
Abstract
Initial studies have found some evidence for transactive response DNA-binding protein 43 (TDP-43) abnormalities after traumatic brain injury (TBI), and the presence of protein inclusions consisting of TDP-43 are a pathological hallmark of amyotrophic lateral sclerosis (ALS), a condition associated with TBI. However, no study has characterized changes in TDP-43 phosphorylation, mislocalization, and fragmentation (i.e., abnormalities linked to hallmark TDP-43 pathology) after TBI, and how these relate to functional outcomes. Further, how TBI affects an individual with a known predisposition to TDP-43 pathology is unknown. Therefore, this study examined the effects of TBI on TDP-43 post-translational processing, localization, and behavioral outcomes in wild-type (WT) mice and mutant TDP-43A315T mice (i.e., mice predisposed to TDP-43 pathology) at 24 h and 1 week after TBI. Post-mortem brain tissue from human patients with acute TBI was also examined. Western blots found that WT mice given TBI had increased TDP-43 phosphorylation, mislocalization, and fragmentation compared with sham-injured WT mice. The TDP-43A315T mice given a TBI had exacerbated TDP-43 abnormalities, worse cell death, and cognitive deficits compared with all other groups. In the human TBI patients, the only significant finding was increased nuclear accumulation of phosphorylated TDP-43 fragments. The discrepancy between the robust mouse findings and the largely non-significant human findings may be due to factors including heterogeneity in clinical TBI, the small group sizes, and temporal complexities with TDP-43 abnormalities. These findings indicate that TBI can induce a number of TDP-43 abnormalities that may contribute to the neurological consequences of TBI, though further research is still needed.
Introduction
Traumatic brain injury (TBI) is a neurodegenerative condition that is induced by biomechanical forces applied to the brain, and is a leading cause of death and morbidity worldwide. 1 TBI pathology is generally categorized as resulting from two types of injury mechanisms, primary or secondary. 1 Primary injury stems from the mechanical forces at the moment of the impact and is considered irreversible. 1 Secondary injuries can take hours to years to manifest, and there is growing evidence that proteopathies may contribute to this process. 2 –5 TBI has also been associated with an increased risk of developing neurodegenerative conditions that involve proteopathies, including Alzheimer's disease, epilepsy, chronic traumatic encephalopathy (CTE), and amyotrophic lateral sclerosis (ALS). 6 –9 Whereas the majority of the research investigating proteopathies after TBI has focused on tau and amyloid beta, 6 –10 few studies have examined changes in transactive response DNA-binding protein 43 (TDP-43).
TDP-43 is a transcriptional repressor involved in the regulation of biological processes by binding to deoxyribonucleic acid (DNA) or ribonucleic acid (RNA) targets. In motor neurons, it has been shown to be a low molecular weight neurofilament messenger RNA (mRNA)-binding protein. 11 TDP-43 is also involved in regulating the splicing of pre-mRNAs. 12 Studies have found that pathological TDP-43 is common in the neurodegenerative conditions ALS and frontotemporal lobar degeneration with ubiquinated inclusions (FTLD-U). 13 In these diseases, TDP-43 can be hyperphosphorylated at the C-terminal sites (e.g., S403/404, S409/410), or cleaved into 35kDa and 25kDa fragments that lack their nuclear localization signal, causing TDP-43 accumulation, aggregation, and dislocation from the nucleus to the cytoplasm in neurons and glial cells. Consequently, this can result in a toxic gain and/or loss of TDP-43 function, and ultimately neurodegeneration. 14 –17
As alluded to above, some initial studies have examined TDP-43 in the context of TBI. Post-mortem examination of brain tissue from 12 CTE patients (i.e., a neurodegenerative condition postulated to be caused by repetitive brain trauma) found that nine cases had evidence for TDP-43 proteinopathy. 18 Another study found increased TDP-43 and its 35kDa fragment in the cerebrospinal fluid (CSF) of 21 patients with severe TBI. 16 Further, an immunohistochemical study in post-mortem brain tissue collected from 62 patients with severe TBI found increased phosphorylation-independent TDP-43 immunoreactivity both acutely and chronically post-injury. 19 In addition to these preliminary TBI patient findings, initial studies in rodent TBI models also suggest TDP-43 abnormalities. Mice given a TBI had increased cytoplasmic TDP-43 and TDP-43 breakdown products. 16 Rats given a TBI had progressive neurodegeneration and motor impairments similar to those in ALS that occurred in the presence of increased phosphorylated and cytoplasmic TDP-43. 20
Although these findings do provide initial support that TDP-43 abnormalities can occur after TBI, a comprehensive understanding of the spectrum of TDP-43 abnormalities, and how they relate to functional outcomes, is still lacking. Moreover, to our knowledge, no study has investigated the effects of TBI on an organism known to be genetically predisposed to TDP-43 pathology, which may provide important insight into the relationship between TBI and the development of neurodegenerative disorders involving TDP-43 (e.g., ALS or CTE). Therefore, this study examined abnormalities in TDP-43 and behavior in mice given an experimental TBI (i.e., a fluid percussion injury; FPI). This was conducted in wild-type (WT) mice, as well as in mutant mice that express human TDP-43 (i.e., TDP-43A315T) and are predisposed to pathological TDP-43 accumulation in the brain. Post-mortem brain tissue from TBI patients was also examined to assess whether similar TDP-43 abnormalities occurred in humans after a TBI.
