
Abstract
Adjudin, a small molecular compound that is used as a male contraceptive, has been reported to play a neuroprotective role in an ischemic stroke injury model. However, its effect on traumatic brain injury (TBI) has not been assessed. Hence, we investigated the effects of adjudin on cerebral edema using a mouse model of TBI and explored the underlying mechanisms. Adult male C57BL/6 mice received controlled cortical impact (CCI) injury, then an injection of adjudin (50 mg/kg). The mice were euthanized 3 days post-CCI injury, and samples were collected for further analysis. Cultured primary mouse astrocytes were used for in vitro experiments. Adjudin treatment significantly attenuated cerebral edema on Day 3 and improved neurobehavioral outcomes on Days 3, 7, and 14 after CCI injury, compared with the vehicle group. Additionally, the evaluation of Evans blue extravasation and expression of tight junction proteins demonstrated remarkable effects of adjudin on blood–brain barrier protection. Further, adjudin treatment significantly decreased the gene and protein expression of aquaporin 4 in post-injury mice and inhibited progression of neuroinflammation in both mice and cultured astrocytes. The Western blot results of the peritraumatic protein samples demonstrated that adjudin significantly blocked the phosphorylation of IKKα, IκBα/β, and NF-κB p65, which resulted in a reduction of NF-κB p65 nuclear translocation. In conclusion, adjudin attenuated the development of TBI-induced cerebral edema at least partly via anti-inflammatory effects and inhibition of the NF-κB pathway. These findings suggest that adjudin is a potential therapeutic intervention preventing the development of cerebral edema after TBI.
Introduction
T
Cerebral edema, which is a fatal pathological phenomenon following TBI, elevates intracranial pressure (ICP) due to an increase in brain volume within the enclosed skull. The increased ICP induces high pressure in the brain parenchyma, hypoxia, and even cerebral hernia, which can quickly lead to death. Cerebral edema occurs mainly during the secondary injury phase post-TBI as a result of several mechanisms, including a hyper-inflammatory response to injury, neuronal excitotoxicity, and mitochondrial dysfunction. 4,5 Among these mechanisms, inflammation plays a key role in inducing secondary injury. 6,7 Many reports have proposed that cerebral edema is associated with the secretion of inflammatory cytokines, and several anti-inflammation methods have reportedly alleviated cerebral edema. 8 –10
Aquaporins (AQPs) are water channel proteins that transport fluids across plasma membranes, and the subtype AQP4 plays a key role in the regulation of cellular water balance. Large amounts of convincing evidence indicate that the formation of cerebral edema is associated with overexpression of AQP4 in the endfeet of astrocytes 11 –13 and AQP4-null mice were found to form less cerebral edema compared with normal mice in a middle cerebral artery occlusion model. Further, in many inflammation-related diseases, AQP4 is upregulated in comparison with the control, which implies an innate relationship between inflammation and upregulation of AQP4. 14 Hence, drugs that reduce neuroinflammation and AQP4 expression may alleviate cerebral edema after brain injury. 8
Adjudin, 1-(2,4-dichlorobenzyl)-1H-indazole-3-carbohydrazide, is a small molecular compound derived from lonidamine. It was initially used as a male contraceptive based on its anti-spermatogenic function. Recently, its other effects, such as sirtuin-3 activating 15 and anti-cancer 16 and anti-oxidative stress 17 functions, have been widely explored. In addition, several studies have demonstrated that adjudin has blood–brain barrier (BBB) protective and anti-inflammatory effects in an ischemic stroke injury model, 18 –20 but the underlying mechanisms require further exploration. In this study, we explored the role of adjudin in the formation of cerebral edema and neurological functional recovery after TBI.
Methods
Experimental design and animals
This study was approved by the Institutional Animal Care and Use Committee of Shanghai Jiao Tong University, Shanghai, China. A total of 276 adult male C57BL/6 mice weighing 20-25 g (Shanghai SLAC Laboratory Animal Corporation, Shanghai, China) were used in this study, and all mice were bred under standard nutritional and environmental conditions. Adjudin was provided by the Mary M. Wohlford Laboratory, Population Council (New York, NY) and was diluted in dimethyl sulfoxide (DMSO; Sigma Aldrich, St Louis, MO); this mixture was then diluted 1:10 in corn oil. Mice were randomly divided into three groups: Group I received no treatment (sham); Group II received controlled cortical impact (CCI) injury to induce TBI followed by an intraperitoneal injection of the vehicle (DMSO mixed with corn oil,1:9); and Group III received CCI injury followed by an intraperitoneal injection of adjudin (50 mg/kg). 19,20 Mice were euthanized 3 days post-CCI injury and brain samples were collected for further analysis.
CCI injury procedure
Prior to inflicting CCI injury, C57BL/6 mice were anesthetized with xylazine (10 mg/kg) and ketamine (75 mg/kg). Then, the mice were fixed to a stereotaxic frame (Stoelting, Wood Dale, IL) with a hot pack placed under the body to maintain body temperature at 37°C. After the scalp was cleaned with ethanol swabs, a 10-mm incision was made over the head. The skin and fascia were then pushed aside, and a craniotomy was performed using a 4-mm trephine over the central aspect of the right parietal bone, 1 mm lateral to the sagittal suture. When performing the operation, care was taken to maintain an intact dura; if dural integrity was breached, that mouse was excluded from the study. After the intact dura was exposed, a contusion injury was created using a 3-mm diameter rounded steel impactor tip of a CCI device (PinPoint Precision Cortical Impactor PCI3000; Hatteras Instruments Inc., Cary, NC), which many labs have employed to simulate TBI. 21,22
Moderate CCI injury was induced by an impact velocity of 1.5 m/sec, deformation depth of 1.5 mm, and dwell time of 100 msec. Sterile cotton was used to control bleeding on the injured cortical surface once the injury was induced, and the cranial opening was sealed with bone wax. After the incision was closed with silk sutures (6-0), the animal was injected with adjudin or vehicle and placed in a heated cage to regain full consciousness, then placed in its home cage. The same procedure was performed in sham animals but without the CCI injury. Drug treatment and CCI injury were performed by the same person to minimize variance.
