells are present within areas of accumulation of caspase-3-cleaved tau, suggesting possible involvement of this cell type in tau cleavage. However, despite the presence of microglial cells in areas of caspase-3-cleaved tau accumulation, our co-localization studies with the microglial marker Iba1 did not detect obvious co-localization of this marker with either cleaved-caspase-3 or caspase-3-cleaved tau, suggesting that microglia are not directly involved in caspase-3-mediated apoptosis. It is possible, however, that microglial activation may play a role in caspase-3-cleaved tau internalization, although its role in tau clearance might be limited. 89 –91
Numerous studies have shown that the gliosis associated with proliferation and activation of reactive astrocytes is a prominent response to TBI. 92 –94 Mechanisms involved in astrocytic caspase-3-mediated apoptosis are not clear. In this study we did not detect the presence of reactive astrocytes in the areas of caspase-cleaved tau accumulation, suggesting that astrocytes are not directly involved in tau cleavage in the corpus callosum at chronic time points.
BBB breakdown is also associated with the infiltration of blood-borne inflammatory cells including macrophages and monocytes, especially evident during the acute period after TBI. 60,95,96 To further investigate the possible involvement of inflammatory cells, we performed immunohistochemical analyses with CD68, a lysosomal glycoprotein expressed primarily by macrophages/monocytes and activated microglia. Macrophages differentiate from circulating monocytes, predominantly in response to inflammation, and are present in virtually all tissues, including brain. 97 Additional data suggest that activated parenchymal microglia/macrophages and infiltrating monocytes are involved in pathophysiological processes following human and experimental TBI. 55,98 Previous experimental reports have provided evidence of the acute accumulation of the CD68-positive microglia/macrophages within the site of injury, as well as a chronic distribution throughout white matter regions including the corpus callosum, which are not directly impacted by experimental TBI. 49,99 In related human stroke studies, caspase-3 upregulation was observed in different types of CD68-positive cells, including infiltrating macrophages and microglia. 100 –102 The co-localization of cleaved-caspase-3 with CD68 shown in our study demonstrates the presence of cleaved-caspase-3-positive macrophages/monocytes. We further reported the co-localization of the CD68-positive structures with punctate BBB extravasation assessed by rat IgG immunohistochemistry at chronic stages of TBI. 49 The results of our study further demonstrated co-localization of IgG-positive puncta with cleaved-caspase-3, suggesting its involvement in chronic BBB damage resulting in extravasation of blood proteins and, possibly, infiltration of blood cells such as macrophages/monocytes and erythrocytes that may contribute to chronic inflammation and excitotoxicity.
Potential cellular cascades involved in chronic caspase-3-mediated changes in the corpus callosum
The results of our study suggest that caspase-3 activation is involved in pathways involving different cell types that may be differentially involved in neurodegenerative processes and may differentially affect chronic TBI outcomes. The temporal profiles of cleaved-caspase-3 activation and caspase-3-cleaved tau accumulation demonstrated distinct progressions of chronic immunohistopathological alterations in cellular and extracellular immunoreactivity in the corpus callosum following experimental TBI. These distinctive morphological changes are differently associated with different glial and inflammatory markers. The temporal profiles of different immunohistochemical and histopathological hallmarks associated with the progression of chronic TBI have been characterized and previous published 49 and, partially, in this study, included chronic changes in reactive astroglia (GFAP) and microglia (Iba-1), infiltration of macrophage/monocytes (CD68) and signs of BBB extravasation (IgG), endothelial dysfunction (intercellular adhesion molecule-1 [ICAM-1]), and accumulation of microbleeds (Prussian blue) (Fig. 6). The data demonstrate a temporal association of cleaved-caspase-3-positive cells with GFAP immunoreactivity, as well as caspase-3-cleaved tau-positive cells, and diffuse aggregates with microglia activation (Iba-1). Chronic BBB extravasation (IgG) chronic endothelial damage (ICAM-1), and accumulation of microbleeds were associated with perivascular aggregation of caspase-3-cleaved tau.
