
Abstract
Spinal cord injury (SCI) frequently results in chronic neuropathic pain (CNP). However, the understanding of brain neural circuits in CNP modulation is unclear. The present study examined the changes of ventral tegmental area (VTA) putative GABAergic and dopaminergic neuronal activity with CNP attenuation in rats. SCI was established by T10 clip compression injury (35 g, 1 min) in rats, and neuropathic pain behaviors, in vivo extracellular single-cell recording of putative VTA gamma-aminobutyric acid (GABA)/dopamine neurons, extracellular GABA level, glutamic acid decarboxylase (GAD), and vesicular GABA transporters (VGATs) were measured in the VTA, respectively. The results revealed that extracellular GABA level was significantly increased in the CNP group (50.5 ± 18.9 nM) compared to the sham control group (10.2 ± 1.7 nM). In addition, expression of GAD65/67, c-Fos, and VGAT exhibited significant increases in the SCI groups compared to the sham control group. With regard to neuropathic pain behaviors, spontaneous pain measured by ultrasound vocalizations (USVs) and evoked pain measured by paw withdrawal thresholds showed significant alteration, which was reversed by intravenous (i.v.) administration of morphine (0.5–5.0 mg/kg). With regard to in vivo electrophysiology, VTA putative GABAergic neuronal activity (13.6 ± 1.7 spikes/sec) and putative dopaminergic neuronal activity (2.4 ± 0.8 spikes/sec) were increased and decreased, respectively, in the SCI group compared to the sham control group. These neuronal activities were reversed by i.v. administration of morphine. The present study suggests that chronic increase of GABAergic neuronal activity suppresses dopaminergic neuronal activity in the VTA and is responsible for negative emotion and motivation for attenuation of SCI-induced CNP.
Introduction
S
Over the course of a decade, the literature has suggested that dopaminergic transmission in the brain is highly correlated with pain behaviors. 9,10 Recently, Voulalas and colleagues documented the decrease in dopaminergic transmission in the periaqueductal gray after spinal lesion with neuropathic pain behaviors. 11 Additionally, fast scan cyclic voltammetry revealed that noxious heat stimulation decreased dopamine (DA) signaling in the nucleus accumbens (NAc), suggesting that mesolimbic dopaminergic transmission is well associated with pain behaviors. 12 The mesolimbic dopamine system in the midbrain is predominantly composed of dopaminergic pathways between the ventral tegmental area (VTA) located in the midbrain and the NAc located in the basal forebrain rostral to the preoptic area of the hypothalamus. 13 However, GABAergic interneurons in the VTA regulate the activity of VTA dopaminergic neurons and determine the final level of DA in the NAc. 14 Consequently, extracellular DA in the NAc characterizes motivation behaviors. 15,16 Therefore, hyperactivity of VTA gamma-aminobutyric acid (GABA) neurons results in DA-dependent behaviors, such as mood disorders, anxiety, and pain. 17,18
In addition, previous reports have demonstrated that activation of the endogenous opioid system contributed to the enhanced dopaminergic neuronal activity in the VTA, followed by the increase of DA levels in the NAc. 19,20 Although recent studies have suggested that the dopaminergic system contributes to pain neurotransmission, the role of VTA GABAergic neurons in SCI-induced CNP has not been clearly investigated. In the present study, we aimed to determine the roles of VTA GABA-dopaminergic pathways in SCI-induced CNP.
Methods
Animals
Male Sprague-Dawley rats (200–225 g, n = 218) were obtained from Harlan Sprague-Dawley Inc (HyoChang Co, South Korea), housed with a light/dark cycle of 12/12 h, and fed ad libitum. The protocol for experiments using animal was reviewed and approved (Protocol#: DHU2015-072) by the Animal Care Committee of Daegu Haany University (Kyungsansi, South Korea), and the experiments were carried out in accord with the NIH Guide for the Care and Use of Laboratory Animal.
Spinal cord injury
SCI at T10 was performed using clip compression (35 g, 1 min) under anesthesia by intraperitoneal (i.p.) injection of sodium pentobarbital (60 mg/kg). Low thoracic laminectomy at the T8/9 vertebral segment exposed the T10 spinal level. In the age-matched control group, sham surgery (no spinal compression) was performed with anesthesia and laminectomy only. Four rats were excluded from the present study because of lack of recovery of locomotion and the presence of urinary inflammation at 3 weeks. Because all SCI rats exhibited mechanical allodynic behaviors, as determined by decrease of mean paw withdrawal thresholds (PWTs; less than 7) to von Frey filaments (VFFs), the SCI-induced pain and nonpain groups were not separated in the present study.
