
Abstract
Blood-brain barrier (BBB) disruption and dysfunction result in brain edema, which is responsible for more than half of all deaths after severe traumatic brain injury (TBI). Fibroblast growth factor 21 (FGF21) has a potential neuroprotective function in the brain. However, the effects and underlying possible mechanism of action on BBB integrity following TBI remain unknown. The purpose of the current study was to determine the effects of FGF21 on BBB protection and TBI treatment. The effects of recombinant human FGF21 (rhFGF21) on BBB integrity and on tight junction (TJ) and adhesion junction (AJ) proteins were investigated both in a TBI mouse model and an in vitro BBB disruption model established with tumor necrosis factor alpha (TNF-α)-induced human brain microvascular endothelial cells (HBMECs). The ability of rhFGF21 to form an FGF21/FGFR1/β-klotho complex was confirmed by in vitro β-klotho small interfering RNA (siRNA) transfection and FGFR1 co-immunoprecipitation. In addition, the specific FGFR1 and peroxisome proliferator-activated receptor gamma (PPARγ) inhibitors PD173074 and GW9662, respectively, were applied to further explore the possible mechanism of rhFGF21 in BBB maintenance after TBI. rhFGF21 markedly reduced neurofunctional behavior deficits and cerebral edema degree, preserved BBB integrity, and recued brain tissue loss and neuron apoptosis in the mouse model after TBI. Both in vivo and in vitro, rhFGF21 upregulated TJ and AJ proteins, thereby preserving the BBB. Moreover, rhFGF21 activated PPARγ in TNF-α-induced HBMECs through formation of an FGF21/FGFR1/β-klotho complex. rhFGF21 protected the BBB through FGF21/FGFR1/β-klotho complex formation and PPARγ activation, which upregulated TJ and AJ proteins.
Introduction
T
The blood–brain barrier (BBB) is responsible for physiologically protecting the brain from exposure to toxins and ill effects, and it is composed of brain microvascular endothelial cells (BMECs) connected by junctional complexes, astrocytes, pericytes, perivascular macrophages, and a basement membrane and is essential for maintenance and regulation of the neural microenvironment. 12,13 Accumulating evidence has demonstrated that cerebral hemorrhage and brain edema caused by BBB breakdown after TBI is a major cause of death in TBI patients. 14 Therefore, development of a drug or biological therapy targeting BBB permeability and integrity is a potential therapeutic strategy for TBI. The endothelial cells (ECs) of the BBB are unique compared with ECs in different tissues because they have continuous intercellular tight junction (TJ) including claudins, occludin, junctional adhesion molecule (JAM)-A, and zona occludens (ZO-1, ZO-2, and ZO-3), and adhesion junction (AJ) proteins, such as VE-cadherin and PECAM-1. 15 –18 Alteration of the expression of intercellular TJ and AJ proteins affect the permeability of cells and destroy BBB integrity. 19 Thus, maintaining the integrity of TJ and AJ proteins is essential.
Fibroblast growth factor 21 (FGF21) has been confirmed as a novel metabolic regulator in non-human primate models. Because of its impressive metabolic benefits and safety profile, FGF21 holds great promise as a drug for diabetes, obesity, and dyslipidemia. More interestingly, due to its low affinity with heparin, FGF21can penetrate the BBB via simple diffusion and has a potential protective effect on the brain. FGF21 has been demonstrated to play an important role in brain metabolism, protection, and cognition. 20
One important downstream effector of FGF21 signaling is peroxisome proliferator-activated receptor gamma (PPARγ). PPARγ is a nuclear receptor that is primarily expressed in adipocytes. It acts as a ligand-dependent transcription factor and regulates gene expression responsible for adipocyte growth and differentiation. It plays a critical role in obesity and diabetes due to its ability to increase insulin sensitivity. 21 Many studies have shown that PPARγ can protect neurovascular units against ischemic injury by reducing inflammation and oxidative stress, improving angiogenesis, and inhibiting neuron apoptosis. 22
Human FGF21 shares a high degree of homology with the murine orthologues (79% of the amino acids). 23,24 Recombinant human FGF21 (rhFGF21) is widely used in treatment of human disease by mouse models, such as diabetic, 25 hepatic steatosis and inflammation. 26 In the present study, we evaluated whether rhFGF21 can attenuate neurofunctional deficits in TBI mice by protecting against BBB breakdown and explored the molecular mechanisms by which exogenous rhFGF21 treatment preserves BBB integrity. We demonstrate that rhFGF21 activated downstream signaling by forming an FGF21/FGFR1/β-klotho ternary complex, and upregulation of PPARγ stimulated by rhFGF21 preserved TJ and AJ proteins through upregulation of their expression. Taken together, our results indicate that rhFGF21 may be an effective and feasible drug development target due to its ability to improve BBB function after TBI both in vivo and in vitro.
Experimental groups
In our experiment, both in vitro and in vivo assays were performed to confirm the protection effects of rhFGF21 on TBI. The experimental groups examined both in vitro and in vivo are presented in Figure 1. For in vitro, tumor necrosis factor alpha (TNF-α)-damaged human brain microvascular endothelial cells (HBMECs) were used to simulate a BBB disruption model. The experimental groups included a control group (without drugs), TNF-α group, TNF-α+rhFGF21 group, as well as a TNF-α+rhFGF21+PD173074 group. The concentrations of TNF-α, rhFGF21, and PD173074 were 50 ng/mL, 50 nM, and 10 nM dissolved in sterile water, respectively. The permeability of the monolayer of HBMECs was detected by endothelial permeability and transendothelial electrical resistance (TEER) assay. To confirm the protective effect of rhFGF21 on TJ and AJ, western blot and immunofluorescence staining were applied. To confirm the possible mechanism of action, western blot, co-immunoprecipitation (co-IP), and small interfering RNA (siRNA) transfection were utilized. Also, PD173074 and GW9662 were used as FGFR1 and PPARγ inhibitors, which were dissolved in sterile water with a concentration of 10 nM and 10 μM, respectively (Fig. 1A). For in vivo, 60 heads of C57BL/6N mice were randomly divided into three groups: sham group (phosphate-buffered saline [PBS] administrated intraperitoneally [i.p.]), TBI group (PBS administrated i.p.), and TBI+rhFGF21 group (administered i.p. 1.5 mg/kg, dissolved in PBS). The protective effect of rhFGF21 on TBI mouse brain was detected by Gracia test, brain water content, and Evans blue (EB) extravasation, as well as histological analysis hematoxylin and eosin (H&E) staining and Nissl staining. The protective effects on mouse brain TJ and AJ proteins were analyzed by western blot and immunofluorescence staining (Fig. 1B). The experimental protocol based on the TBI mouse model was illuminated in Figure 1C. For the TBI+rhFGF21 group, rhFGF21 was administrated i.p. immediately after TBI surgical procedures. The sham and TBI group mice were given an equal volume of PBS by i.p. injection. After recovery for 24 h, the mice were sacrificed and used for detecting the protective effects of rhFGF21 by various assay methods.