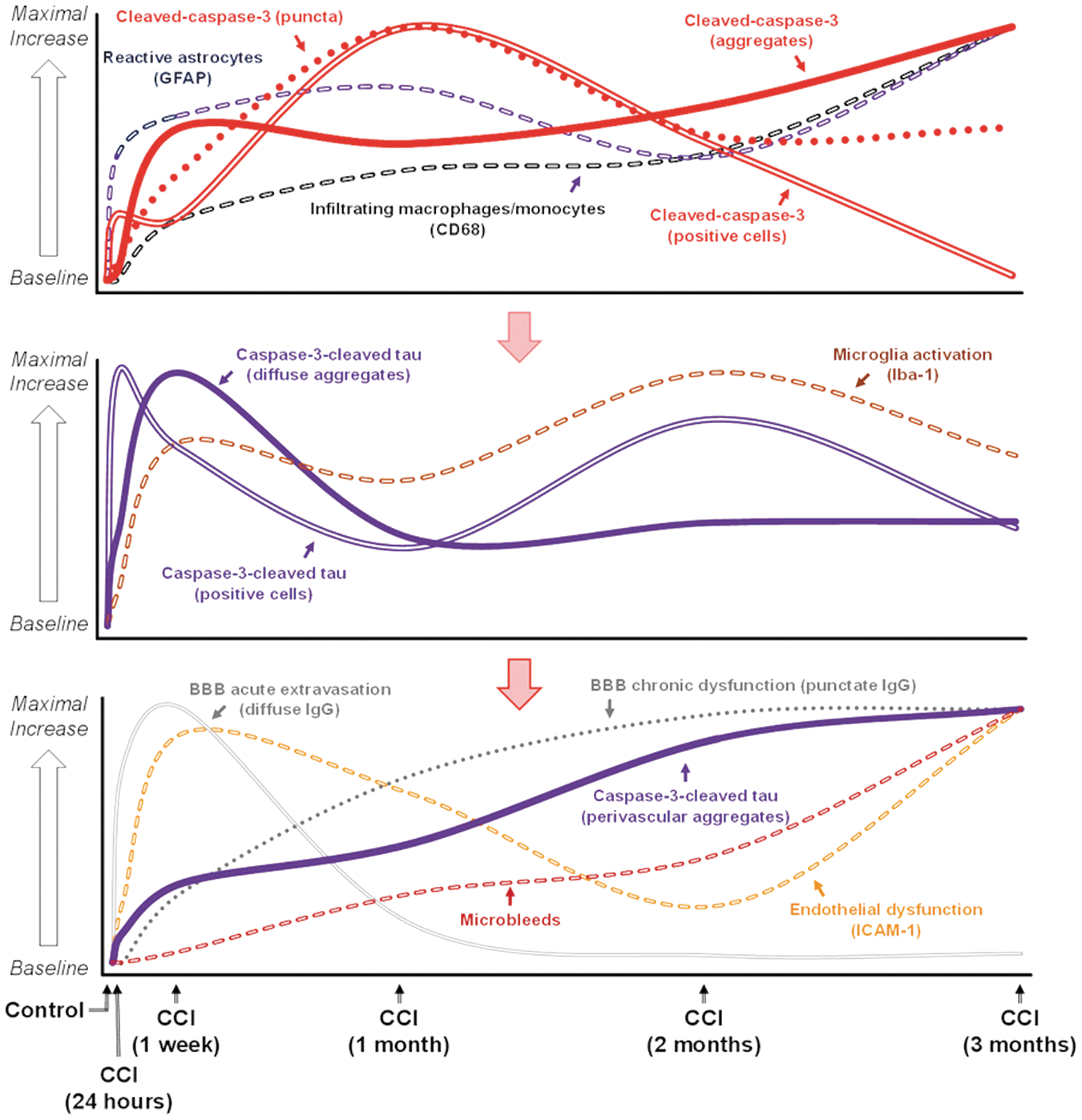
Temporal association of cleaved-caspase-3 and caspase-3-cleaved tau with biomarkers of reactive astrogliosis, microglia activation, endothelial dysfunction, blood–brain barrier (BBB) extravasation, and accumulation of microbleeds. The graphs represent a schematic time course representation of immunohistochemical and histopathological changes above the baseline (i.e., control) based on the relative values of different morphologically distinct cleaved-caspase-3 and caspase-3-cleaved tau-immunopositive structures (e.g., immunopositive cells, extracellular aggregates, and puncta) and corresponding inflammatory and BBB damage biomarkers that were measured in the present study and previously reported in our recent study 49 utilizing the same controlled cortical impact (CCI) model. Values are shown as a percentage of their maximal expression.