Methods
Mice
Eight-week-old male mice (n = 83) were used in this study. TDP-43A315T mice 21,22 and WT littermate controls were obtained from colonies at the Florey Institute of Neuroscience and Mental Health, The University of Melbourne. TDP-43A315T mice express a human TDP-43 transgene containing the A315T mutation seen in familial ALS patients, under the control of the mouse prion protein promoter. 22 All procedures were approved by the Howard Florey Institute Animal Ethics Committee (14-007UM) and within the guidelines of the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes by the Australian National Health and Medical Research Council.
Surgery and FPI
FPI and sham procedures were performed as previously described. 23,24 Mice in the FPI groups were given a single injury with a force of 1–1.5 atm. 23,24 Ten mice died immediately after the FPI (21% mortality rate) and were therefore not included in the study. The mice in the sham groups did not receive the FPI but were treated identically otherwise.
Apnea, duration of unconsciousness, and self-righting reflexes were monitored to ensure consistent injury severities between the groups (see Table 1). 23,24 FPI worsened all acute injury measures as indicated by a significant effect of injury on apnea (F1,65 = 203.965, p < 0.001), pain reflex (F1,65 = 488.048, p < 0.001), and self-righting reflex times (F1,65 = 411.400, p < 0.001). There were no statistically significant findings related to strain or assigned recovery time.
Acute Injury Measures
Mice administered TBI had worse acute injury outcome measures compared with sham-injured mice, regardless of strain or assigned recovery time, as indicated by significantly (p < 0.05) increased apnea, pain reflex, and self-righting times (Mean ± standard error [SE]).
TBI, traumatic brain injury; TDP-43, transactive response DNA-binding protein 43.
After the injury, mice were randomly assigned to receive either a 24-h recovery time (n/group: WT + SHAM = 4; TDP-43A315T + SHAM = 4; WT + TBI = 4; TDP-43A315T + TBI = 4) or a 1-week recovery time (n/group: WT + SHAM = 14; TDP-43A315T + SHAM = 14; WT + TBI = 14; TDP-43A315T + TBI = 15). Because TDP-43A315T mice develop motor deficits by 12–16 weeks of age and typically die between 16 and 20 weeks of age, 22 longer recovery periods were not included to avoid confounding motor deficits and mortality.
Mouse western blots
After the 24-h or 1-week recovery times, mice were euthanized, their brains were rapidly removed, the parietal cortex directly under the craniotomy was dissected and divided in half (i.e., half for TDP-43 analysis, half for α-fodrin analysis), and the tissue was frozen in liquid nitrogen and stored at −80°C. Four mice/group/recovery time were used for western blot analyses. As such, all of the 24-h recovery mice were included in the western blot analysis, whereas the 4 mice/group from the 1-week recovery groups were randomly selected. To examine TDP-43 phosphorylation, mislocalization, and fragmentation, western blotting was used to assess total, cytosolic, and nuclear protein levels for anti-phosphoTDP-43 (S403/404), anti-phosphoTDP-43 (S409/410), and anti-TDP-43 (full-length TDP-43) antibodies. 25,26 Western blotting was also used to measure α-fodrin cleavage as a marker for cell death. α-Fodrin can undergo calpain-dependent cleavage (150/145kDa) and caspase-dependent cleavage (120kDa) after TBI. All western blotting procedures were conducted by a researcher blinded to experimental conditions.