Measurement of gamma-aminobutyric acid levels in the ventral tegmental area
In order to measure GABA levels in the VTA (SCI n = 6 and sham n = 6), the guide cannula implantation was performed under anesthesia induced by administration of sodium pentobarbital (60 mg/kg, i.p.). Each rat was placed on a stereotaxic frame (Kopf Instruments, Tujunga, CA) and restrained by the head fix with blunt ear bars and mouth/nose clamp. Frontal and parietal skull bones were exposed by incision of the scalp at the midline. Three surgical screws were implanted into the skull. A CMA/12 guide cannula (CMA microdialysis AB, Torshamnsgatan, Kista, Sweden) and 26-gauge stainless steel guide cannula (Plastics One, Roanoke, VA) were implanted for microdialysis and -injection, respectively, and fixed using dental cement. The surgical coordinates were as follows: VTA (guide cannula): anterior-parietal (AP) −5.8 mm, medial-lateral (ML) −0.6 mm, and dorsoventral (DV) −7.4 mm from the skull. At the end of the experiments, rats were sacrificed and whether the guide cannula was placed correctly was verified. In order to measure GABA levels in the VTA, 20 μL of microdialysis samples were mixed with 15 μL of the working reagent (o-phthaldialdehyde [OPA]/2-mercaptoethanol [βME]). The mixed sample was separated using a reversed-phase column (Waters Xterra MS C18, 3.0 × 50 mm, 2.5 μm; Waters Corporation, Milford, MA), and isocratic measurement was performed using high-performance liquid chromatography (HPLC) with an electrochemical detector (ECD; ESA, Coulochem II, Model 5200A). A stock solution of the OPA/βME reagents was prepared by dissolving 27 mg of OPA in 1 mL of HPLC-grade methanol and adding 5 μL of βME and 9 mL of 0.1 M of tetraborate buffer (pH 9.3). The working OPA/βME reagent, prepared daily before performing HPLC analysis, was prepared by diluting 1 mL of the OPA/βME reagent with 8 mL of 0.1 M of tetraborate buffer. The mobile phase contained 0.1 M of Na2HPO4, 20% methanol, 3.5% acetonitrile (pH 7.4) with phosphate acid, and perfused at a rate of 0.6 μL/min. ECD cell voltage complied with the guard cell electrode (650 mV), detector electrode 1 (150 mV), and detector electrode 2 (550 mV). To obtain consistent retention times, the column was heated to 30°C.
Western blot analysis for gamma-aminobutyric acid synthesis enzyme
In order to isolate the VTA region, 20 rats (SCI n = 10 and sham n = 10) were transcardially perfused with heparinized saline under deep anesthesia with inhalation of isoflurane. Rats were decapitated, and the brain was immediately removed. Using the rat brain mold (BS-Z4000C; Braintree Scientific Inc., Braintree, MA), the VTA area was isolated and punctured on the ice. Tissues were immediately homogenized in radioimmunoprecipitation assay buffer (Thermo Scientific, Rockford, IL) containing protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany) using a glass homogenizer. Total protein concentrations were determined using the bicinchoninic acid protein assay kit (Sigma-Aldrich, St. Louis, MO). Lysates (30 μg of protein/lane) were electrophoresed in 10% sodium dodecyl sulfate polyacrylamide gel and then electrophoretically transferred to a nitrocellulose membrane. After incubation in a blocking solution of 5% skimmed milk in Tris-buffered saline containing 0.1% Tween 20 for 1 h at room temperature (RT; 20–22°C), the membrane was probed with anti–glutamic acid decarboxylase (GAD)65 or -GAD67 antibody (antirabbit, 1:500; Santa Cruz Biotechnology Santa Cruz, CA) or monoclonal anti-β-actin antibody (1:5000; Sigma-Aldrich) overnighted at 4°C, followed by incubation with horseradish peroxidase (HRP)-conjugated antirabbit immunoglobulin G (IgG; 1:5000, Cell Signaling Technology, Danvers, MA) or HRP-conjugated antimouse IgG (1:10,000, Cell Signaling Technology) for 4 h at 4°C. Antigen-antibody complexes were visualized by incubation in an enhanced chemiluminescence western blotting analysis system (GE Healthcare, Little Chalfont, UK) for 1 min. Membranes were then exposed to a medical x-ray film (AGFA, Mortsel, Belgium) for 1–10 min. Density of each band was measured using a computer-assisted image analysis system (LabWorks Image acquisition and Analysis software; BioImaging Systems, UVP, Cambridge, UK).