Caspase-3 activation was first detected morphologically as extracellular puncta within 24 h, and was sustained over the course of study. By 1 week, puncta had resolved into extracellular aggregates, which were also sustained over 3 months. Intracellular caspase-3 activity was detected only transiently at 1 month and was accompanied by a significant increase in caspase-3-negative cells 3 months after injury. Caspase-3-cleaved tau was detected at 1 week and was sustained for the duration of the study: a time course consistent with the presence of extracellular puncta and aggregates. Therefore, the rapid presence of extracellular caspase-3 could contribute to caspase-3 cleavage of tau. Both intracellular and extracellular accumulation were sustained over 3 months, but perivascular accumulation continued steadily over this period and was maximally detected at 3 months: a time point associated with inflammation and BBB alterations. 49
Increases in caspase-3-cleaved tau in combination with immunohistochemical experiments with MBP and CNPase (Fig. S2), and our published data with Luxol Fast Blue at chronic time points 49 are consistent with evolving processes contributing to delayed axonal damage in the corpus callosum. Perivascular accumulation of caspase-3-cleaved tau aggregates and their co-localization with microglia in combination with co-localization of selected direct and indirect markers of BBB damage (i.e., IgG and CD68) suggest a possible association of these pathways with chronic BBB damage. However, the detailed mechanisms and injury pathways contributing to evolving and prolonged white matter degeneration following TBI remain unclear. Taking into account that the cellular organization of the corpus callosum primarily consists of neuronal axons and glial cells 103 (Fig. S1), it is possible that delayed axonal pathology could result from delayed Wallerian degeneration of projecting axons not primarily affected by injury, 87 but damaged by later apoptotic processes. The hypothetical pathways involved in the chronic sequelae of TBI including caspase-3 activation are summarized in Figure 7. The presence of these putative associations of apoptotic cascades warrants further study to elucidate interactions between different cell types and their neuropathological implications.
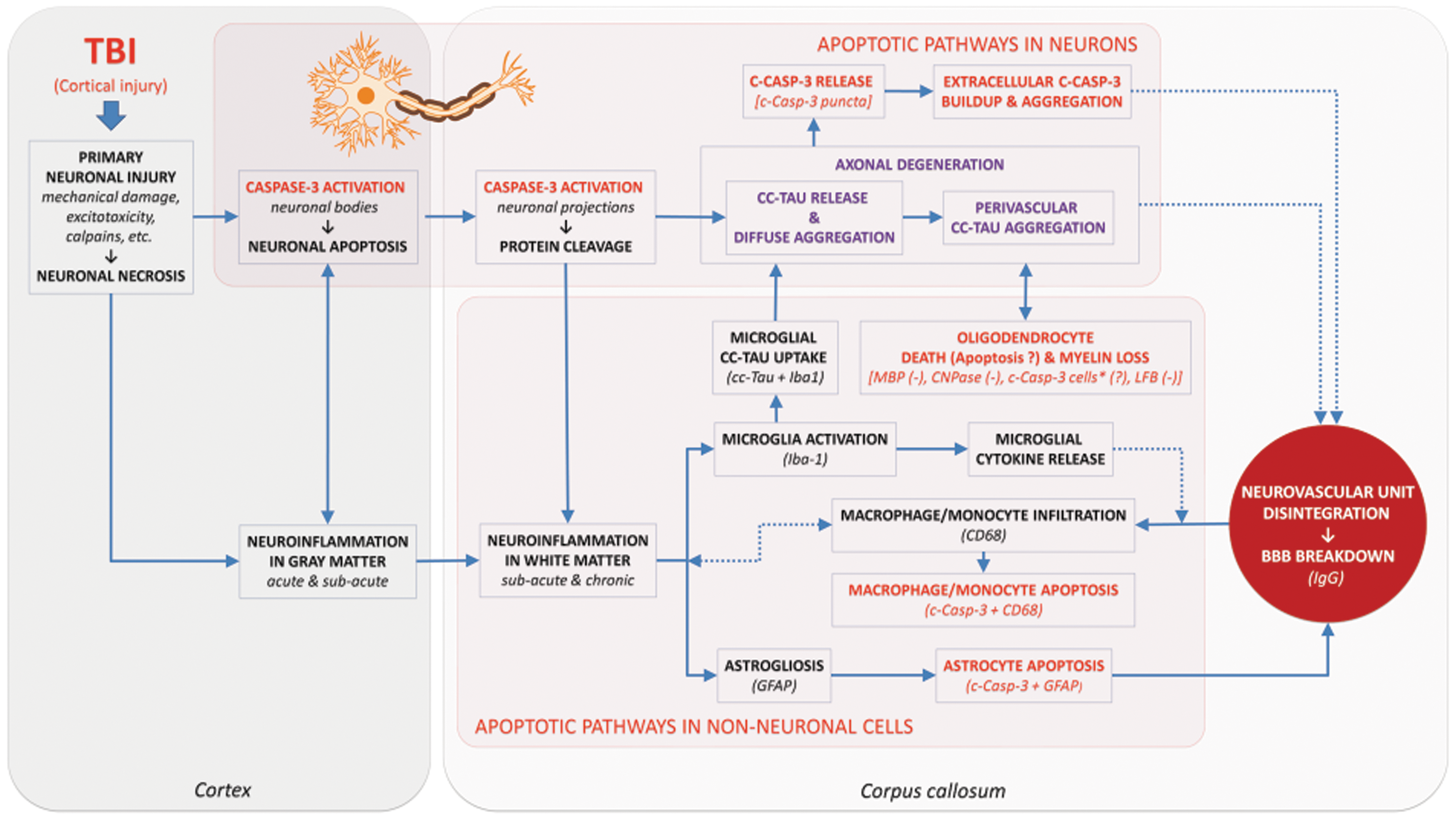
The putative role of caspase-3-mediated pathways in chronic axonal degeneration blood–brain barrier (BBB) damage. Text in red represents features of caspase-3 activation and caspase-3-mediated apoptosis. Text in purple indicates features of caspase-3-mediated tau cleavage. Dotted lines denote indirect associations. Text in parentheses denotes LFB decrease in values of this specific marker. *Indicates an assumption of the possible presence of a fraction of apoptotic oligodendrocytes (cleaved-caspase-3-positive cells) that are not co-localized with other markers used in the study. c-Casp-3, cleaved-caspase-3; cc-tau, caspase-3-cleaved tau; LBF, luxol fast blue.