Measurement of cerebral edema
The wet–dry method was used to estimate cerebral edema by measuring water content in the brain tissue, as described in our earlier study. 23 –25 Briefly, water content was estimated from a 3-mm coronal tissue section of the ipsilateral cortex (or corresponding contralateral cortex), centered on the impact site. Once removed, the sample was immediately weighed, then dried in an oven (100°C) for 24 h. Based on the wet and dry weights, tissue water content (%) was calculated as follows: (wet weight–dry weight)/(wet weight × 100). To eliminate the effect of gender on the result, we additionally performed wet-dry weighting test on single-female mice. In addition, several doses of adjudin were used in this test to choose an optimal dose for our subsequent studies.
Magnetic resonance imaging (MRI) also was used to evaluate cerebral edema. At 24 h after the CCI procedure, each mouse brain was scanned using a mouse MRI scanner (Simenz Signa EXCITE 3.0 T; Buffalo Grove, IL). The T2-weighted MRI parameters were as follows: repetition time = 3670 msec; echo time = 97 msec; thickness = 0.8 mm; field of view = 70 × 70 mm; number of excitations = 1.5. To further verify the effect of adjudin on cytotoxic edema, apparent diffusion coefficient (ADC) maps were performed on MRI diffusion weighted imaging sequence by Bruker Biospec 7.0 Tesla 20 cm horizontal bore scanner (Bruker Biospin MRI GmbH, Germany). Throughout the process, a bot pack was used to maintain the body temperature at 37.6 ± 0.5°C. All MRI images were evaluated by a proficient neuroimaging doctor who was blinded to the study design. The cerebral edema lesion, including the contusion area, was evaluated based on T2-weighted images using National Institutes of Health (NIH) ImageJ software (Bethesda, MD).
Evans blue extravasation
BBB permeability was evaluated by measuring the extravasation of Evans blue (EB) dye (Sigma Aldrich) in brain tissue 3 days post-CCI injury. Mice were anesthetized 2 h after injection of EB dye (2%, 4 mL/kg) and were perfused transcardially with phosphate-buffered saline (PBS) through the left ventricle of the heart for sufficient elimination of the intravascularly localized dye. Once the brain was removed, it was divided into two hemispheres and weighed. Each sample was soaked in 1 mL 50% trichloroacetic acid solution and homogenized. After the homogenate was centrifuged at 12,000 × g for 20 min, it separated into two layers, and the supernatant was transferred and diluted 1:3 with ethanol. A spectrophotometer (BioTek, Winooski, VT) was used to detect the absorbance of the mixture at 610 nm. The quantity of dye was calculated with reference to a standard curve and expressed in micrograms per gram of brain tissue.
Cell culture
Primary astrocytes were obtained from newborn C57BL/6 mouse brains. After careful removal of the skull and meninges, the cortices were separated from the brain under a dissecting microscope. The cortices were then homogenized mechanically and digested in 0.25% trypsin for 8-12 min at 37°C. After centrifuging, cells were resuspended in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS). The cells (10 × 106) were then plated onto 10-cm dishes to produce confluent cell monolayers at 10–12 days following plating. Cells were cultured at 37°C in an incubator in a 5% CO2 air-humidified atmosphere.
CCK8 assay
The cell toxicity induced by adjudin was assessed using the CCK8 assay (Dojindo, Tokyo, Japan). Initially, astrocytes were seeded in 96-well plates at 1 × 104/well (three wells per group) in 100 μL DMEM (containing 10% FBS and 0, 10, 30, 60, 70, 80, 90, or 110 μM adjudin). After incubating the cells in 5% CO2 at 37°C for 24 h, 10 μL CCK-8 solution were added to each well. After a 2 h incubation, the absorbance at 450 nm was recorded using a spectrophotometer (BioTek).
Relative real-time polymerase chain reaction
Total RNA was isolated from brain tissues and astrocytes using TRIzol Reagent (Invitrogen, Carlsbad, CA). After the RNA concentration was measured using a spectrophotometer (NanoDrop1000; Thermo, Wilmington, DE), real-time polymerase chain reaction (PCR) was performed using the One Step SYBR® PrimeScriptTM PLUS RT-PCR Kit (Takara Bio Inc., Shiga, Japan). The primer sequences used to amplify the target genes were as follows: • IL-1β: 5′-GCAACTGTTCCTGAACTCAACT-3′ (forward), 5′-ATCTTTTGGGGTCCGTCAACT-3′ (reverse); • IL-6: 5′-TAGTCCTTCCTACCCCAATTTCC-3′ (forward), 5′-TTGGTCCTTAGCCACTCCTTC-3′ (reverse); • Tumor necrosis factor (TNF)-α: 5′-CCCTCACACTCAGATCATCTTCT-3′ (forward), 5′-GCTACGAGTGGGCTACAG-3′ (reverse); • IL-4: 5′- GGTCTCAACCCCCAGCTAGT -3′ (forward), 5′- GCCGATGATCTCTCTCAAGTGAT-3′ (reverse); • IL-10: 5′- GCTCTTACTGACTGGCATGAG -3′ (forward), 5′- CGCAGCTCTAGGAGCATGTG -3′ (reverse); • AQP4: 5′-CTTTCTGGAAGGCAGTCTCAG-3′ (forward), 5′-CCACACCGAGCAAAACAAAGAT-3′ (reverse); and • GAPDH: 5′-AGGTCGGTGTGAACGGATTTG-3′ (forward), 5′-TGTAGACCATGTAGTTGAGGTCA-3′ (reverse).
Relative messenger RNA (mRNA) expression levels were calculated using SDS software (Applied Biosystems, HT7900, CA).