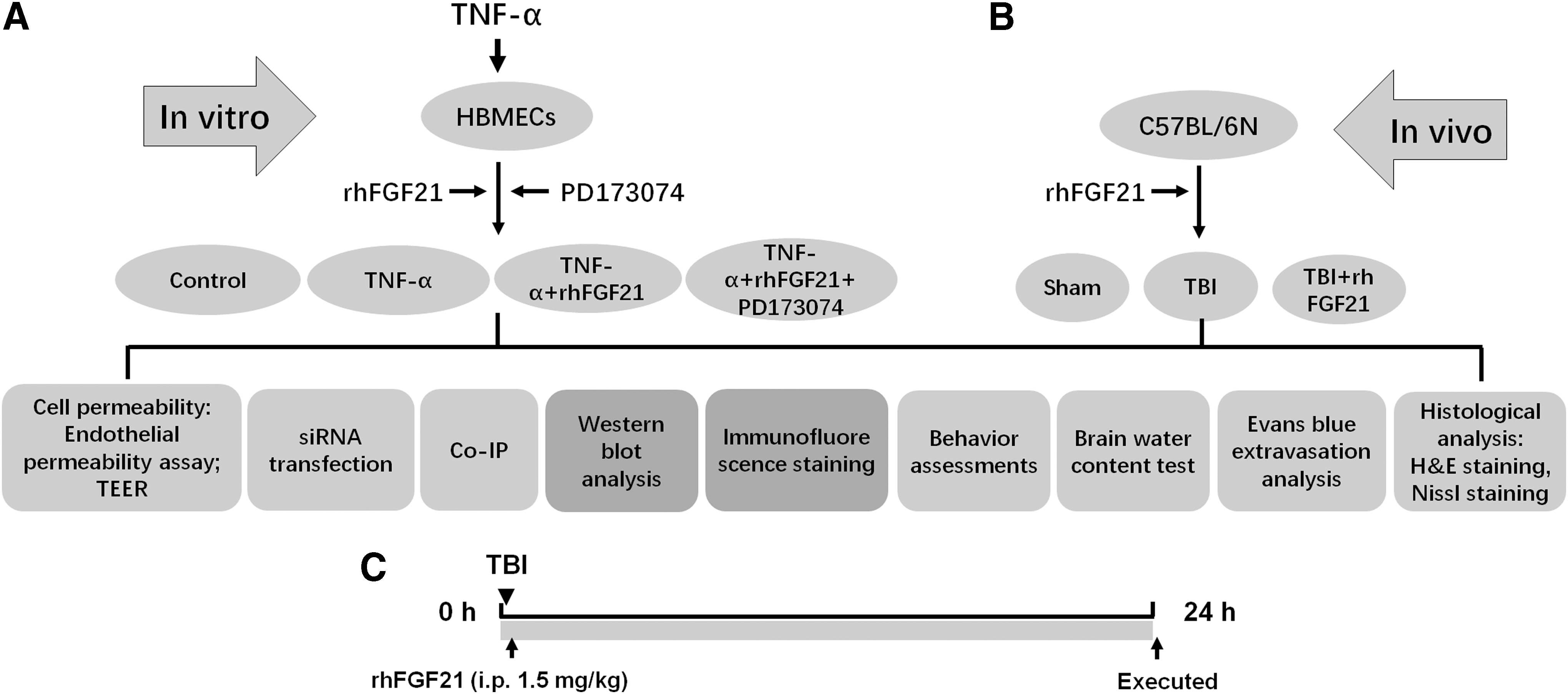
The schematic diagram showing the experimental protocol and experimental groups.
Methods
Reagents and antibodies
rhFGF21, FGFR1 inhibitor PD173074, PPARγ inhibitor GW9662, FITC-dextran, and EB were purchased from Sigma (Sigma-Aldrich, St Louis, MO). TNF-α was obtained from R&D Systems (Minneapolis, MN). The primary antibodies applied in this study were as follows: anti-FGFR1 antibody (Cell Signaling Technology, Danvers, MA); anti-pFGFR1, anti-β-klotho, anti-ZO-1, anti-VE-cadherin, anti-claudin-5, and anti-PPARγ antibodies (Abcam, Cambridge, MA); and anti-occludin antibody (Invitrogen, Carlsbad, CA). The secondary antibodies used in this study were goat anti-rabbit immunoglobulin G (IgG) H&L (HRP) (Abcam) and goat anti-mouse IgG-HRP (Santa Cruz Biotechnology, Dallas, TX).
Animal and TBI model surgical procedures and drug administration
Male C57BL/6N mice (20–25 g) were purchased from the Animal Center of the Chinese Academy of Sciences. The animal use and care protocol conformed to the Guide for the Care and Use of Laboratory Animals from the National Institutes of Health and was approved by the Animal Care and Use Committee of Wenzhou Medical University. The surgical procedures to construct the controlled cortical impact (CCI) mouse TBI model were performed as previously described. 27 In short, 8- to 10-week-old mice were anesthetized with 4% choral hydrate (10 mL/kg, i.p.). The head of the mouse was fixed in a stereotactic frame after the hair between the ears was shaved, and the shaven area was sterilized with 10% iodine. A longitudinal incision was made in the middle of the head and the skin was removed. Then, a 4-mm-diameter circle was drilled midway between lambda and bregma 0.5 mm away from the midline using forceps. Vertically directed CCI was performed using a pneumatic impactor with a 3-mm flat tip. The impact speed, tissue displacement, and impact duration were set at 3 m/sec, 0.75 mm, and 300 msec, respectively. After the impact, the craniotomy was immediately closed. According to the previous study, FGF21 can cross the BBB by simple diffusion, and in the periphery system FGF21 can reach the brain directly. 20,28 Moreover, a high level of plasma FGF21 was detected due to the exogenous FGF21 administration (0.1 mg/kg, i.p.). 29 Our previous study showed the FGF21 concentration in the cerebrospinal fluid of mice collected at 4 h after administration of rhFGF21 (0.5 mg/mL, i.p.), was significantly increased, indicating rhFGF21 can cross the BBB. 30 A dose of 1.5 mg/kg rhFGF21 was injected i.p. after induction of the TBI model used as the TBI+rhFGF21 group. To further explore whether rhFGF21 activates FGFR1/β-klotho and increases the expression of PPARγ, FGFR1 and PPARγ-specific inhibitors, PD173074 (26 μg/kg) and GW9662 (0.4 mg/kg), were intracerebroventricularly injected at a rate of 2 μL/min before induction of the TBI model, as the TBI+rhFGF21+PD173074 group and the TBI+rhFGF21+GW9662 group, respectively. The TBI group and the TBI+rhFGF21 group each received the same volume of DMSO by intracerebroventricular injection, when compared with TBI+rhFGF21+PD173074, and TBI+rhFGF21+GW9662. After the surgical incisions were closed, the mice were allowed to recover for 24 h. Mice were maintained in identical conditions at room temperature (25°C) and 60% humidity under a 12-h light-dark cycle and given free access to water and food.
Evans blue extravasation analysis
For analysis of cerebrovascular permeability after TBI, EB extravasation assays were performed 24 h after TBI as previously described. 31 Briefly, mice were injected with 250 mL of 2% EB via the tail vein. After a circulation period of 2 h, animals were anesthetized and transcardially perfused with saline. The brains were removed, and then, the injured hemispheres were dissected out and weighed. Each injured hemisphere of TBI and drug treatment group was then incubated with N,N-dimethyl formamide for 3 days at 72°C and centrifuged for 45 min at 15,000 rcf, and the corresponding hemisphere of sham group used as control. The supernatants were collected and EB extravasation was detected with a spectrophotometer at 610 nm excitation, 680 nm emission wavelengths. The EB dye content in the brain tissue was quantitatively calculated using a standard curve. 32
Behavior assessments
The sensorimotor Garcia test was performed as previously described with slight modification and was used to assess neurofunctional deficits in mice 24 h after surgery. 33 The Garcia test comprises six individual tests: spontaneous activity, symmetry in the movement of four limbs, forepaw outstretching, climbing, body proprioception, and response to vibrissae touch. A score of 0 (worst performance) to 3 (best performance) was prescribed for each individual test, and the total score was calculated as the sum of all six sub-tests (maximum score of 18).
Brain water content test
The wet-dry weight method, which is a prevalently used and simple method, was applied to evaluate brain edema after brain injury in experimental animals. At 24 h post-injury, mice were sacrificed, and the injured hemispheres were quickly collected. Brain tissues were immediately weighed to obtain the wet weight, dehydrated at 100°C for 48 h, and then reweighed to obtain the dry weight. The percentage of brain water content was calculated using the following equation:
Histological analysis
H&E staining was performed, as described previously. 35 Nissl staining was performed to detect neuron damage in the brain after TBI. Briefly, brains were removed and kept in 4% paraformaldehyde (PFA) for a post-fix overnight, and the paraffin-embedded brain tissues were then sectioned into 5-μm thickness sections by an HM 340E rotary microtome (LEICA, Germany). The sections were dried at 40°C, followed by deparaffinization, and were rehydrated by a series of ethanol solution. The rehydrated sections were subjected to H&E solution or Nissl solution based on the manufacture's protocol. After the slides were gently washed with tap water, and subsequently rinsed in distilled water, and they were mounted with mounting medium. Finally, the sections were observed and imaged using a Nikon ECLIPSE 80i fluorescence microscope (Nikon, Japan).