Study limitations and future directions
Assays of activation of caspase-3 in tissue were not performed in this study. Such information would provide more rigorous biochemical clarification of the temporal profiles of morphological and quantitative immunohistochemical changes related to caspase-3 activity. Future studies will examine the diagnostic applications of the biomarkers associated with caspse-3 activation and the utility of caspase-3 inhibitors as prospective targets for treatment of TBI. Future studies of spatiotemporal profiles of other important caspase-3-mediated protein cleavage products associated with brain injuries, including CCCK-1821 and SBDPs (e.g., SBDP120 and SBDP150) 29 –31 are also warranted to provide further insights into potential cell-specific involvement of caspase-3-mediated pathways in brain injury, thereby extending their potential diagnostic and therapeutic applications.
Conclusions
In conclusion, our data provide the first experimental evidence that activation of caspase-3 following experimental TBI is closely linked to cleavage of tau, which could ultimately contribute to delayed axonal damage, misfolding of tau, and subsequent neurodegeneration. In addition, the results suggest that caspase-3-mediated apoptosis is accompanied by chronic inflammatory responses presumably involving reactive astrocytes and activated microglia/macrophages, which may contribute to microvascular damage and chronic BBB breakdown. Our results indicate that the chronic accumulation of caspase-3-cleaved tau aggregates may represent a critical mechanism contributing to delayed and potentially enduring white matter degeneration and development of neurodegenerative disease following TBI. Therefore, caspase-3-mediated pathways might be promising targets for development of novel therapeutic strategies.
Footnotes
Acknowledgments
The authors thank Danny Johnson for his technical assistance with in vivo CCI experiments and immunohistochemistry, Marda Jorgensen, at the University of Florida, MBI CTAC Core, and the UF Molecular Pathology Core for assistance with histology.
Author Disclosure Statement
Ronald L. Hayes owns stock, receives compensation from, and is an executive officer of Banyan Biomarkers, Inc. and as such may benefit financially as a result of the outcomes of this research or work reported in this article. Alexander V. Glushakov and Olena Y. Glushakova are cofounders of Single Breath, Inc. and may benefit financially as a result of the outcomes of this research or work reported in this article. The other authors have nothing to disclose.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.