Western blot analysis
Western blotting was performed as described previously. 24 Briefly, astrocytes and brain samples (at 72 h after CCI) were collected and added to lysis buffer. To separate the nuclear and cytoplasmic proteins, a nuclear and cytoplasmic protein extraction kit (Beyotime Institute of Biotechnology, Nan Tong, China) was used. Samples that contained equal amounts of protein were loaded onto a resolving gel (Promoton, Shanghai, China) for electrophoresis, then transferred onto a polyvinylidene difluoride membrane (Whatman, Piscataway, NJ) and blocked with 5% nonfat milk. The membranes were incubated overnight at 4°C with primary antibodies against ZO-1, occludin, AQP4 (1:500; Santa Cruz Biotechnology, Santa Cruz, CA), phospho-IKKα/β, IKKα, phospho-IκBα/β, IκBα/β, phospho-NF-κB p65, NF-κB p65 (1:500; Cell Signaling Technology, Beverly, MA), β-tubulin, histone H3, and β-actin (1:1000; Cell Signaling Technology). After washing, the membranes were incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature and then reacted with an enhanced chemiluminescence substrate (Pierce, Rockford, IL). Chemiluminescence was detected using Quantity One image software (Bio-Rad, Hercules, CA), and the relative intensity was calculated using Gel-Pro Analyzer software (Media Cybernetics, Bethesda, MD).
Electrophoretic mobility shift assay (EMSA)
DNA–protein interactions were measured using the LightShift Chemiluminescent EMSA Kit (Pierce) as follows. A biotin-labeled NF-κB probe 26 (5′-TCGACATGTGGGATTTTCCCATGAC-3′) was incubated with nuclear extracts (25 μg) at 25°C for 20 min, and the protein–DNA complexes were resolved on a 6% polyacrylamide gel, transferred to nylon membranes (Millipore, Bedford, MA), and cross-linked via exposure to ultraviolet light at 312 nm. Membranes were subsequently incubated with streptavidin–horseradish peroxidase for 15 min. Biotin-labeled DNA signals were visualized using an enhanced chemiluminescence detection system.
Immunostaining
Double immunostaining of ZO-1 (1:100; Invitrogen)/CD31 (1:200; BD Biosciences, San Jose, CA) or occludin (1:100; Invitrogen)/CD31 (BD Biosciences) was performed as described previously. 23,24 For immunostaining of AQP4, ionized calcium-binding adapter molecule 1 (iba-1) and glial fibrillary acidic protein (GFAP), mice were deeply anesthetized and perfused with saline and 4% paraformaldehyde (PFA), then the brains were removed and dehydrated in 30% sucrose after overnight post-fixation in 4% PFA. During immunostaining, brain sections (20-μm thick) were blocked with 10% bovine serum albumin (BSA) for 1 h, then incubated overnight at 4°C with primary antibodies: rabbit anti-AQP4 (1:100; Santa Cruz Biotechnology), mouse anti-GFAP (1:100; Millipore), and Iba-1 (1:100). After washing, the brain sections were incubated with the appropriate secondary antibodies for 1 h at 37°C. Three fields in the peritraumatic area in each slice were imaged with confocal microscope (Leica, Solms, Germany), and three evenly spaced slices were used per mouse brain. Photomicrographs were taken using the same parameters. The number of activated microglia cells and astrocytes were counted with Image J software (Version1.51e, NIH) by an investigator who was blind to the identity of each image using.
Neurological severity score determination
The neurological status of each animal was evaluated at 1, 3, 7, and 14 days after CCI. A modified neurological severity score (mNSS) test was performed, which included motor, sensory, balance, and reflex tests (normal score, 0; maximal deficit score, 14). 8 In the motor test, forelimb flexion was evaluated after raising the mouse by the tail (0-3), and the mouse was placed on the floor to evaluate its gait (0-3). A balance beam test was performed to assess the posture of the mouse on a beam post-injury (0-6). In the test for reflex absence (0-2), the pinna and corneal reflexes were tested.
The rotarod test was used to examine the motor coordination of the mice. Before CCI injury, the mice were trained for 3 days (three sessions/day, 5 min/session), and with each session, the speed was accelerated from 0 to 40 rounds/min. After the training, only mice that could stay on the rod for at least 5 min were selected for the surgical procedure. After CCI injury, each mouse was subjected to the rotarod test on Days 1, 3, 5, 7, and 14, and the time when the mouse fell off was recorded.
The Morris water maze (MWZ) test was used to evaluate spatial learning and memory from the 14th-19th day after CCI procedure. Briefly, the water maze apparatus consisted of a circular tank (100 cm in diameter) with water maintained at 25°C. Extra-maze visual cues were hung on the walls surrounding the pool and a hidden platform (10 cm in diameter) was submerged 1 cm below the surface of the water. Each training day consisted of four trials, and the start positions were randomly changed each day. The mice were allowed for 90 sec to find the platform and remain on it for 15 sec. Mice that did not locate the platform within 90 sec were placed on the platform for 15 sec, and the latency time was recorded as 90 sec. On the 19th day, a probe trial was conducted in which the platform was removed, and the mice were allowed to swim freely for 60 sec, and the time spent in goal quadrant was recorded. Results were expressed as latency to platform and time spent in the goal quadrant.
Statistical analysis
All data are presented as means ± standard deviation. Power analysis was performed by PASS 11 (PASS software, Kaysville, Utah). Equality of variance was assessed by Levene's test. Student's t-test was used to compare means between groups. The p values <0.05 were considered statistically significant. Graphical representations of the data were produced using Graph Pad Prism 6 (GraphPad Software, San Diego, CA). SPSS 20.0 for Windows (SPSS Inc., Chicago, IL) was used for the statistical analyses.
Results
Adjudin reduced cerebral edema and improved neurological function in a mouse model of CCI injury
To evaluate the severity of cerebral edema in mice 3 days after CCI injury, brain tissue samples from the same set of mice were weighed and dehydrated. As shown in Figure 1A, mice exposed to the CCI procedure exhibited a significant increase in the water content of the ipsilateral cortical tissue samples (81.2%) compared with the sham group (78.8%, p < 0.01). Mice treated with adjudin (50 mg/kg) post-CCI injury exhibited a significant reduction in the CCI injury-induced increase in brain water content compared with the vehicle group (79.8% vs. 81.2%, p < 0.01). Further, a similar result was obtained in single female mice (Supplementary Fig. 1; see online supplementary material at

Adjudin reduced traumatic brain injury–induced cerebral edema and improved neurological function.