TDP-43
Frozen brain tissue was homogenized in 10 uL × the sample weight of cell lysis buffer (CLB; 10 mm HEPES, 10 mm NaCl, 1 mm KH2PO4, 5 mm NaHCO3, 5 mm ethylene diamine tetraacetic acid [EDTA], 1 mm CaCl2, 0.5 mm MgCl2) + 1% Triton X-100 + protease inhibitors (Complete, Mini, EDTA-free; Protease Inhibitor Cocktail Tablets; Roche) + phosphatase inhibitors (PhosSTOP™, Roche). The homogenate was incubated on ice for 10 min. After the incubation period, 15 uL of the homogenized tissue was collected.
To isolate the cytoplasmic and nuclear homogenates, 27 the total tissue homogenate was centrifuged at 6300 × g at 4°C for 10 min and the cytoplasmic supernatant and nuclear pellet were collected. The nuclear pellet was resuspended in 1 mL Tris/sucrose/EDTA (TSE) buffer (10 mm Tris, 300 mm sucrose, 1 mm EDTA, 0.1% IGEPAL-CA 630 v/v, pH 7.5) and recentrifuged at 4000 × g at 4°C for 5 min. The resulting pellet was then resuspended in TSE buffer and used as the nuclear sample. The cytoplasmic supernatant was centrifuged at 107,000 × g for 30 min. The resulting supernatant was collected and used as cytosolic sample. After measuring protein concentrations, the samples were mixed (5:1 [v/v] ratio) with sample buffer, boiled, centrifuged, and stored at −20°C. 27
For western blotting, proteins were separated with SDS-polyacrylamide gel electrophoresis (SDS-PAGE), and the bands of proteins were electro-blotted onto polyvinyl difluoride (PVDF) membranes. The blots on PVDF were developed with the following primary antibodies: anti-phosphoTDP-43 S403/404 (monoclonal; Cosmo Bio Co. Ltd.; 1:2000); anti-phosphoTDP-43 S409/410 (polyclonal; Cosmo Bio Co. Ltd.; 1:2000); anti-TDP-43 (rabbit, polyclonal; Proteintech, #10782-2-AP; 1:2000); anti-GAPDH (monoclonal; Proteintech; 1:5000); anti-Lamin B1 (polyclonal; Abcam; 1:5000). GAPDH was used as the loading control for the cytosolic samples, 28 whereas Lamin B1 was used as the loading control for the nuclear samples. 29 Blots were visualized by enhanced chemiluminescent substrate and exposure to X-ray films. 30 The mean intensity of the blots was quantified using Image J software (National Institutes of Health, Bethesda, MD).
α-Fodrin
Brain tissue was homogenized in 1 mL of ice-cold phosphate buffered saline (PBS) containing protease inhibitors (Complete, Mini, EDTA-free; Protease Inhibitor Cocktail Tablets; Roche) and phosphatase inhibitors (PhosSTOP, Roche). Soluble and insoluble fractions were separated by centrifugation and protein content determined by using the BCA protein assay kit (Pierce). Soluble fractions were then prepared for electrophoresis by the addition of 5 × loading sample buffer (0.5 M Tris-HCl pH 6.8, 20% [v/v] glycerol, 10% [w/v] SDS, 0.01% [w/v] bromophenol blue, and 25% [v/v] ß-mercaptoethanol) then heated at 100°C for 5 min. Proteins (10 μg) were electrophoresed on 6% SDS-PAGE gels and transferred onto PVDF membranes (Bio-Rad) at 100 V for 80 min. Membranes were blocked in Tris buffered saline and Tween-20 (TBS-T) containing 5% skim milk powder, and then incubated with α-fodrin primary antibody (Chemicon) overnight at 4°C. Membranes were rinsed in TBS-T and incubated in secondary antibody for 1 h at room temperature. Signals were detected by chemiluminescence with Immobilon™ Western Reagent (Millipore) and imaging using a ChemiDoc MP Imaging system and Image Lab software package (Bio-Rad). Data were normalized to ß-tubulin as a loading control.