Immunohistochemical analysis
In order to evaluate c-Fos (a marker of neuronal activation; SCI n = 6 and sham n = 7) and vesicular GABA transporters (VGATs; SCI n = 8 and sham n = 8) expression in the VTA, rats were transcardially perfused with physiological saline followed by 4% paraformaldehyde under anesthesia induced by sodium pentobarbital (100 mg/kg, i.p.). Brains were immediately removed and subjected to post-fixation processing in the same fixative. The following day, the VTA regions were removed using Rodent Brain Matrix (RBM-3000C for rat; ASI Instruments, Warren, MI) on ice and were cryoprotected in a 30% sucrose solution for a minimum of 48 h. Brains were cryosectioned into 30-μm-thick sections, and every fifth section was collected. Brain sections were incubated in a blocking solution containing 5% normal goat serum and 0.3% Triton X-100 in 0.1 M of phosphate-buffered saline (PBS) for 2 h at RT. After rinsing in PBS, sections were incubated with primary antibody for c-Fos (1:200; Sigma-Aldrich) overnight at 4°C. Coexpression of VGAT (1:200; EMD Millipore, Temecula, CA)/neuronal nuclei (NeuN; 1:500; EMD Millipore) was also examined in the VTA. Sections were then processed with secondary antibodies; donkey anti-rabbit Alexa Fluor 568 (red; 1:500, Thermo Fischer Scientific, MA, USA) and 488 (green; 1:500) for 3 h at RT. All tissues were mounted on a gelatin-coated glass slide and coverslipped with mounting media and 4’,6-diamidino-2-phenylindole (DAPI). Immunofluorescence images of VGAT were captured and analyzed using a confocal laser scanning microscope (LSM700; Carl Zeiss, Wetzlar, Germany) and Zen 2009 programs.
Measurement of spontaneous pain in naïve groups
In order to measure spontaneous pain induced by SCI, the audible ultrasound vocalizations (USVs) was measured. Before USV measurement, an individual naïve rat was placed in a Plexiglass rat cage and adapted (30 min) to the USV test box (70 × 50 × 60 cm) for 3 consecutive days to avoid stresses by environmental changes. USVs from individual rats were measured using high-performance microphones (Ultrasonic USB Microphone 250k; Dodotronic, Rome, Italy) attached by a hole at the center of the ceiling and positioned 30 cm above the head. Recording and analysis of USVs was performed using Abisoft-SASLap Pro (Version 5.2; Avisoft Bioacoustics, Glienicke, Germany). Pain-related USV frequency ranges (18–35 kHz) were measured in accord with previous reports. 21,22 The USV test box with air circulation was shielded from auditory and visual stimulation. In order to monitor the internal environment during recordings in USV boxes in a naïve group (n = 5), temperature, humidity, and CO2 concentration were measured with a CO2/humidity/temperature data recorder (MCH-383SD; Lutron Electronic, Coopersburg, PA). Spontaneous pain behaviors induced by formalin were assessed in a separate naïve group (n = 19) according to previous study. 23 Briefly, subcutaneous injection of formalin (1% and 5%, 30 μL) in the hind paw was performed using a 32-gauge needle and held for 1 min to prevent backdraft after injection. Formalin-induced pain behaviors, such as licking, flinching, biting, and lifting, were recorded for 1 min in 3-min intervals. In addition, these spontaneous pain behaviors were recorded to evaluate the feasibility of the USV box in a separate naïve group (formalin n = 7 and vehicle n = 5).
Recovery of locomotion
The return of spontaneous locomotion was assessed using the open field test (SCI n = 12 and sham n = 10) for a score of 0–21 on the Basso, Beattie, and Bresnahan (BBB) locomotor scale. This scale allows for validation of hindlimb locomotion and weight-bearing ability in order to evaluate somatosensory function. BBB scores 0–7 rank movement of the primary three joints (hip, knee, and ankle), scores 8–13 rank weight-bearing stance to coordinated stepping, and scores 14–21 rank the return of toe clearance, paw position, and trunk stability. 24 Scores were determined for each hindlimb (left and right) and combined because there were no significant differences between the right and left hindlimb. BBB scores were assessed every 3 days for 30 days.