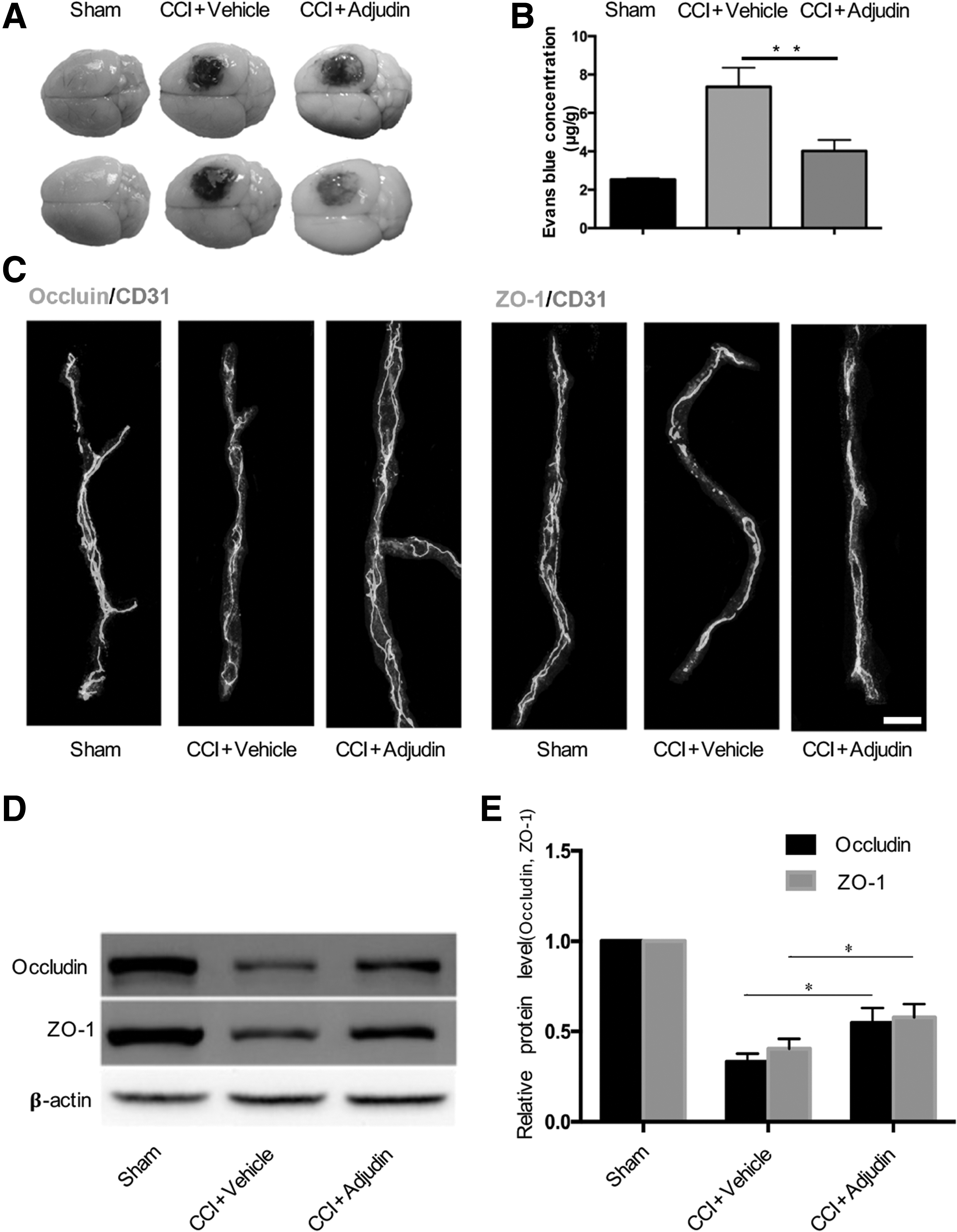
Adjudin alleviated blood–brain barrier disruption in a mouse model of controlled cortical impact (CCI) injury.
The MRI evaluations of cerebral edema were consistent with the wet–dry method (Fig. 1B, 1C; p < 0.01 vs. vehicle group). The ADC maps showed that ADC values in the edema area were lower than that in the contralateral non-edema area (Supplementary Fig. 3A; see online supplementary material at

Adjudin reduced the gene and protein expression of aquaporin 4 (AQP4) in mice after controlled cortical impact (CCI) injury.
To explore the effects of adjudin on neurological functional recovery, mNSS was used to examine the motor, balance, and reflex functions of mice post-CCI injury. The results showed that mice in the adjudin-treated group had significantly lower scores at 3 days (p < 0.05), 7 days (p < 0.05) and 14 days (p < 0.01) post-CCI injury, compared with vehicle group (Fig. 1D).
In the rotarod test, the duration that mice stayed on the rod was remarkably reduced after CCI injury (Fig. 1E). In the adjudin-treated group, behavioral outcomes were significantly better at 3, 7, and 14 days post-CCI injury compared, with the vehicle group (Fig. 1E; p < 0.01).
In the training trails of the MWZ test, the latency to find the platform was longer in mice that underwent CCI injury than those in the sham group (Fig. 1F), and in the probe trail, CCI mice spent less time in goal quadrant than sham group mice (Fig. 1G, 1H). The mice in the adjudin-treated group displayed significantly shorter latency at 16, 17, and 18 days post-CCI injury compared with vehicle group in the training trails (Fig. 1F; p < 0.01), and adjudin-treated mice spent significantly more time in the goal quadrant (Fig. 1H; p < 0.01, vs. vehicle group).
Adjudin alleviated BBB disruption in a mouse model of CCI injury
The EB extravasation test was performed to assess BBB permeability after CCI injury. EB leakage was remarkably reduced in the injured ipsilateral cortex of the adjudin-treated group, compared with the vehicle group, at 3 days post-CCI injury (Fig. 2A). Further analysis indicated that adjudin significantly alleviated CCI injury-induced BBB disruption at 3 days post-injury (Fig. 2B; p < 0.01 vs. vehicle group).