Cell culture and in vitro BBB disruption model
Primary HBMECs (ACBRI376, Cell Systems Corporation, Kirkland, WA) were grown in EBM-2 supplemented with 10% fetal bovine serum (FBS) and penicillin-streptomycin in cell culture plates coated with fibronectin. The plates were incubated at 37°C in a humidified atmosphere of 5% CO2 and 95% air. Recombinant human TNF-α, which is a well-established cytokine mediator of vascular leakage, was used to construct the BBB disruption model described in a previous study. 36 Briefly, after the cells reached approximately 90% confluence, they were treated with TNF-α (50 ng/mL), with or without rhFGF21 (50 nM), for 24 h. To further illustrate the effect of FGFR1 activation after TNF-α treatment, cells were treated with the FGFR1 inhibitor PD173074 (10 nM). After 24 h, the cells were harvested for further analysis.
Endothelial permeability assay
Endothelial permeability assays were performed via quantification of FITC-dextran (70 kDa) extravasation across HBMECs seeded in a transwell chamber as previously described. 31 In brief, HBMECs were seeded at 1 × 105 cells/well in 200 μL of EBM-2 on polycarbonate 24-well transwell chambers with 0.4-mm pores (Corning). After 48 h, cells were cultured until they reached a compact density and then were treated with or without TNF-α, rhFGF21, and PD173074. After 24 h, the medium was replaced with medium containing 1% FITC-dextran (10 mg/mL), and the cells were incubated for 4 h. Then, the fluorescence of each well in the 24-well chamber was detected with an EnSpire Manager (PerkinElmer Company) multimode microplate reader at 485 nm excitation and 520 nm emission wavelengths.
Transendothelial electrical resistance (TEER)
The TEER value was determined to estimate the permeability across ECs in vitro. The TEER value of an HBMEC monolayer was detected with an EndOhm-6 chamber and EVOM resistance meter (World Precision Instruments, Sarasota, FL) as previously described. 37 Briefly, a 6-mm transwell chamber containing cultured HBMECs was transferred into an EndOhm-6 chamber containing 0.1 M KCl. The culture medium within the transwell chamber was also replaced with 0.1 M KCl. The EndOhm cap was inserted on the top of the chamber and then connected with the chamber using a connector cable. Resistance was then measured using the EVOM resistance meter. A blank transwell chamber containing only medium was used as a blank control.
siRNA transfection
To assess whether β-klotho is necessary for rhFGF21-mediated activation of FGFR, transient transfection with siRNA was performed using Lipofectamine 2000 (Invitrogen) as described in a previous study. 37 One day before transfection, cells were cultured on a six-well plate at 2 × 105 cells/well in 1 mL of EBM-2. Briefly, 100 pmol of siRNA and 3.5 μL of oligofectamine were added to separate tubes containing 250 μL of serum-free EBM-2. The contents of the two tubes were mixed together and incubated for 20 min at room temperature for complex formation. Subsequently, 500 μL of serum-free EBM-2 was added to the complex medium, and the entire mixture (1 mL) was added to each well. Cells were assayed 24 h after transfection.
Co-immunoprecipitation (co-IP) assay
To confirm whether FGF21 effected formation of the FGF21/FGFR1/β-klotho receptor complex, a co-IP assay was performed according to the kit manufacturer's protocol (Life Technologies, Carlsbad, CA). In brief, the total protein of cells treated with or without TNF-α and rhFGF21 was extracted in cell lysis buffer. FGFR1 antibody was added to Dynabeads® in a tube and incubated with rotation for 10 min at room temperature. Subsequently, the Dynabead-FGFR1 antibody complexes were separated from the supernatant with a magnet, and the supernatant was removed. Then, the protein lysate was added to the Dynabead-FGFR1 antibody complexes and resuspended with a pipette. After 10 min of incubation at room temperature, the Dynabead-FGFR1 complexes were collected with a magnet, mixed with elution buffer and pre-mixed NuPAGE® LDS sample buffer, and heated for 10 min at 70°C to elute the FGFR1 complexes. The eluted samples were then subjected to western blot analysis of β-klotho and FGFR1. IgG was considered the negative control, and β-klotho with input was used as the positive control.
Western blot analysis
Total protein from brain tissue and HBMECs was purified using protein extraction reagents containing 1% protease and phosphatase inhibitors. Membrane proteins from HBMECs were purified using a Plasma Membrane Proteins Extraction Kit. The protein content of the samples was determined with a Bradford Protein Assay Kit. An equivalent amount of protein (80 μg for in vivo and 60 μg for in vitro) was separated on an SDS-PAGE gel and then transferred onto a polyvinylidene difluoride (PVDF) membrane. After being blocked, the membranes were further incubated with primary antibodies overnight at 4°C (1:300 dilution of anti-ZO-1, a 1:400 dilution of anti-PPARγ, and a 1:1000 dilution of anti-VE-cadherin, anti-occludin, anti-claudin-5, and anti-β-klotho). After three washes with tris-buffered saline and polysorbate 20 (TBST), the membranes were incubated with a 1:10,000 dilution of goat anti-rabbit IgG secondary antibody for 1 h at room temperature. Finally, the immunoreactive protein bands were developed and visualized with an enhanced chemiluminescence (ECL) kit, and the band densities were quantified using Image Lab 3.0 software (Bio-Rad).
Immunofluorescence staining
To determine the expression of ZO-1, VE-cadherin, occludin, and claudin-5, immunofluorescence staining was performed as described previously. 31 In brief, for in vitro protein evaluation, HBMECs were plated in a six-well cell culture plate. The cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and incubated with 5% bovine serum albumin (BSA) to block non-specific binding. Then, HBMECs were incubated with primary antibodies against ZO-1, VE-cadherin, occludin, and claudin-5 with 1:200 dilution at 4°C overnight, followed by incubation with AlexaFluor 488 donkey anti-rabbit secondary antibody (1:1000) at 37°C for 1 h. Then, the nuclei were stained with DAPI. For in vivo protein evaluation, paraffin-embedded sections were deparaffinized and rehydrated as described above and then washed three times with PBS and incubated with 3% H2O2 in methanol for 15 min. Antigen retrieval was then performed in citric acid solution at a high temperature and pressure. The sections were blocked and incubated with primary antibody followed by secondary antibody for immunofluorescence staining of cells. The stained sections were observed and imaged using a Nikon ECLPSE 80i fluorescence microscope at × 400 magnification.
Statistical analysis
The data are expressed as the mean ± standard error of the mean (SEM). Statistical differences among data from multiple groups were evaluated with one-way analysis of variance (ANOVA) followed by Tukey-Kramer's test using GraphPad Prism, version 7.1 (GraphPad Software, Inc., San Diego, CA). P < 0.05 was considered statistically significant.
Results
rhFGF21 reduces neurofunctional deficits after TBI in mice
To assess the efficacy of rhFGF21 on TBI, we evaluated neurofunctional behavior deficits, cerebral edema degree, and BBB disruption. The Garcia test was used to assess the level of neurofunctional deficit. The Garcia neuroscores of mice markedly decreased after TBI, and rhFGF21 treatment significantly ameliorated neurofunctional behavior deficits (Fig. 2A). Then, the brain water content analysis, which was determined to evaluate the degree of cerebral edema, showed that the brain water content of mice subjected to TBI markedly increased 24 h after TBI compared with the sham group, whereas rhFGF21 treatment significantly rescued the water content compared with the TBI group (Fig. 2B). This result indicates that BBB breakdown caused by TBI results in upregulated permeability, cerebral edema, and secondary neuronal injury, and FGF21 treatment alleviates these harmful changes. As an indicator of BBB injury, an EB leakage experiment showed that EB leakage through the BBB was dramatically increased after TBI, whereas treatment with rhFGF21 significantly reduced leakage (Fig. 2C–F). The results demonstrate that rhFGF21 treatment ameliorated neurofunctional behavior, cerebral edema degree, and BBB disruption after TBI.