Pain measurement in spinal cord injury groups
Time courses of USVs and evoked pain were assessed in the locomotion test group. USV assessment (SCI n = 6 and sham n = 5) was performed every week up for 6 weeks. Mechanically evoked pain (SCI n = 6 and sham n = 5) was measured by the changes of mean PWTs to six applications of calibrated VFFs (beginning with 4.31, and the series of VFFs was 3.61 [0.45 g], 3.84 [0.74 g], 4.08 [1.26 g], 4.31 [2.04 g], 4.56 [3.31 g], 4.74 [5.50 g], 4.93 [8.32 g], and 5.18 [14.45 g]; Semmes-Weinstein monofilaments; Stoelting Co., Wood Dale, IL). PWTs were determined by 50% withdrawal mechanical threshold using the formula: log (50% threshold) = 10(Xf+κδ)/10000. Xf = value of the final VFF (log unit), κ = correction factors (from calibration table), and δ = mean differences of log units between stimuli. The 18-g pressure of PWTs (according to the up-down method) was selected as the cut-off value. 21 To test whether pain behaviors were influenced by opioids, morphine (0.5, 1, and 5 mg/kg; 0.5 mL, intravenous [i.v.]; SCI n = 6; sham n = 6 in each group), and saline (control, n = 5) were delivered over 2 min by a pre-implanted jugular vein catheter using a stereotaxic injector. The jugular vein catheter was implanted using PE10 tubing (10 cm) under mild anesthesia using isoflurane (1.5%) inhalation 5 days preceding morphine administration. A 2-cm section of the free end was left exposed at the nape of the neck.
In vivo ventral tegmental area putative GABAergic and dopaminergic neuronal activity
The spontaneous activity of the putative GABAergic and dopaminergic neurons in the VTA was investigated using in vivo extracellular single-cell recording techniques. In order to carry out head adjustment on the stereotaxic frame, rats were anesthetized with sodium pentobarbital (60 mg/kg, i.p.). A single-unit putative GABAergic (SCI n = 10 and sham n = 6) and dopaminergic (SCI n = 5 and sham n = 5) neuronal activity recording at the VTA (AP, −5.8 to −6.2 mm; ML, 0.4–1.0 mm; DV, −6.9 to −8.2 mm from the skull) was performed using a single glass microelectrode (internal solution, 2 M of NaCl; resistance, 10–25 MΩ) under anesthesia by inhalation of isoflurane (1.0–1.5%). Owing to the variable action potential frequencies (5–60 Hz in GABAergic neurons and 0.5–10.0 Hz in dopaminergic neurons), only putative GABAergic and dopaminergic neurons with <1- and >2-ms durations, respectively, were included. In this study, GABAergic and dopaminergic neurons were not clearly characterized in the VTA by immunohistochemical analysis, and thus neurons were paired with putative GABAergic and dopaminergic neurons according to their in vivo electrophysiological properties. 25,26 After identification of single-unit activity, the spontaneous activity was recorded over 30 min. Unit activity was amplified and filtered (low filter, 300 Hz; high filter, 10 kHz; voltage gain, 104; ISO-80; WPI, Sarasota, FL) and fed directly into the data acquisition unit (microCED-1401; Cambridge Electronic Design, Cambridge, UK). The data were stored on computer in order to construct the waveforms or plot peristimulus time histograms (spikes/1-sec bin width; Spike2 software). In order to assess whether neuropathic pain attenuation was influenced by the putative VTA GABAergic (n = 5 in each group) and dopaminergic (n = 6 at 1 mg/kg) neuronal activity, morphine (0.5–5.0 mg/kg; 0.5 mL, i.v.) and saline (for control, n = 5) were delivered over 2 min by a pre-implanted jugular vein catheter using a stereotaxic injector. To ensure that single-unit activity was held for the duration of the recording experiment, the Spike2 program was used to confirm matching action potential shape and amplitude.
Statistical analysis
Data are expressed as means ± standard error. Behavioral data analyses were performed using repeated one- or two-way analysis of variance (ANOVA), and the Student-Newman-Keuls method was used for multiple comparisons. Electrophysiological and bioassay data analyses were performed using the t-test for comparison. An alpha level of significance was set at 0.05 for all statistical tests using SigmaStat program (Ver 3.1).
Results
Microdialysis
Microdialysis-ECD detection identified the elevation of VTA GABAergic tone at 40 days post-injury. Figure 1A represents the typical detection of extracellular GABA in the VTA as measured by microdialysis-ECD and statistical analysis. Extracellular GABA (50.5 ± 18.9 nM; n = 6) levels in the VTA were significantly increased compared to the sham control group (10.2 ± 1.7 nM; n = 6; p < 0.05).