To further evaluate the integrity of the BBB after CCI injury, the positions of tight junction (TJ) proteins were examined via in situ double immunostaining for ZO-1/CD31 and occludin/CD31 at 3 days post-CCI injury. As shown in Figure 2C, occludin and ZO-1 were stained consistently along the vessels in the sham group, while gaps were present after CCI injury, and the adjudin-treated group showed fewer gaps than did the vehicle-treated group. Western blot analysis was used to analyze the expression of TJ proteins, and less reduction in the expression of ZO-1 and occludin was seen in the adjudin-treated group, compared with the vehicle-treated group, at 3 days post-CCI injury (Fig. 2D, 2E; p < 0.05 vs. vehicle group). Adjudin reduced the gene and protein expression of AQP4 in mice after CCI injury
AQP4 is thought to play a key role in cytotoxic cerebral edema, because it is expressed in the cytomembrane where it allows water to flow into the brain from the blood. 27 To investigate the effects of adjudin on AQP4 expression, mice from all three groups were euthanized at 3 days post-injury; mRNA and proteins were extracted, and brain sections were prepared for immunostaining.
PCR was used to detect AQP4 expression in the mouse brains. As shown in Figure 3A, the expression of AQP4 mRNA was remarkably elevated at 3 days post-CCI injury compared with sham Group. A significant reduction in the CCI injury-induced increase in AQP4 mRNA expression was seen in the adjudin-treated group at 3 days post-CCI injury (Fig. 3A; p < 0.01 vs. vehicle group).
The AQP4 protein level was examined by Western blot analysis and immunofluorescence staining. The results of both analyses revealed that the protein levels of AQP4 were clearly elevated in the peritraumatic area at 3 days post-CCI injury (Fig. 3B, 3D). In adjudin-treated mice, this elevation was significantly alleviated (Fig. 3C; p < 0.01 vs. vehicle), implying that adjudin plays a role in reducing AQP4 expression in the brain after TBI.
Adjudin reduced the activation of microglia and astrocytes in the peritraumatic area of mouse brain after CCI injury
To evaluate the inflammation generated in the peritraumatic area of mouse brains post-CCI injury, brain sections obtained at 3 days post-injury were immunostained for Iba-1 and GFAP, which are specific markers of microglia and astrocytes. As shown in Figure 4A, microglia and astrocytes were assembled or activated after CCI injury, causing swelling. Fewer swollen microglia and astrocytes were found in the adjudin-treated group than in the vehicle group (Fig. 4A-C; p < 0.01).
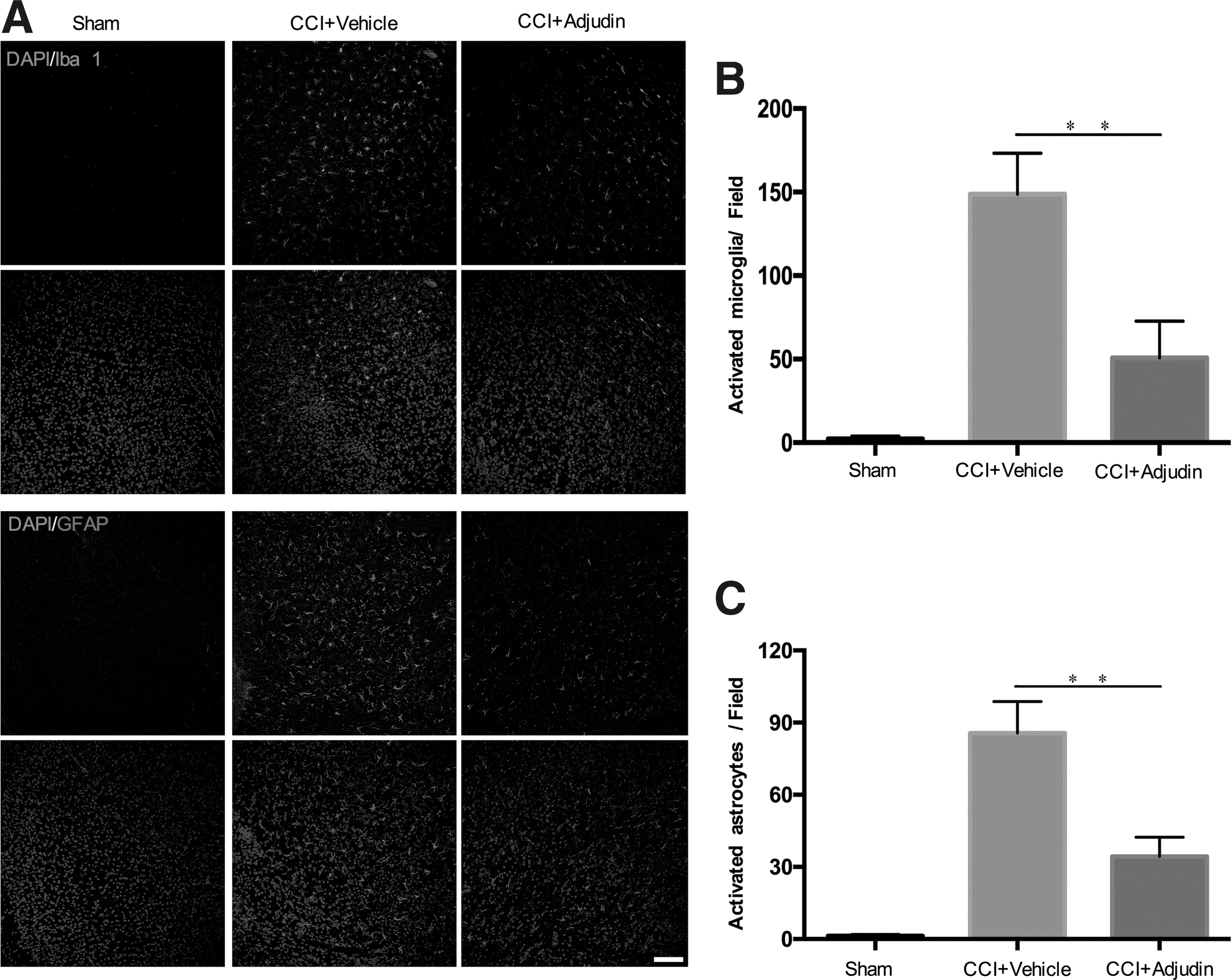
Adjudin reduced the activation of microglia and astrocytes in the peritraumatic area of mouse brains after controlled cortical impact (CCI) injury.