Exogenous rhFGF21 (1.5 mg/kg) improves neurofunctional deficits, and rescues TBI parameters in mouse brain.
Further, we used H&E staining and Nissl staining to examine brain tissue status. The histological brain sections stained with H&E exhibited remarkable tissue loss in the cortex after TBI, and rhFGF21 treatment significantly decreased the brain tissue damage (Fig. 2G–L). Nissl staining shows that the number of neurons was significantly decreased in the TBI group, and rhFGF21 treatment increased the number of neurons present (Fig. 2M–R).
rhFGF21 rescues TBI-induced BBB breakdown by upregulating junction proteins
In our previous study, we found that the expression of TJ and AJ proteins can affect BBB integrity. Consequently, the effect of rhFGF21 on junction protein levels after TBI was determined. The data show that TBI significantly reduced the levels of ZO-1, occludin, VE-cadherin, and claudin-5, whereas rhFGF21 observably rescued the expression of these proteins (Fig. 3A–E).
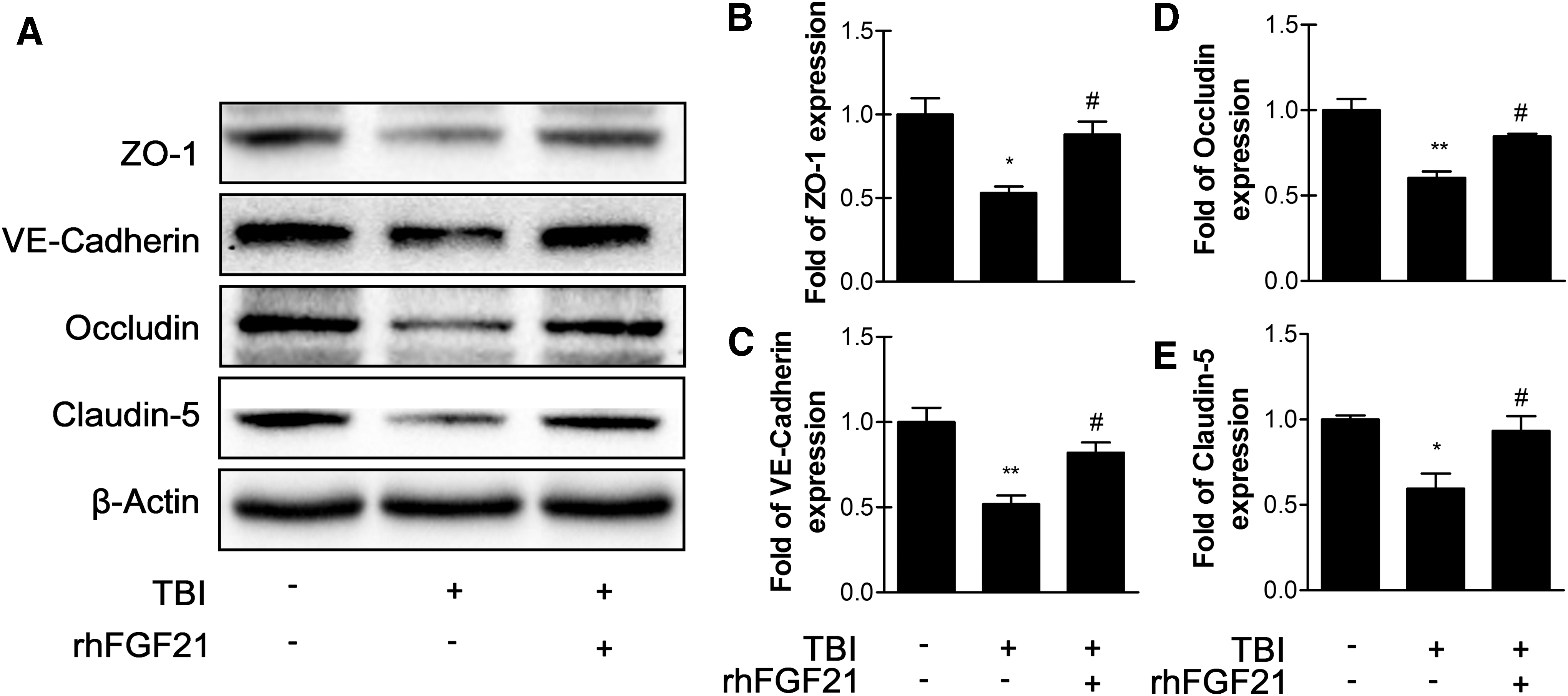
Exogenous rhFGF21 (1.5 mg/kg) recues ZO-1,VE-cadherin, occludin, and claudin-5 expression at 24 h in mouse brain after TBI.
Consistent with the western blot results, immunofluorescence confirmed that ZO-1, occludin, VE-cadherin, and claudin-5 were markedly upregulated after rhFGF21 treatment compared with the TBI group (Fig. 4A–D). Based on the above data, we assume that rhFGF21 treatment protected BBB integrity after TBI at least in part by ameliorating the expression of TJ and AJ proteins.
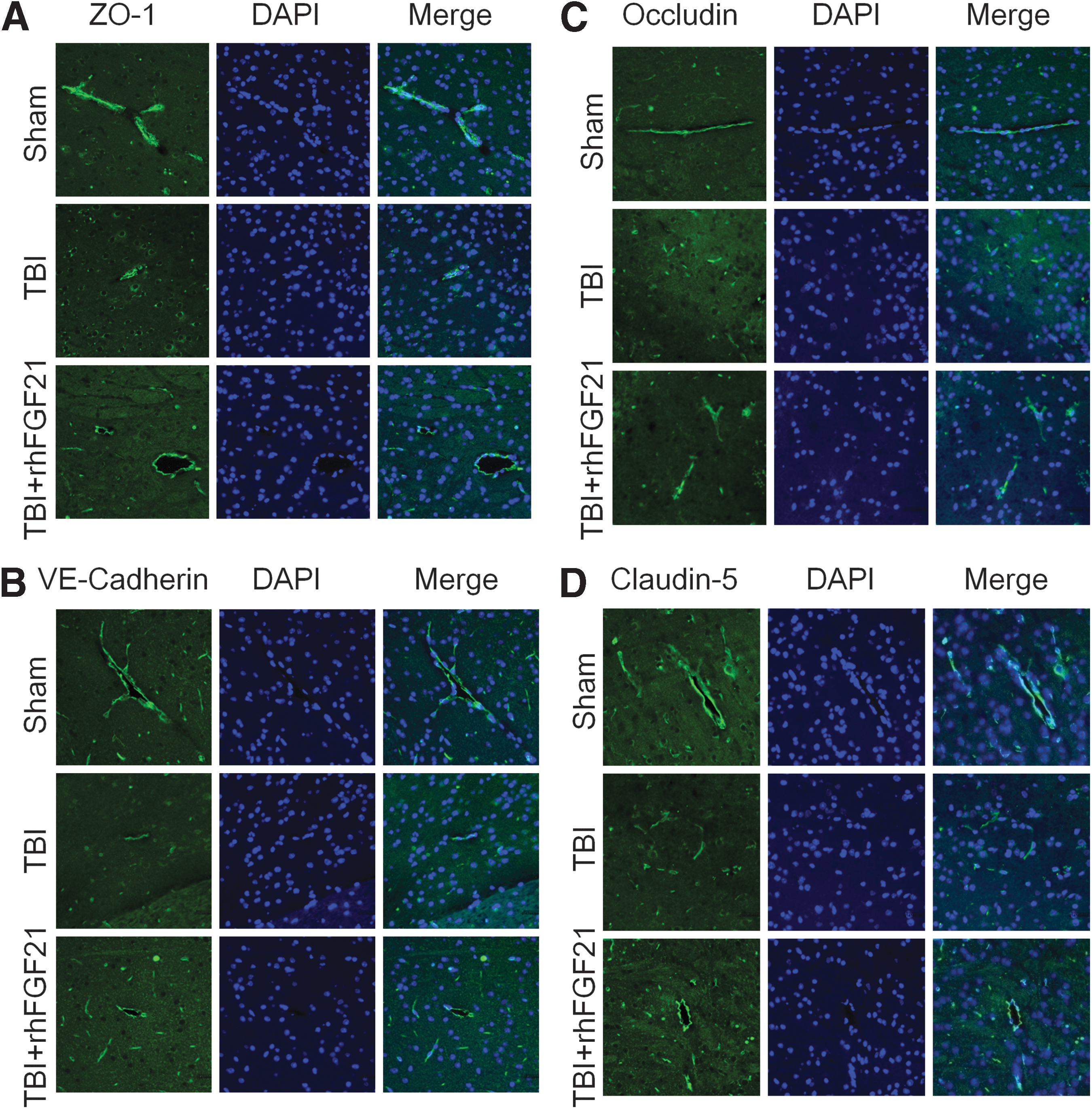
Exogenous rhFGF21 (1.5 mg/kg) recues ZO-1,VE-cadherin, occludin, and claudin-5 activity at 24 h in mouse brain after TBI.