Increase of VTA GABAergic tone post-SCI. (
Western blot analysis
To assess the mechanism underlying the increase of GABA levels in the VTA, the GABA synthase enzyme, GAD, was measured at 40 days post-injury. Densities of GAD65 and GAD67 in the SCI (n = 10) and sham control (n = 10) groups were 1.64 ± 0.12 and 1.90 ± 0.1, and 1.32 ± 0.1 and 1.64 ± 0.12, respectively, and showed significant differences (p < 0.05; Fig. 1B).
Immunohistochemical analysis
To measure the changes in neuronal activation and VGAT expression in the VTA at 40 days post-injury, immunostaining to evaluate the expression of c-Fos and VGAT was performed. Figure 2A demonstrates that the SCI group showed the increase of c-Fos immunopositive cells in the VTA (52.7 ± 8.4; n = 6) compared to the sham control group (24.5 ± 5.6; n = 7; p < 0.05). Because excess GABA is regulated by GABA transporters, expression of VGAT in the VTA was determined. VGAT intensity was significantly increased in the SCI group (14.5 ± 1.8; n = 8) compared to the sham control group (7.4 ± 1.3; n = 8; p < 0.05; Fig. 2B).
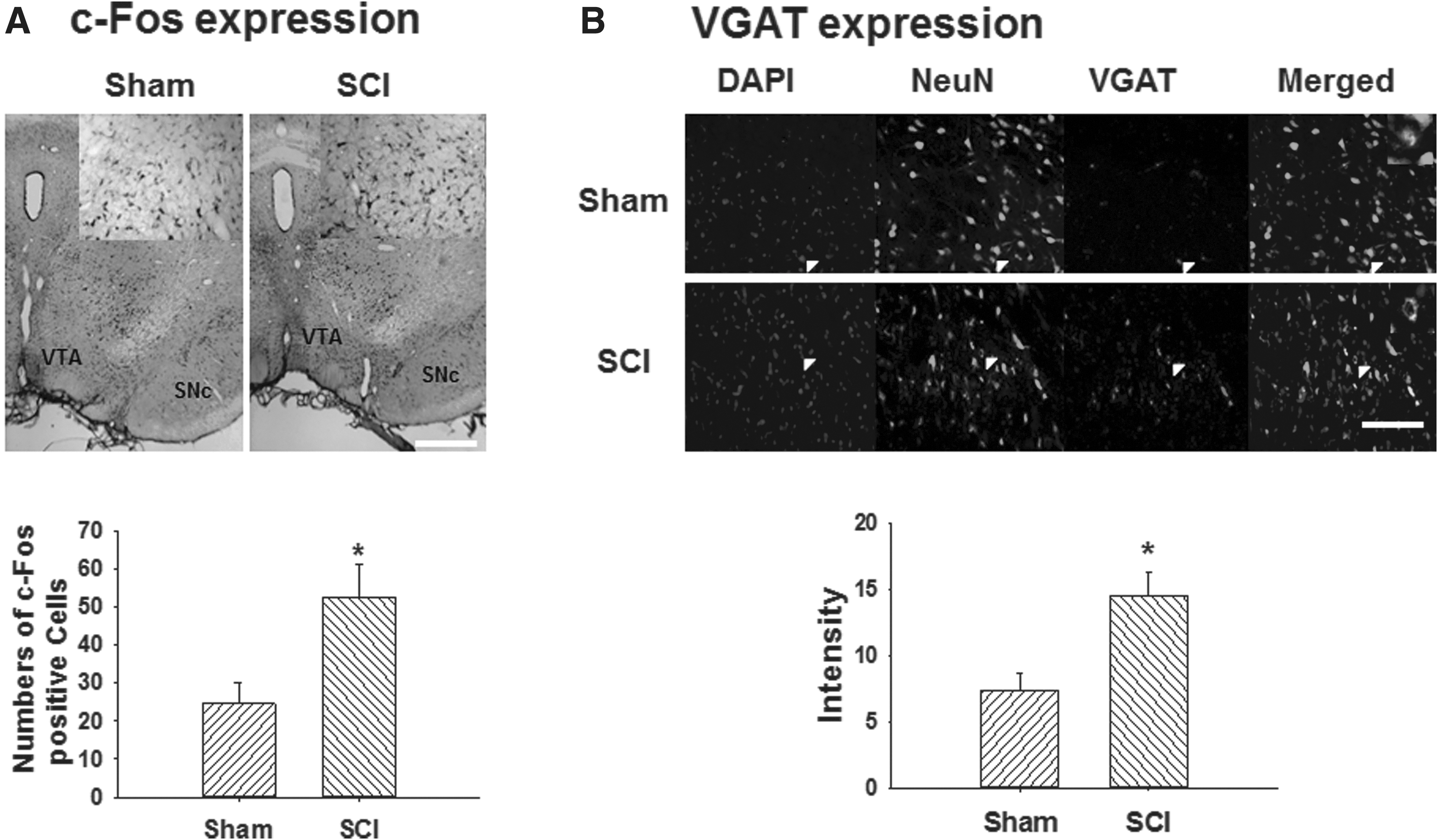
Neuronal activation and expression of GABA transporter in the VTA post-SCI. (
Behaviors
USVs were measured to determine whether SCI caused spontaneous pain. The internal environment of the USV test box was first examined to ensure that no stress-inducing environmental changes occurred. Figure 3A depicts the levels of the various environmental measures during the test. Over the course of 1 h, the USV test box maintained constant levels of CO2, humidity (%), and temperature (°C; the first naïve group, n = 5). In addition, spontaneous pain caused by formalin injection was measured in a second naïve group in order to confirm the feasibility for the in vivo spontaneous pain measurement with the USV box. Formalin injection (1% and 5%, 30 μL, subcutaneous, n = 19) at the hind paw demonstrated dose-dependent and typical biphasic pain responses, including licking, biting, lifting, and flinching (Fig. 3B). In addition, the USV test (the third naïve group, n = 7) revealed biphasic patterns similar to the response patterns observed in formalin-induced pain behaviors, whereas the vehicle group (n = 5) did not exhibit a significant response (Fig. 3C).