Adjudin reduced IL-1β, IL-6, and TNF-α production both in vivo and in vitro
To assess the anti-inflammatory effects of adjudin, levels of the pro-inflammatory cytokines (interleukin [IL]-1β, IL-6, and TNF-α) and anti- inflammatory (IL-4 and IL-10) were detected by PCR in vivo and in vitro. Tissue samples were collected from each group of mice at 3 days post-CCI injury for the in vivo analysis, and astrocytes were treated with 1 μg/mL lipopolysaccharide (LPS) for 24 h to induce inflammation for the in vitro analysis. Figure 5 shows that after CCI injury in vivo or after LPS treatment in vitro, the production of IL-1β, IL-6, and TNF-α were enhanced markedly, and adjudin treatment (50 mg/kg in vivo, 60 μM in vitro) significantly decreased these levels (Fig. 5; p < 0.01 vs. vehicle). In addition, adjudin treatment could significantly enhance the production of IL-4 (p < 0.05) and IL-10 (p < 0.01) after CCI injury in vivo (Supplementary Fig. 4A; see online supplementary material at
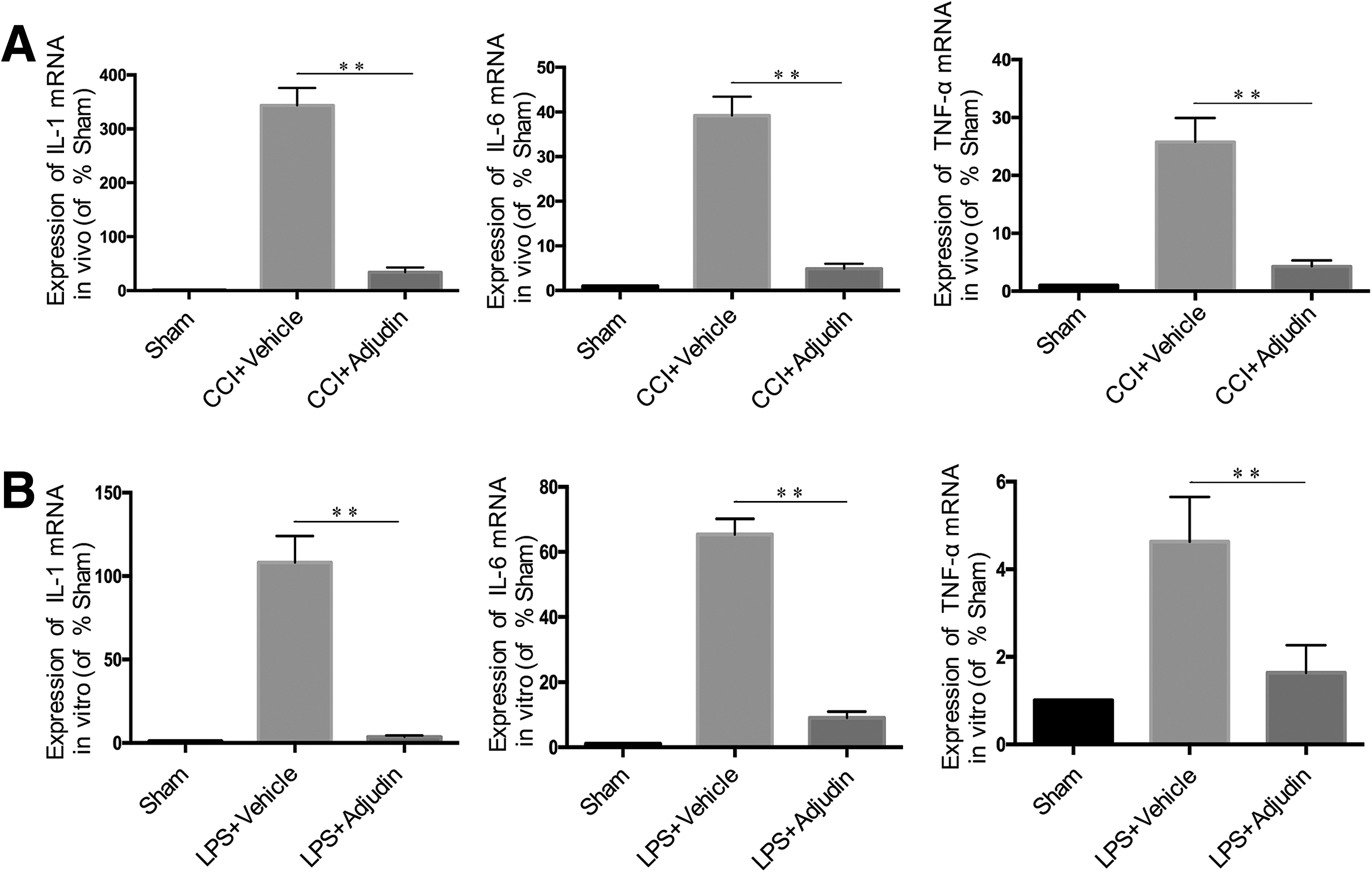
Adjudin reduced the production of interleukin (IL)-β, IL-6, and tumor necrosis factor (TNF)-α both in vivo and in vitro. Bar graph of the messenger RNA levels of IL-β, IL-6, and TNF-α in peritraumatic brain tissues at 3 days post–controlled cortical impact injury
Adjudin inhibited activation of the NF-κB pathway in mice after CCI injury
The NF-κB pathway is a major signaling pathway involved in inflammation in many disease models. To determine whether TBI and adjudin affect NF-κB activation, Western blot analysis was used to demonstrate markedly increased levels of NF-κB p65, IKBα, and IKKα phosphorylation in the peritraumatic area at 3 days post-CCI injury (Fig. 6A). However, adjudin treatment significantly alleviated these increased levels (Fig. 6B-6D; p < 0.01, p < 0.05, p < 0.01, vs. vehicle). For further assessment, nuclear proteins were extracted and Western blot analysis was performed; the results showed elevated nuclear NF-κB p65 levels in peritraumatic brain tissue at 3 days post-CCI injury, compared with the sham group (Fig. 7A). Additionally, adjudin treatment significantly reduced the nuclear translocation of NF-κB p65 (Fig. 7A; p < 0.01, vs. vehicle). EMSA suggested that CCI injury induced strong NF-κB DNA-binding activity, which was markedly inhibited by adjudin. The binding specificity was verified by incubating excess unlabeled specific competitor or nonspecific oligonucleotide probe with nuclear extracts from peritraumatic brain tissue at 3 days post-CCI injury.