rhFGF21rescues TBI-induced BBB breakdown by increasing PPARγ via FGFR1/β-klotho
The formation of the FGF21/FGFR1/β-klotho ternary complex activates FGFR1 trans-phosphorylation and its downstream signaling pathway, including PPARγ, which has a neuroprotective effect. Thus, we examined whether rhFGF21 activates FGFR1/β-klotho and increases the expression of PPARγ. To further evaluate this effect, FGFR1 inhibitor PD173074 and PPARγ inhibitor GW9662 were applied. Neurofunctional deficits, EB extravasation analysis, β-klotho, and PPARγ protein level were analyzed. PD173074 and GW9662 co-administration with rhFGF21 reversed the neuroprotection effects of rhFGF21 as shown by the Garcia test. As predicted, PD173074-, GW9662-, and rhFGF21-treated mice showed significant neurofunctional deficits compared with rhFGF21-treated mice (Fig. 5A). Further, PD173074-, GW9662-, and rhFGF21-treated mice exhibited significantly more EB dye extravasation than did rhFGF21-treated mice (Fig. 5B,C). The western blot data showed that rhFGF21 improved p-FGFR1 and β-klotho expression (Fig. 5D–F) and PPARγ activation (Fig. 5G,H). As expected, PD173074 significantly reversed the rhFGF21-increased p-FGFR1/FGFR1 protein level, and decreased the β-klotho expression level without significant difference (Fig. 5D–F). GW9662 co-administration decreased rhFGF21 and increased PPARγ protein level but without significant difference (Fig. 5G,H). Taken together, all of these results suggest that p-FGFR1 and β-klotho increased with rhFGF21 treatment protects against BBB breakdown after TBI, at least partially through PPARγ activation.
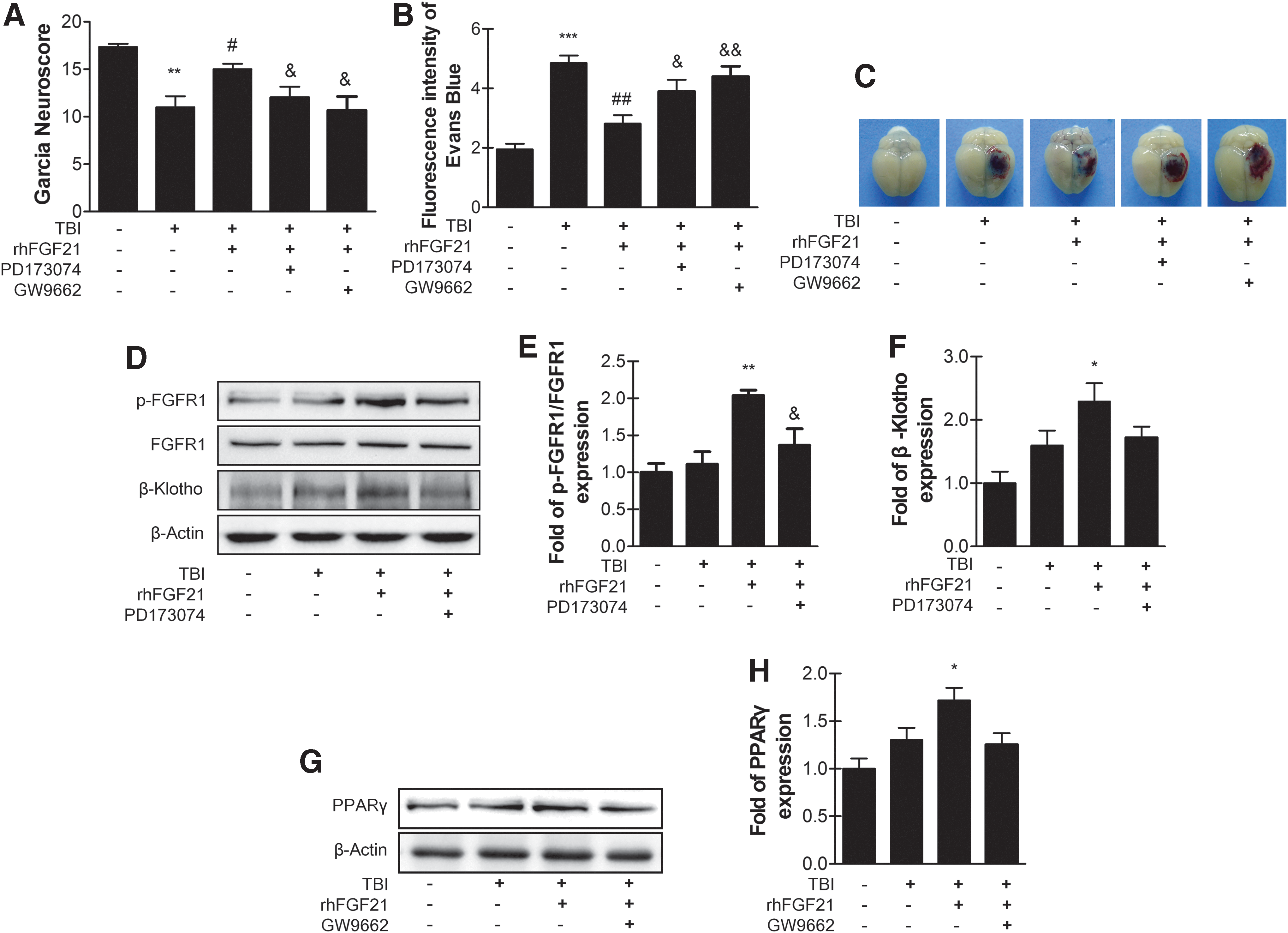
Exogenous rhFGF21 recues TBI-induced BBB breakdown by increasing PPARγ via FGFR1/β-klotho. FGFR1 inhibitor PD173074 and PPARγ inhibitor GW9662 reversed the neuroprotective effects of rhFGF21 at 24 h after TBI.
rhFGF21 protects against TNF-α-induced BBB injury via FGFR1/β-klotho
To further verify the hypothesis that rhFGF21 protects against BBB injury in a cellular model, HBMECs were exposed to TNF-α followed by treatment with rhFGF21 or rhFGF21 combined with the FGFR1 inhibitor PD173074.
Paracellular permeability of HBMECs enhanced significantly after TNF-α treatment, and rhFGF21 administration observably reduced the FITC-dextran permeation. However, the FGFR1 inhibitor PD173074 reversed the protective effect of rhFGF21 (Fig. 6A). The resistance of ECs is also an important indicator of paracellular permeability; high resistance indicates low paracellular permeability. Consistent with the dextran extravasation, the TEER data suggest that TNF-α decreased endothelial cell resistance. rhFGF21 treatment ameliorated the low resistance, but PD173074 reversed the rhFGF21 effect (Fig. 6B). Our results illustrate that rhFGF21 treatment ameliorated TNF-α-induced endothelial permeability via FGFR1/β-klotho.