Feasibility of ultrasound vocalizations (USVs) in in vivo pain measurement (
Pain behaviors critically depend on the locomotion ability of the hind paw. Therefore, the BBB scores of the hind paws were determined to confirm the recovery of motor functions that were affected by SCI. Before injury, the mean BBB score in the SCI group (n = 12) was 21 ± 0.0. At day 1 post-injury, the mean BBB score was significantly lower in the SCI group (0.03 ± 0.03; n = 12) compared to the pre-injury scores and the sham control group (n = 10; p < 0.05; Fig. 4A). However, the mean BBB score at 28 days post-injury in the SCI group was 10.59 ± 1.02, indicating weight-bearing, posture, stepping, and withdrawal responses to mechanical stimulation. To determine the time courses of spontaneous and evoked pain behaviors, the locomotion test groups underwent USV and evoked pain tests. Two weeks post-injury, USVs exhibited significant increases (maintained over a 1-month period) in the SCI group (n = 6) compared to the sham control group (n = 5; p < 0.05; Fig. 4B). Evoked pain was also increased over a 1-month period (Fig. 4C). To avoid multiple tests being performed on the same group, different groups underwent the assessment of morphine effects on spontaneous and evoked pain behaviors. At 40 days post-injury, USVs were significantly increased in the SCI-saline group (24.7 ± 6.7; n = 6) compared to the sham control group (3.6 ± 0.7; n = 5; p < 0.05). However, USVs were significantly attenuated in the SCI group after morphine treatment at 0.5 mg/kg (7.8 ± 3.3; n = 6) and 1 mg/kg (8.9 ± 3.4; n = 6) compared to the SCI-saline group (24.7 ± 6.7; n = 6; p < 0.05; Fig. 4D). At 40 days post-injury, mean PWTs were significantly decreased before morphine treatment in the SCI group (2.78 ± 0.6 g; n = 12) compared to sham controls (15.5 ± 1.4 g; n = 6; p < 0.05). However, PWTs were significantly increased in the SCI group receiving 1 mg/kg of morphine (7.5 ± 0.9 g; n = 6) compared to before treatment (4.4 ± 0.4 g). PWTs did not significantly change in the SCI group receiving 0.5 mg/kg of morphine (n = 6) compared to before treatment and the 1-mg/kg morphine group (p < 0.05; Fig. 4E).