Adjudin inhibited activation of the NF-κB pathway in mice after controlled cortical impact (CCI) injury.
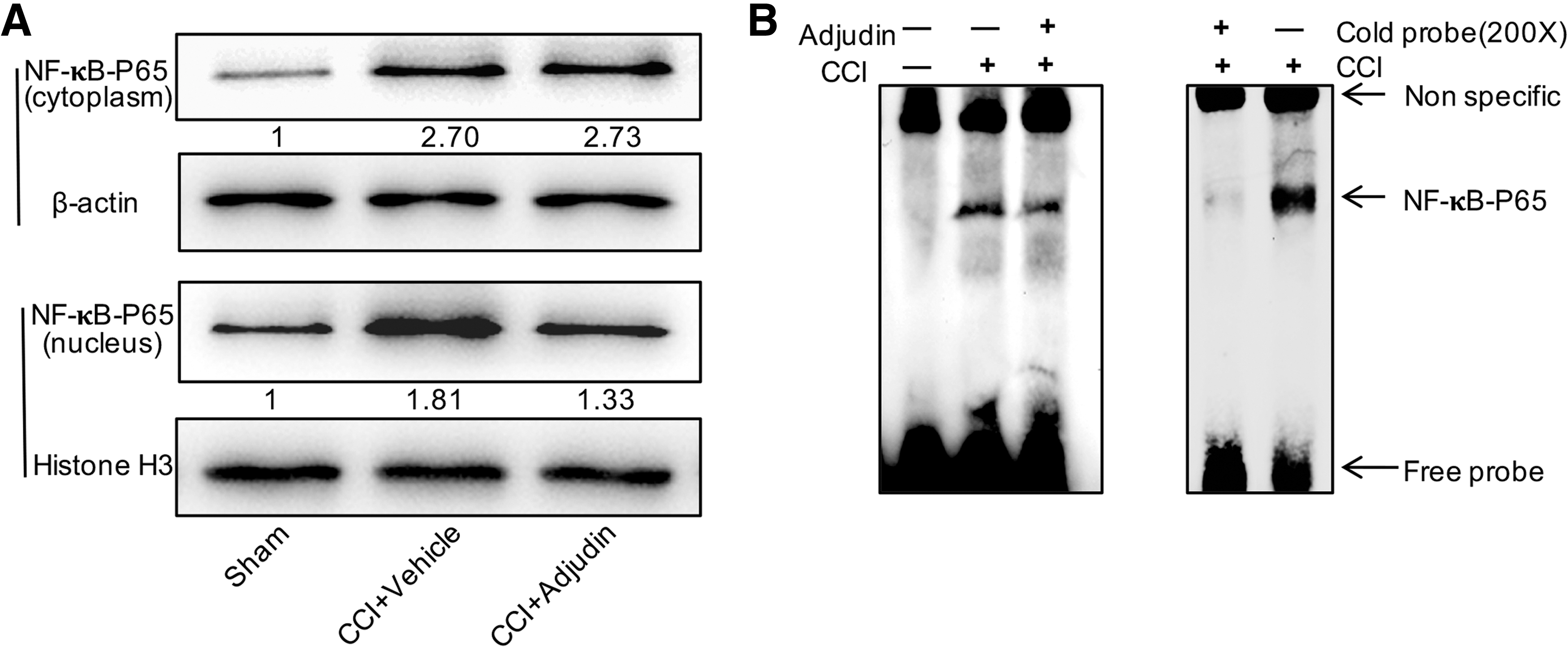
Adjudin reduced the nuclear translocation of NF-κB p65 in mice at 3 days post–controlled cortical impact injury.
Discussion
In this study, we demonstrated that adjudin, a small molecular compound, attenuated cerebral edema and reduced neurological deficits in mice after CCI injury, which is a model of TBI. These functions may be associated with anti-inflammation caused by attenuating activation of the NF-κB pathway. We further revealed that the anti-edematous effect of adjudin may be mediated by reducing the AQP4 overexpression in the endfeet of inflammation-activated astrocytes and alleviating BBB disruption after CCI injury.
Cerebral edema, a common complication of TBI, cerebral ischemia, hemorrhage, and liver failure, 28 –31 contributes to additional ischemic injuries, cerebral hernia-related deaths, and severe neurological deficits. It is classified mainly into vasogenic edema and cytotoxic edema. The former occurs primarily in the center of the lesion and soon spreads to the peritraumatic area as a result of disruption of the BBB. The latter occurs mainly in the peritraumatic area as a result of glial cell and neuronal swelling. 28 According to recent studies, cytotoxic edema plays a more important role than does vasogenic edema in many TBI models at the 3rd day post-injury, 28,32 which was also verified by our ADC mapping of MRI. In this study, we assessed the role of adjudin in alleviating vasogenic edema and cytotoxic edema, and the astrocytes were mainly focused on in cytotoxic edema. Astrocytes comprise the majority of all neural cells, and astrocytic swelling is considered a hallmark of cellular edema. 9 Further, astrocytic swelling is a result of glial activation, which mirrors the neuroinflammatory response in TBI and other disorders of the central nervous system (CNS). 33 We chose Day 3 as the time-point to detect cerebral edema by MRI and by the wet–dry method, because Day 3 was believed to be the peak of cerebral edema after CCI injury in mice. 23,24 We found that adjudin injection after CCI injury attenuated cerebral edema and improved performance in the rotarod, mNSS, and Morris water maze tests, which is consistent with other reports on adjudin treatment in mouse models of ischemic stroke injury. These results strongly imply that adjudin has therapeutic potential for patients post-TBI.