Behavioral testing
All behavioral procedures were conducted by a researcher blinded to experimental conditions. Mice assigned to the 1-week recovery groups underwent behavioral testing to assess motor function, cognition, and anxiety-like behavior. On day 5 after injury, anxiety-like behavior was assessed using the elevated plus maze as previously described. 31 Mice were first placed in the center of the elevated plus maze facing an open arm and allowed to explore the maze freely for 5 min. A percentage score for the time spent in the open arm was used to measure anxiety-like behavior, and entries into the closed arm was calculated as a measure of locomotion. 23
On day 5 and 6 after injury, the rotarod was used to assess motor function as previously described. 32 Rotarod assessment was performed over 2 consecutive days (training and testing), with each day consisting of three trials. For each trial, the mouse was placed on the rotating barrel, the speed was accelerated from 4 to 40 rpm over a period of 5 min, and the time that the mouse was able to maintain its balance was recorded.
On day 6 after injury, Y-maze testing to assess cognitive ability was conducted as previously described. 23 Prior to Y-maze testing, mice were given a 15-min training trial. For the training trial, one arm (i.e., novel arm) was blocked. The mouse was then placed at the distal end of one of the remaining arms (i.e., start arm), and allowed to freely explore the start arm and “other” arm. After a 2-h inter-trial interval, a 5-min test trial was conducted where the novel arm was unblocked, and the mouse was placed in the same start arm and allowed to freely explore all three arms.
Human TBI patient western blots
Post-mortem frontal cortical tissue samples from TBI patients (n = 6) and non-neurological controls (n = 9; i.e., no history of brain trauma or other neurological/psychiatric disorder and no significant neuropathology) were obtained from the Australian Brain Bank Network (see Table 2). All procedures were conducted in accordance with the Australian National Health and Medical Research Council's National Statement on Ethical Conduct in Human Research, the Victorian Human Tissue Act 1982, the National Code of Ethical Autopsy Practice, and the Victorian Government Policies and Practices in Relation to Post Mortem. The brain samples were stored at −80°C until use. Frozen samples were then homogenized at 150 mg wet weight per mL of PBS +1% Triton™ X-100 + protease and phosphatase inhibitors. Consistent with the mouse TDP-43 analysis, western blotting was used to assess total, cytosolic, and nuclear protein levels for anti-phosphoTDP-43 (S403/404), anti-phosphoTDP-43 (S409/410), and anti-TDP-43 antibodies (i.e., the same antibodies and concentrations used in the mouse studies) to examine TDP-43 phosphorylation, mislocalization, and fragmentation in the human samples.
Human TBI and Control Sample Details
Clinical and epidemiological details of TBI and non-TBI patients.
IHD, ischemic heart disease; MVA, motor vehicle accident; TBI, traumatic brain injury.
Statistical analysis
Mouse data were analyzed using two-way or three-way analysis of variance (ANOVA) with either strain (i.e., TDP-43A315T and WT), injury (i.e., FPI and sham), and recovery time (i.e., 24 h and 1 week) as the between-subject variables. Bonferroni post hoc comparisons were carried out when appropriate. Human data were analyzed with t tests. All analyses were performed using SPSS 23.0 software (IBM Corp., Armonk, NY). Statistical significance was set at p < 0.05.
Results
Phosphorylated TDP-43 (S403/404)
Nuclear fractions
There was a significant strain × time × injury interaction (F1,24 = 111.333, p < 0.001; Fig. 1A) for the accumulation of nuclear 25kDa TDP-43 fragment in injured parietal cortex tissue. Post hoc analyses indicated that the WT + TBI group had increased levels of 25kDa TDP-43 fragment than their sham counterparts at both the 24-h and 1-week recovery times (p < 0.001), and the 1-week TDP-43A315T + TBI group had increased levels of 25kDa TDP-43 fragment compared with all other groups regardless of recovery time (p < 0.001).