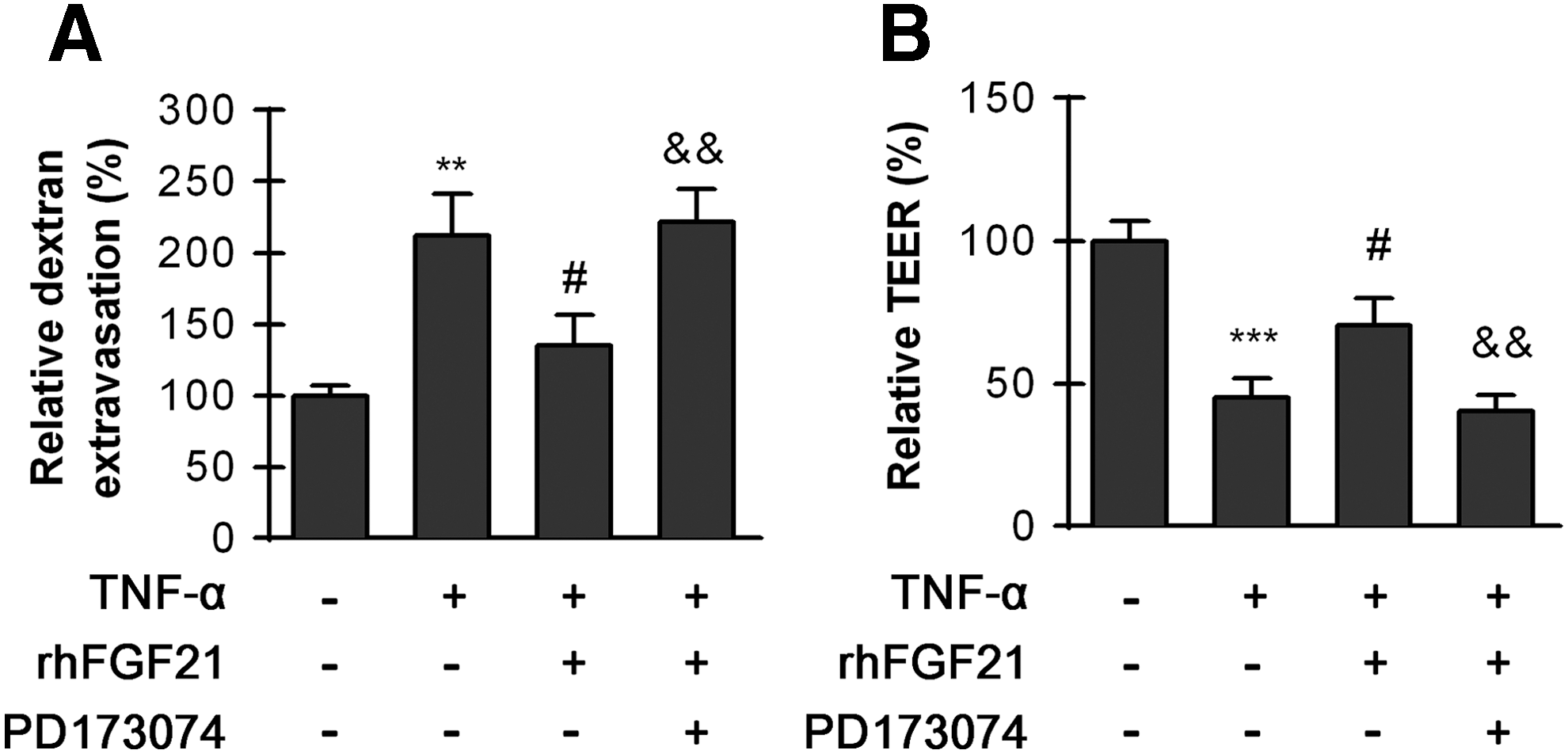
HBMECs were pre-treated with 50 ng/mL rhFGF21 with or without the inhibitor PD173074 (10 nM) for 1 h and then treated with TNF-α for 24 h. The FGFR inhibitor PD173074 reversed the protective effects of FGF21.
FGF21 typically functions through FGF receptor 1 and the β-klotho receptor complex. We used FGFR1 co-IP kits and measured the expression level of β-klotho using western blotting to confirm whether FGF21 could form anFGF21/FGFR1/β-klotho receptor complex. The data demonstrate that FGF21 formed a trimeric FGF21/FGFR1/β-klotho complex to activate p-FGFR1, and the effect of rhFGF21 treatment is likely dependent on this complex (Fig.7A,B). We then used β-klotho siRNA to further confirm the effect of rhFGF21 though the trimeric FGF21/FGFR1/β-klotho complex. The results demonstrate that the expression of p-FGFR1 significantly decreased after β-klotho siRNA transfection (Fig. 7C–E). These results illustrate that FGF21 could not activate FGFR1 when the β-klotho gene is silenced.

HBMECs were treated with or without rhFGF21 under TNF-α conditions. The FGFR1/β-klotho complex was separated by co-IP with an FGFR1 antibody, and then, the β-klotho protein level was measured by western blot. rhFGF21 protects against TNF-α-induced BBB injury via FGFR1/β-klotho formation.
These results demonstrate that FGF21/FGFR/β-klotho was co-localized and that phosphorylation of FGFR1 was activated by FGF21. FGFR1 activation and β-klotho expression after exposure to FGF21 were also measured. The data show that the p-FGFR1/FGFR1 and β-klotho protein level did not significantly change after TNF-α treatment, but rhFGF21 dramatically improved the protein levels. Moreover, the FGFR1 inhibitor PD173074 blocked FGFR1 activation (Fig. 7C–E). PD173074 is also a potent inhibitor of VEGFR; however, in the cultured cells, PD 173074 is a potent and selective inhibitor of FGFR1 with an IC50 value from 1 nM to 5 nM, while inhibiting VEGFR with 100-fold greater IC50 of 100–200 nM. 38,39 At the concentration of 10 nM, PD173074 can significantly inhibit FGFR1 effectively rather than VEGFR.
rhFGF21 protects against TNF-α-induced BBB injury by upregulating junction proteins via FGFR1/β-klotho
Endothelial TJ and AJ proteins are important markers that regulate endothelial paracellular permeability. Consequently, we assumed that TNF-α-induced modulation of TJ and AJ proteins likely increased the paracellular permeability of HBMECs. Total protein levels of ZO-1, VE-cadherin, occludin, and Claudin-5 induced by TNF-α treatment were detected. The data show that TNF-α markedly reduced the protein levels of ZO-1, VE-cadherin, occludin, and claudin-5, whereas rhFGF21 rescued the levels of these proteins. Addition of PD173074 inhibited the effects of rhFGF21 (Fig. 8A–E).