Attenuation of pain behaviors by treatment with intravenous morphine. (
Electrophysiology
To measure the VTA putative GABAergic neuronal activity, in vivo extracellular single-cell recording was performed, as demonstrated in Figure 5A. Figure 5B,C represents the typical waveforms and peristimulus histograms of VTA putative GABAergic neurons in the sham control and SCI groups, respectively. At 40 days post-injury, GABAergic neuronal firing rate was significantly increased in the SCI group (13.7 ± 1.7 spikes/sec; n = 10) compared to the sham control group (7.3 ± 1.1 spikes/sec; n = 6; p < 0.05; Fig. 5D). In order to evaluate the effects of GABAergic activity on dopaminergic signaling, dopaminergic neuronal activity was measured. Figure 5E represents the diagram of in vivo extracellular single-cell recording of VTA putative dopaminergic neurons. Figure 5F,G represents the activity of VTA putative dopaminergic neurons in the sham control and SCI groups, respectively. At 40 days post-injury, VTA putative dopaminergic neuronal firing rates were significantly decreased in the SCI group (1.2 ± 0.5 spikes/sec; n = 5) compared to the sham control group (2.7 ± 0.7 spikes/sec; n = 5; p < 0.05; Fig. 5H).

In vivo extracellular recording of VTA neurons. Diagram of in vivo extracellular single-cell recording for the VTA putative GABAergic neurons (
To assess whether VTA putative GABAergic neuronal activity was influenced by the opioid system, VTA putative GABAergic and dopaminergic neuronal activity were evaluated with administration of 0.5–5.0 mg/kg of morphine. Figure 6 represents the real-time waveforms and peristimulus histograms for the VTA putative GABAergic and dopaminergic neuronal firing rates with morphine treatment. Before morphine injection (1 mg/kg, i.v.), VTA putative GABAergic neuronal activity was 13.4 ± 2.5 spikes/sec (n = 5) and was significantly decreased at 30 min after morphine treatment (8.1 ± 1.1 spikes/sec; p < 0.05, Fig. 6A). Before morphine injection (1 mg/kg, i.v.), the VTA putative dopaminergic neuronal firing rate was 1.3 ± 0.8 spikes/sec (n = 6) and was significantly increased at 30 min after morphine treatment (5.4 ± 0.8 spikes/sec; p < 0.05; Fig. 6B).

In vivo extracellular recording of VTA putative GABA and dopamine neurons with morphine (MOR) treatment. (
Discussion
In the present study, we suggested that SCI resulted in the increase of VTA putative GABAergic neuronal activity followed by the decrease of putative dopaminergic transmission. In addition, biochemical and electrophysiological changes of VTA GABAergic tone were correlated with CNP behaviors post-SCI. Our results support previous reports that hypodopaminergic tone and reduced responsiveness of the mesolimbic dopamine system were developed in the chronic pain condition. 27 It has been well documented that the decrease of VTA dopaminergic activity followed by depletion of dopamine levels in the NAc triggered negative emotions, such as pain. 20,28 Therefore, we suggest that the CNP condition has a similar mechanism of negative emotions, and dopamine therapies are useful therapeutic tools for CNP attenuation post-SCI. 29
In human SCI studies, chronic pain conditions indicated disruption of VTA GABA-dopaminergic circuits and strong motivation to avoid such pain. For example, fibromyalgia-induced pain patients exhibited disruption of GABA/dopaminergic circuits at the VTA-NAc and enhanced pain transmission. 30,31 In brain neural circuits, VTA neurons receive glutamatergic synaptic input from areas such as the prefrontal cortex (PFC). Previous reports have demonstrated that SCI caused cortical hyperexcitability with neuropathic pain by activation of glutamate receptors. 32,33 In addition, neuropathic pain induced by peripheral nerve injury showed enhanced synaptic currents of NMDA and AMPA in the medial PFC with linear correlation of pain thresholds. 34 Although no studies have suggested the presence of direct circuits between glutamate inputs of the cortex to the VTA, glutamatergic input to VTA GABAergic neurons has been speculated. For example, optogenetic stimulation of the lateral habenula (LHb) efferent terminals in the RMTg (the rostromedial tegmental nucleus) and posterior VTA inhibited firing of VTA dopaminergic neurons, and was blocked by glutamate receptor antagonist. In addition, VTA GABAergic neurons received glutamatergic input from the LHb that resulted in GABA activation-induced inhibition of dopamine neuronal activity. 35 Therefore, we suggest that one mechanism for the enhancement of VTA GABAergic neuronal activity may be mediated by glutamatergic inputs from the LHb. Thus, glutamatergic input would be an important factor for the increase of VTA GABAergic neuronal activity. However, it has been noted that the glutamatergic input from the lateral dorsal tegmentum activates VTA DA neurons, 5 suggesting that the glutamatergic circuits between the cortex and the VTA GABA/dopamine neurons remain a target for further study in SCI-induced neuropathic pain. Consequently, the decrease in extracellular dopamine levels in the NAc may trigger dopamine-dependent negative emotional behaviors, such as pain, that may induce motivation for pain attenuation. 36 In addition, western blot and double immunofluorescence investigations in the present study demonstrated the increase of GABA synthase enzymes, such as GAD65 and GAD67, c-Fos, and VGAT, expression in the VTA in SCI-induced CNP. It is well known that high levels of extracellular glutamate induce the activation of glutamate-GABA shunts, resulting in increased GAD expression. 37 However, it is notable that VGAT expression was increased in the VTA post-SCI. Previous studies have suggested that the downregulation of VGAT contributed to the pathological pain. 38 However, others have reported the upregulation of GABA transporter 1 (expressed in the presynaptic terminal) in the spinal dorsal horn of paclitaxel-induced neuropathic pain 39,40 and in the gracile nucleus of neuropathic pain caused by peripheral nerve injury. 40 Although the specific mechanism is not clear, the present results suggested that the upregulation of VGAT consistently contributed to the enhanced GABA neuronal activity in the VTA.