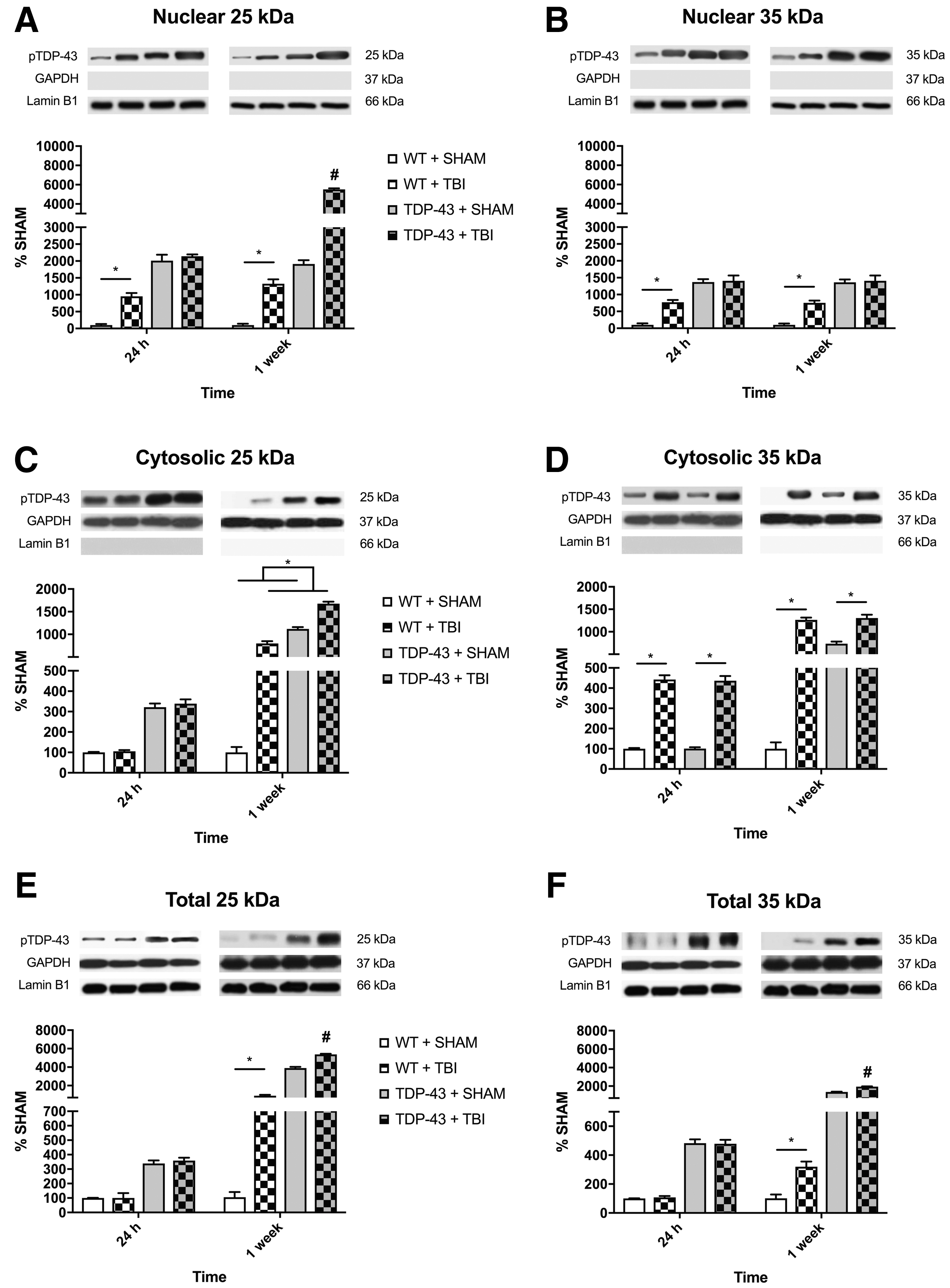
Representative western blots of nuclear, cytosolic, and total phosphorylated TDP-43 (S403/404) for 25kDa and 35kDa fragments.
For nuclear 35kDa TDP-43 fragment, there was a significant injury × strain interaction (F1,24 = 20.102, p < 0.001; Fig. 1B). Post hoc analyses revealed that the WT + TBI group had increased accumulation of 35kDa TDP-43 fragment compared with the WT + SHAM at both the 24-h and 1-week recovery times (p < 0.001), whereas theTDP-43A315T groups did not differ.
Cytosolic fractions
There was significant injury × time (F1,24 = 205.727, p < 0.001; Fig. 1C) interaction for the accumulation of cytosolic 25kDa TDP-43 fragment. There was also a significant strain × time (F1,24 = 281.007, p < 0.001) interaction. Post hoc analyses indicated that the TBI groups had increased levels of 25kDa TDP-43 fragment compared with shams at 1-week in both genotypes, but not 24-h, post-injury (p < 0.001).