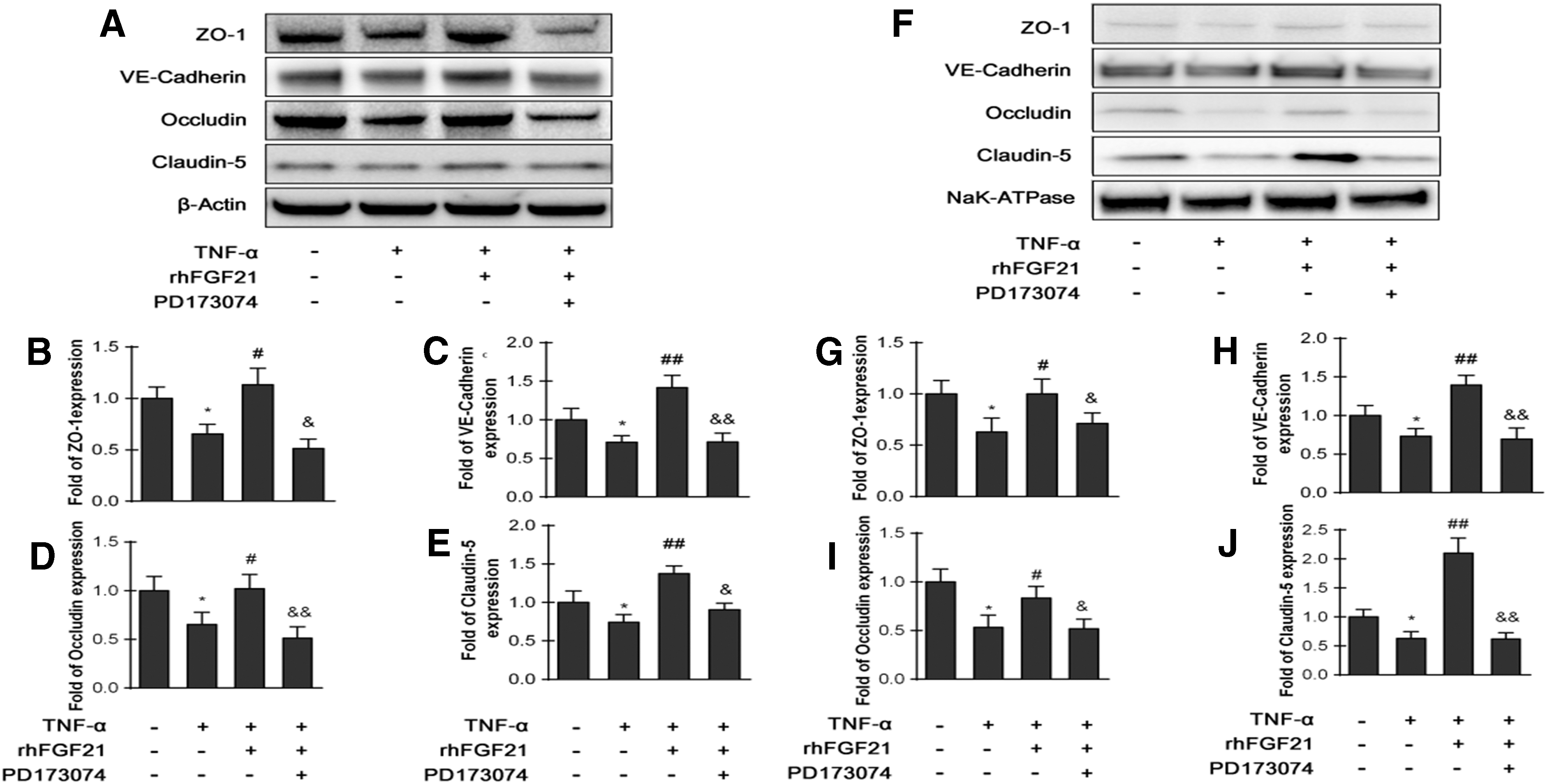
HBMECs were pre-treated with 50 ng/mL rhFGF21 with or without the inhibitor PD173074 (10 nM) for 1 h and then treated with TNF-α for 24 h. rhFGF21 recued both total and membrane TJ and AJ protein expression.
These junction proteins are known to be primarily distributed in the membrane. Therefore, we further detected the levels of these junction proteins in cell membranes by separating the membrane fractions. NaK-ATPase was used as an internal marker. The data suggest that TNF-α significantly decreased the membrane protein levels of VE-cadherin and claudin-5, whereas rhFGF21 administration dramatically rescued these protein levels. PD173074 blocked the effect of rhFGF21. In addition, the ZO-1 and occludin protein levels in the membrane were not significantly altered by either TNF-α or rhFGF21 (Fig. 8F–J).
We also performed immunostaining to analyze the levels of these junction proteins. Consistent with the western blotting results, the immunostaining data show that TNF-α treatment decreased the levels of ZO-1 and VE-cadherin, whereas rhFGF21 administration markedly improved this reduction. PD173074 inhibited the effect of rhFGF21 on TJ and AJ protein expression (Fig. 9A–D). The above results confirm that rhFGF21 likely recovered TNF-α-induced BBB damage by increasing TJ and AJ protein levels via the FGF21/FGFR1/β-klotho signaling system.

HBMECs were pre-treated with 50 ng/mL rhFGF21 with or without the inhibitor PD173074 (10 nM) for 1 h and then treated with TNF-α for 24 h. rhFGF21 increased the activity of the TJ proteins ZO-1 and VE-cadherin in confluent HBMEC monolayers. The FGFR1 inhibitor PD173074 reversed these effects.
rhFGF21 protects against TNF-α-induced BBB injury by upregulating PPARγ through the FGF21/FGFR1/β-klotho receptor complex
A previous study showed that FGF21 activated PPARγ via the FGF21/FGFR1c/β-klotho signaling system. 21 Considering that PPARγ has a neuroprotective effect, we tested whether rhFGF21 can change the level of PPARγ in nuclei of HBMECs using Histone H3 as a nuclear protein marker. The data show that rhFGF21 treatment significantly improved the PPARγ protein level, whereas PD173074 blocked the upregulation effect of rhFGF21 (Fig. 10A,B). To examine the effect of PPARγ on endothelial monolayer permeability, the PPARγ inhibitor GW9662 was applied. The results show that GW9662 reversed the protective effect of rhFGF21 on permeability and endothelial resistance (Fig. 10C,D). These results further confirm that the protective effect of rhFGF21 on BBB integrity is mediated by upregulating PPARγ via FGFR1 and the β-klotho receptor complex.
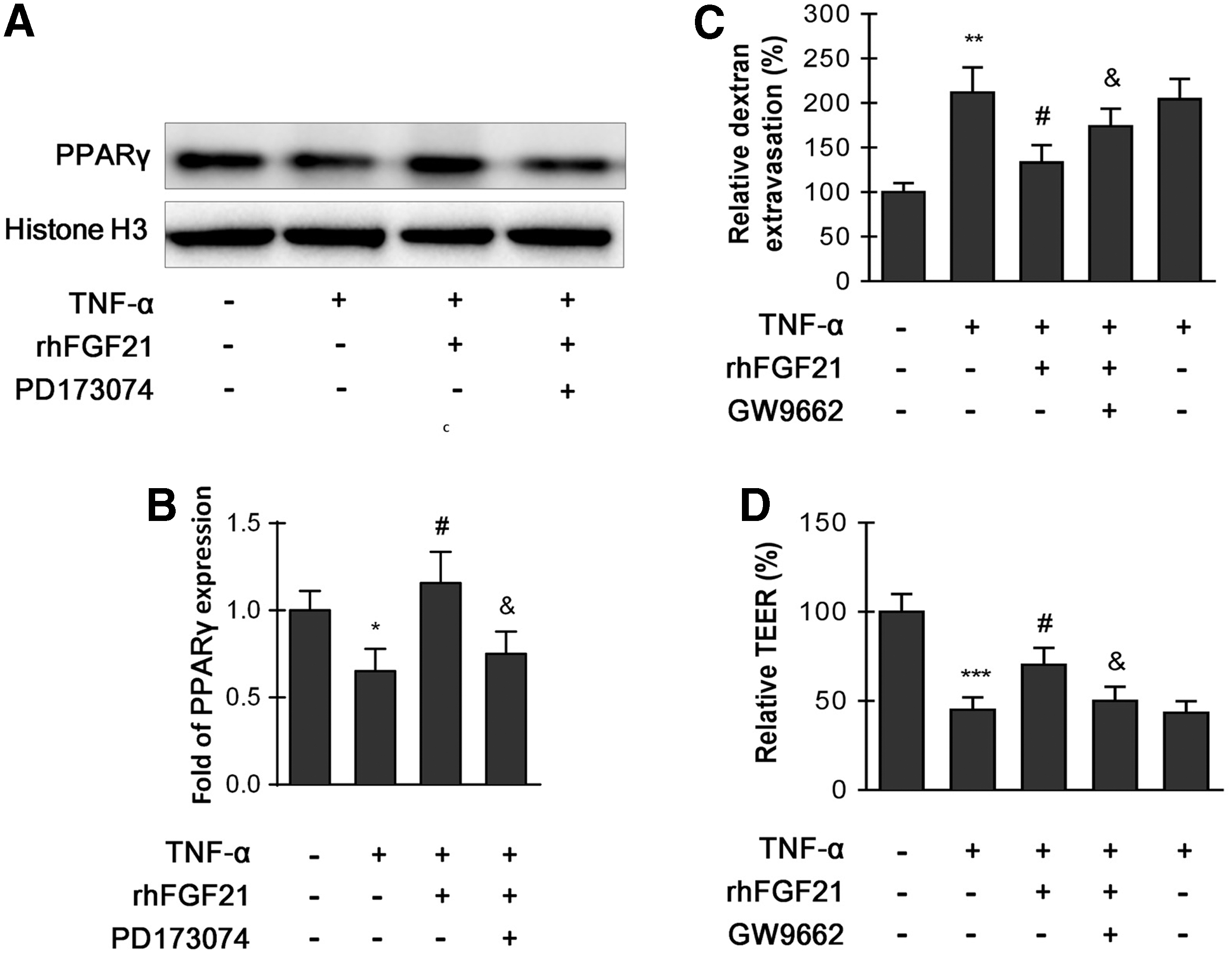
HBMECs were pretreated with 50 ng/mL rhFGF21 with or without inhibitor PD173074 (10 nM) for 1 h, and then treated with TNF-α for 24 h. rhFGF21 alleviates HBMEC monolayer disruption through PPARγ activation.