In the VTA, the majority of VTA GABAergic neurons express the μ-opioid receptor, suggesting that activity of μ-opioid is critical to the modulation of GABAergic neuronal activity. 41 –43 In previous SCI studies, spinal intrathecal administration of morphine resulted in significant attenuation of evoked pain, such as in mechanical allodynia induced by SCI. 44 To evaluate the correlation between opioid and VTA GABA-dopaminergic circuits in the VTA on the CNP, we administered morphine intravenously and observed the decrease of GABAergic neuronal activity and the increase of dopaminergic neuronal activity in the VTA. In a previous study, morphine treatment increased the dopaminergic neuronal activity by inhibition of GABAergic neuronal activity in the VTA. 45 Inflammatory pain also demonstrates the relationship between pain development and loss of morphine (MOR) function in the mesolimbic system. 46 In addition, post-surgical pain models indicated that pain attenuation was well correlated with activation of VTA dopaminergic neurons and increase of extracellular dopamine levels in the NAc. 47 However, studies have reported that activation of μ-opioid receptors on the tail of the VTA/RMTg, which project dense GABA inputs into the VTA, modulated dopamine neuronal firing rates. 48,49 This suggested that further study was required for the elucidation of the role of the RMTg in the modulation of VTA GABAergic tone in SCI-induced CNP. Taken together, these data suggest that chronic increase of VTA GABAergic neuronal activity may cause prolonged decrease of dopaminergic activity and contributes to negative emotion in CNP condition.
It has been well documented that the decrease of GABAergic tone is critically involved in neurological brain disorders 50 and neuropathic pain post-SCI. 51,52 We also demonstrated that the decrease of spinal GABAergic tone contributed to neuropathic pain and neuronal hyperexcitability in the spinal dorsal horn post-SCI. 53 However, the present study indicated that the GABAergic activity increase in the VTA was correlated with SCI-induced CNP. Although there is discrepancy surrounding the role of GABA in CNP, it is notable that GABA exhibits regional specific inhibitory functions in the brain. 54 For example, patients with migraine demonstrated increased GABA levels that correlated with pain symptoms. 55 In addition, sciatic nerve injury-induced increase of GABA concentration in the hippocampus resulted in the impairment of spatial learning and memory in neuropathic pain conditions. 56 Further, GABAergic activation in the rostral ventromedial medulla increased pain behaviors by disinhibition of spinal enkephalinergic/GABAergic interneurons. 57 Based on the findings of the present and other previous reports, we suggest that brain GABA has multiple roles in pain modulation depending on the temporal and spatial differences, and the adequate treatment of GABA is essential to pain therapy.
The present study suggested that SCI-induced chronic activation of GABA neurons resulted in the prolonged decrease of dopaminergic transmission in the VTA that may influence on the negative emotion and motivation to avoid CNP post-SCI. However, emerging literature suggested that dysregulation of negative emotion induced by prolonged decrease of dopaminergic transmission also decreased motivation to pain avoidance. 60 Therefore, more longitudinal study is required to elucidate the changes of motivation for the pain avoidance.
Footnotes
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF-2014R1A1A4A01004179, NRF-2017R1D1A3B03035303, and NRF- 2016R1D1A1B03935206).
Author Disclosure Statement
No competing financial interests exist.