For the cytosolic 35kDa TDP-43 fragment, there was a significant strain × time × injury interaction (F1,24 = 28.751, p < 0.001; Fig. 1D). Post hoc analyses indicated that the WT and TDP-43A315T TBI groups had increased levels of 35kDa TDP-43 fragment than their sham counterparts at both the 24-h and 1-week recovery times (p < 0.001). Further, the WT + TBI, TDP-43A315T + SHAM, and TDP-43A315T + TBI groups all had increased levels of 35kDa TDP-43 fragment at 1-week versus 24 h post-injury (p < 0.05).
Total sample
There was a significant strain × time × injury interaction (F1,24 = 14.693, p < 0.001; Fig. 1E) for the accumulation of 25kDa fragment. Post hoc analyses indicated that the WT + TBI mice had increased accumulation of 25kDa TDP-43 than their sham counterparts at 1-week, but not 24-h post-injury (p < 0.001). Further, the 1-week recovery TDP-43A315T + TBI mice had significantly increased levels of 25kDa TDP-43 fragment compared with all other groups at both recovery times (p < 0.001).
Similar to the 25kDa fragment findings, there was also a significant strain × time × injury interaction (F1,24 = 25.547, p < 0.001; Fig. 1F) for the levels of 35kDa fragment. Post hoc analyses indicated that the WT + TBI mice had increased levels of 35kDa fragment than their sham counterparts at 1-week, but not 24-h post-injury. Further, the 1-week recovery TDP-43A315T + TBI mice had significantly increased accumulation of 35kDa TDP-43 fragment compared with all other groups at both recovery times (p < 0.001).
Phosphorylated TDP-43 (S409/410)
Nuclear fractions
There was a significant strain × time × injury interaction (F1,24 = 151.358, p < .001; Fig. 2A) for the accumulation of nuclear 25kDa fragment. Post hoc analyses indicated that the WT + TBI group had increased accumulation of nuclear 25kDa fragment than their sham counterparts at both the 24-h and 1-week recovery times (p < 0.001), and the 1-week recovery TDP-43A315T + TBI group had increased accumulation of nuclear 25kDa fragment compared with all other groups regardless of recovery time (p < 0.001).
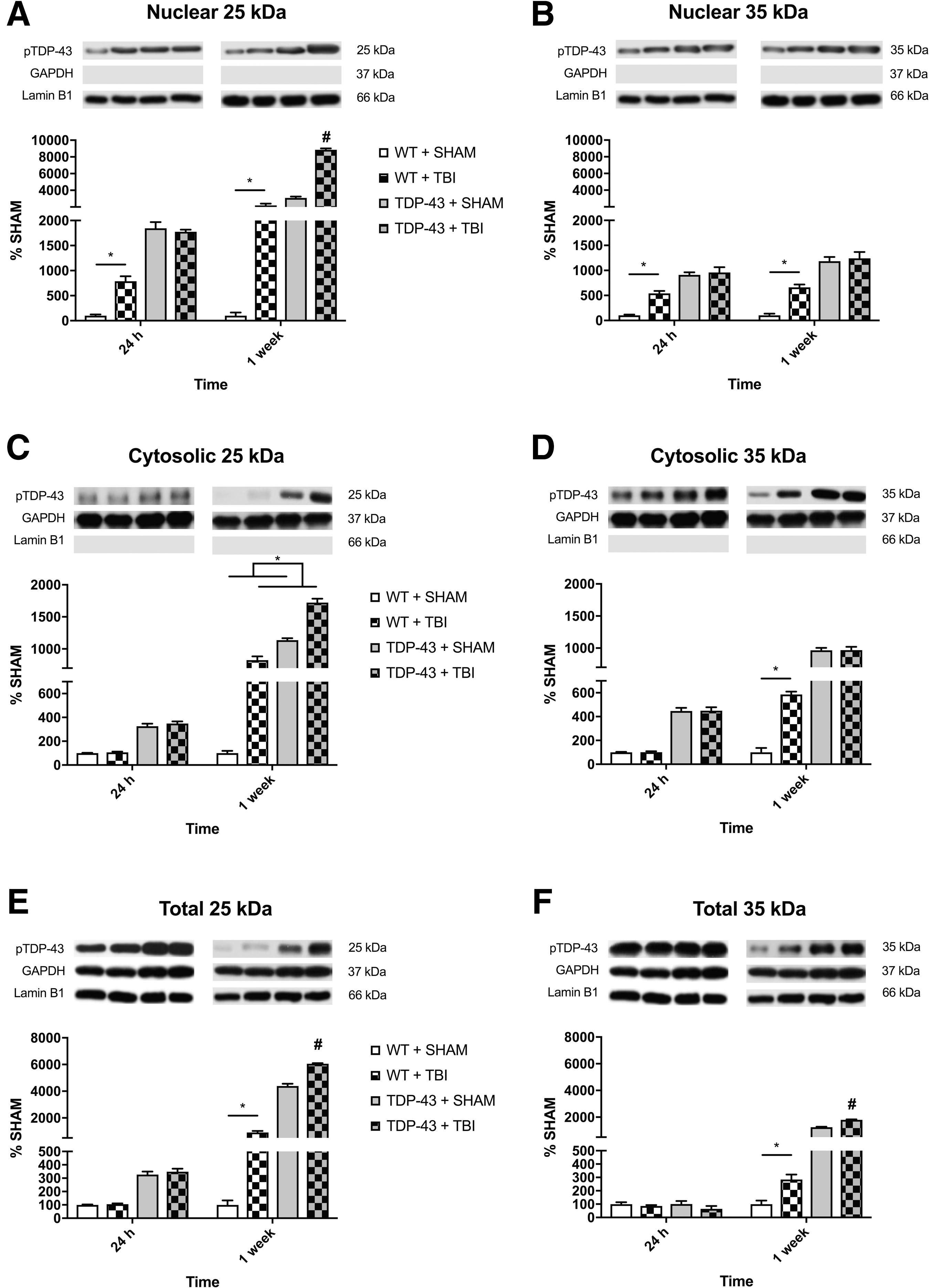
Representative western blots of nuclear, cytosolic, and total phosphorylated TDP-43 (S409/410) for 25kDa and 35kDa fragments.
For nuclear 35kDa fragment, there was a significant injury × strain interaction (F1,24 = 18.023, p < 0.001; Fig. 2B). Post hoc analyses revealed that the WT + TBI group had increased accumulation of nuclear 35kDa fragment compared with the WT + SHAM at both the 24-h and 1-week recovery times (p < 0.001), whereas the TDP-43A315T groups did not differ.