Discussion
Therapeutic strategies for BBB integrity protection following TBI could have many beneficial downstream effects. In the current study, we confirmed that rhFGF21 attenuates brain edema and BBB disruption and improves neurofunctional deficits in mice after TBI. In addition, TNF-α-induced HBMECs were used to establish an in vitro BBB model, and rhFGF21 ameliorated the TNF-α-induced increased permeability of HBMECs. Both in vivo and in vitro experiments demonstrate that rhFGF21 protected the BBB and upregulated the expression of the TJ and AJ proteins ZO-1, VE-cadherin, occludin, and claudin-5 by activating PPARγ through formation of the rhFGF21/FGFR1/β-klotho complex. These data suggest that rhFGF21 may be used as a potential therapeutic agent for BBB protection and TBI treatment.
Disruption and dysfunction of the BBB results in the formation and development of brain edema, which is responsible for more than half of all deaths after severe TBI. 38 Protection and stabilization of the BBB following TBI may be a potential approach to reduce secondary brain insult, including inflammation, excitotoxicity, oxidative stress, and neurodegeneration. 11,40,41 Several studies have reported that certain agents can protect BBB integrity and reduce brain edema. Glibenclamide, rhein, and Satureja khuzistanica Jamzad essential oil have been reported to exhibit protective effects on the BBB dysfunction and brain edema. 41 –43 The present study is the first to reveal the capacity of FGF21 to preserve the BBB after TBI in mice and the mechanism of action of this potential protective ability.
TNF-α, a pro-inflammatory cytokine, is involved in various central nervous system (CNS) disorders, including TBI. 44 The pathophysiological effect of TNF-α in brain injury, especially in the acute stage, has been well reviewed. 40,45 TNF-α dose-dependently reduced ZO-1 expression and occludin co-association in HBMECs and successfully established an in vitro BBB model. 31,36 To confirm the protective effect of rhFGF21 on BBB disruption and explore the mechanisms underlying this protective effect on BBB damage, a BBB model was simulated in vitro with TNF-α-treated HBMECs. Previous studies have demonstrated that HBMECs subjected to TNF-α exhibited BBB integrity disruption and increased permeability, which is consistent with our study. 46
FGF21 is a member of the FGF family with potential therapeutic benefits for obesity and diabetes treatment. 47 Previous studies have demonstrated that FGF21 functions as a metabolic regulator through a cell-surface receptor complex that consists of FGFR and the single-transmembrane protein β-klotho. 48,49 Although the definite mechanism of action of the interaction and activation between FGF21 and the FGFR1c/β-klotho complex is not fully clarified, recent studies have shown that β-klotho may work as a structural scaffold that induces FGF21 binding to FGFR, followed by dimerization and activation of FGFR. 50 In the current study, both in vitro and in vivo experiments demonstrated that rhFGF21 increased the levels of β-klotho, thus improving FGFR1 activity. These findings are consistent with previous reports showing that β-klotho expression could be stimulated by special pathways, such as high glucose conditions. 51,52 To further confirm the activity of β-klotho, β-klotho siRNA knockdown and co-IP experiments were performed and demonstrate that β-klotho is necessary for rhFGF21 to activate FGFR.
PPARγ activation can reduce the release of pro-inflammatory cytokines and suppress the inflammation response. PPARγ agonists have been demonstrated to exert positive effects on a variety of diseases, including TBI, stroke, and spinal cord injury in animal models. 53 –56 Although no studies have been published regarding the relationship among FGF21, the FGFR1c/β-klotho complex, and subsequent PPARγ activity, a recent study has reported that PPARγ activation increased β-klotho expression and the FGF21 sensitivity and signaling response that is involved in lipid metabolism. 57 There is very little information regarding the effects of FGF21 following TBI. However, as members of the FGF family, aFGF and bFGF exhibited therapeutic properties in preserving BBB integrity after TBI by regulating the PI3K/Akt/Rac-1 signaling pathway. In the present study, both in vivo and in vitro, rhFGF21 activated PPARγ and upregulated TJ protein expression. To further confirm these effects, a FGFR1-specific inhibitor, PD173074, and PPARγ-specific inhibitor, GW9662, were supplied to a BBB in vitro and in vivo model. In vitro, PD173074 blocked the upregulation effect of rhFGF21 on FGFR1 phosphorylation and β-klotho expression; after GW9662 treated, the BBB permeability was increased, indicating that rhFGF21 may have a potential role in maintaining BBB integrity via PPARγ activation, which is consistent with previous research. 57 In addition, our in vivo study demonstrates FGFR1 and PPARγ-specific inhibitor PD173074 and GW9662 co-administration with rhFGF21 reverses the protective effects on neurofunctional deficits and EB extravasation. Overall, FGF21 markedly reduced brain damage and edema and preserved BBB integrity following TBI partially due to formation of the FGF21/FGFR1/β-klotho complex and activation of PPARγ (Fig. 11).

Schematic representation of the role of rhFGF21 in BBB function following TBI. In vivo, TJ and AJ protein expression levels were decreased, and TBI induced the BBB disruption in a mouse model after TBI. TNF-α, a pro-inflammatory cytokine, is involved in various CNS disorders, including TBI. In vitro, TNF-α-treated stimulated HBMECs' damage were used to exhibit in vitro a BBB disruption model. The specific FGFR1 and PPARγ inhibitors PD173074 and GW9662, respectively, were also applied. In conclusion, rhFGF21 upregulated TJ and AJ protein expression and protects BBB functions through the FGFR1/β-klotho complex via inducing PPARγ activation. AJ, adhesion junction; BBB, blood–brain barrier; CNS, central nervous system; HBMECs, human brain microvascular endothelial cells; PPARγ, peroxisome proliferator-activated receptor gamma; rhFGF21, recombinant human fibroblast growth factor 21; SEM, standard error of the mean; TJ, tight junction; TNF-α, tumor necrosis factor alpha; TBI, traumatic brain injury.
In conclusion, the current study revealed that exogenous rhFGF21 treatment markedly ameliorated neurofunctional deficits, BBB breakdown, and brain edema in a mouse model of TBI. Moreover, both in vivo and in vitro studies demonstrate that rhFGF21 activated PPARγ by forming an FGF21/FGFR1/β-klotho complex and rescued the expression of junction proteins. The present data suggest that FGF21 protects the BBB through FGF21/FGFR1/β-klotho complex formation and PPARγ activation. Taken together, these results suggest that FGF21 may be a potential therapeutic approach for TBI treatment. Due to the promising therapeutic strategy of rhFGF21 for BBB injury and the beneficial downstream effects, more detailed studies are needed to explore and understand the downstream effects induced by BBB protection.
Footnotes
Acknowledgments
This study was supported by research grants from the Public Welfare Technology Application Research Foundation of Zhejiang Province (No.2017C33080), National Natural Science Foundation of China (Grant No. 81771284), and the Opening Project of Zhejiang Provincial Top Key Discipline of Pharmaceutical Sciences.
Author Disclosure Statement
No conflicting interests exist.