AQP proteins, which are integral to membrane pores, facilitate the diffusion of water molecules, and the distribution, number, and permeability of these pores control the permeability of the CNS. Hence, edema is thought to be a result of AQP dysfunction in which the balance between preventing and facilitating water movement is lost. 34 In the CNS, eight AQP proteins were detected—AQP1, AQP3, AQP4, AQP5, AQP7, AQP8, AQP9, and AQP11—of which AQP4 exhibits the highest expression. 35,36 AQP4 is reportedly expressed mainly on the endfeet membranes of astrocytes, and because they are brain–liquid interfaces, these membranes express 10- to 15-fold higher AQP4 protein levels, compared with non-endfeet membranes. 35 Several studies have revealed reduced intracranial pressure and better survival and neurological outcomes in response to cytotoxic edema in AQP4-null mice or AQP4-pharmacologically inhibited mice compared with controls. 37,38 Filippidis and colleagues 34 even argued that worse cytotoxic edema can occur only if AQP4 is present. There is still controversy over the role of AQP4 in ischemic injury-induced cerebral edema. Researchers observed that AQP4 expression is decreased during the 48 h following hypoxia. 39,40 However, in TBI models, most reports found that AQP4 was overexpressed following injury and that treatments to inhibit AQP4 were effective, 34 supporting the results of our study. This is partly because cytotoxic brain edema is predominant in TBI-induced brain edema. 32 In this study, AQP4 gene and protein overexpression induced by CCI injury, in comparison with the sham group, was significantly reduced by adjudin treatment. This result suggests adjudin to be an AQP4-inhibiting drug that may also play a role in alleviating cerebral edema after TBI injury. We also found large numbers of swollen astrocytes in the edematous areas of brain sections after GFAP immunostaining, further suggesting that AQP4 is highly expressed on the membranes of astrocyte end feet, to allow entry of water molecules. The reduced astrocyte swelling in the adjudin-treated group also supports this function of adjudin on AQP4. Adjudin treatment reduced formation of gaps in TJs and showed a protective effect on BBB integrity after TBI, implying that adjudin also plays a role in inhibition of vasogenic edema.
A growing body of evidence has demonstrated that neuroinflammation plays a vital role in secondary injury responses following TBI and can cause post-injury pathogeneses such as cerebral edema, hemorrhage, and ischemia, which further exacerbate neuronal injury. 23,41 Neuroinflammation refers mainly to the activation of resident cells in the CNS, such as microglia, astrocytes, and neurons, and their release of inflammatory mediators, including cytokines, chemokines, and reactive oxygen/nitrogen species. 42 In the TBI process, tissue at the injury site is damaged by mechanical impact and release. Damage-associated molecular patterns (DAMPs) are induced by a number of endogenous factors such as RNA, DNA, and heat shock proteins. 41,43 Then, the NF-κB and MAPK pathways are activated, resulting in release of a variety of pro-inflammatory molecules, including cytokines (IL-1β, IL-6, TNF-α), chemokines, and immune receptors, from microglia, astrocytes, cerebrovascular endothelial cells, peripheral immune cells and even neurons. 44 –46 This inflammatory response is signaled by rapid increases in cytokine levels. 41 which are pivotal mediators in TBI pathologies. IL-1β, IL-6, and TNF-α were the most elevated cytokines post-TBI. In our study, mRNA expression of these cytokines was 10- to 100-fold higher in peritraumatic brain tissue samples post-CCI injury compared with the sham group, which was consistent with previous studies. In the in vitro study, primary astrocytes were cultured and stimulated by LPS, as a cell model of inflammation, to investigate the anti-inflammatory effects of adjudin. Remarkable elevations in IL-1β, IL-6, and TNF-α were also detected after LPS stimulation. More importantly, in both the in vivo and in vitro studies, adjudin significantly reduced the increased mRNA levels of IL-1β, IL-6, and TNF-α. To further demonstrate the anti-inflammatory function of adjudin, the mRNA levels of several anti-inflammatory markers (IL-4 and IL-10) were detected. The in vivo results suggested that adjudin could significantly increase the expression of anti-inflammatory markers, but in single cultured LPS-stimulated astrocytes, no significant result was obtained, which implies that it seems just as likely, if not more likely, that astrocytes are not the source of the changes in vitro. In conclusion, our results suggest that adjudin inhibits neuroinflammation and alleviates neuroinflammation-induced astrocytic swelling, which is considered a hallmark of cellular edema.
NF-κB, an important transcription factor that controls the expression of pro-inflammatory cytokines, has been widely demonstrated to play a role in the pathology of neuroinflammation following TBI. In recent years, accumulating evidence has confirmed that NF-κB may be responsible for the swelling of astrocytes in several models of neurological disease, 47,48 including TBI. Jayakumar and colleagues 49 found that the NF-κB inhibitor BAY 11-7082 significantly prevented the swelling of astrocyte cultures induced by fluid percussion injury (a TBI model), and in another study, astrocyte cultures obtained from NF-κB-inactivated transgenic mice showed no swelling after fluid percussion injury, while the wild-type group exhibited high amounts of astrocyte swelling. These results highlight the important role of NF-κB in TBI. Before activation, NF-κB is stored in the cytoplasm and bound to IκBα and IκBβ, inhibitory proteins of NF-κB. When IκB is phosphorylated and degraded rapidly, NF-κB is released and phosphorylated, then translocates from the cytoplasm into the nucleus. 50 The activated form of NF-κB consists of two proteins, a p50 subunit and a p65 subunit, which play important roles in triggering the transcription of certain genes. 51 In the TBI process, upon stimulation of DAMPs, specific serine residues of IκB proteins were phosphorylated by activated IKK complexes, mainly comprising IKKα, IKKβ, and IKKγγ 52 . In our study, Western blot analysis showed that adjudin partly prevented activation of the NF-κB pathway by TBI. This implied that the anti-edematous function of adjudin may be executed via inhibition of the NF-κB pathway.
Conclusion
The results of this study suggest that adjudin attenuates the development of TBI-induced cerebral edema by inhibiting the expression of AQP4. This function of adjudin was associated with its anti-inflammatory effect, which acts at least partly through the NF-κB signaling pathway. Adjudin is an effective anti-inflammatory agent and a useful novel inhibitor of AQP4, and therefore, it may play a role in the treatment of TBI.
Footnotes
Acknowledgments
This work was supported by grants from National Natural Science Foundation of China (No 81471245, 81671207, 81701895, 81773115 and 81501048), Shanghai Jiao Tong University Medicine-Engineering Research Fund (YG2016QN20), and Ministry of Science & Technology (2013CB945604). The authors would like to extend their sincere thanks to Dr. C. Yan Cheng for providing adjudin for the research.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.