Cytosolic fractions
There was a significant injury × time interaction (F1,24 = 181.642, p < 0.001; Fig. 2C) for the level of cytosolic 25kDa fragment. There was also a significant strain × time interaction (F1,24 = 237.773, p < 0.001). Post hoc analyses indicated that the TBI groups had increased levels of 25kDa fragment at 1-week compared with their 24-h recovery counterparts (p < 0.05).
For the cytosolic 35kDa fragment, there was a significant strain × time × injury interaction (F1,24 = 30.712, p < 0.001; Fig. 2D). Post hoc analyses indicated that the WT + TBI group had increased levels of 35kDa fragment compared with the WT + SHAM group at the 1-week recovery time (p < 0.001). Further, the WT + TBI, TDP-43A315T + SHAM, and TDP-43A315T + TBI groups all had increased levels of 35kDa fragment at 1-week versus 24-h post-injury (p < 0.05).
Total sample
There was a significant strain × time × injury interaction (F1,24 = 15.254, p < 0.001; Fig. 2E) for the levels of 25kDa fragment. Post hoc analyses indicated that the WT + TBI mice had increased levels of 25kDa fragment than their sham counterparts at 1-week, but not 24-h post-injury (p < 0.001). Further, the 1-week TDP-43A315T + TBI mice had significantly increased levels of 25kDa fragment compared with all other groups at both recovery times (p < 0.001).
Similar to the 25kDa TDP-43 fragment findings, there was also a significant strain × time × injury interaction (F1,24 = 27.165, p < 0.001; Fig. 2F) for the levels of 35kDa fragment. Post hoc analyses indicated that the WT + TBI mice had increased levels of 35kDa fragment than their sham counterparts at 1-week (p < 0.01), but not 24-h post-injury. Further, the 1-week TDP-43A315T + TBI mice had significantly increased levels of 35kDa fragment compared with all other groups at both recovery times (p < 0.001).
Full-length TDP-43
Nuclear fractions
There was a significant strain × time × injury interaction (F1,24 = 108.980, p < 0.001; Fig. 3A). Post hoc analyses indicated that the WT + TBI group had increased expression compared with the WT + SHAM group at 24-h and 1-week post-injury (p < 0.001). Further, the TDP-43A315T + TBI mice had increased expression of nuclear TDP-43 at 1-week post-injury compared with all other 24 h and 1-week recovery groups (p < 0.001).
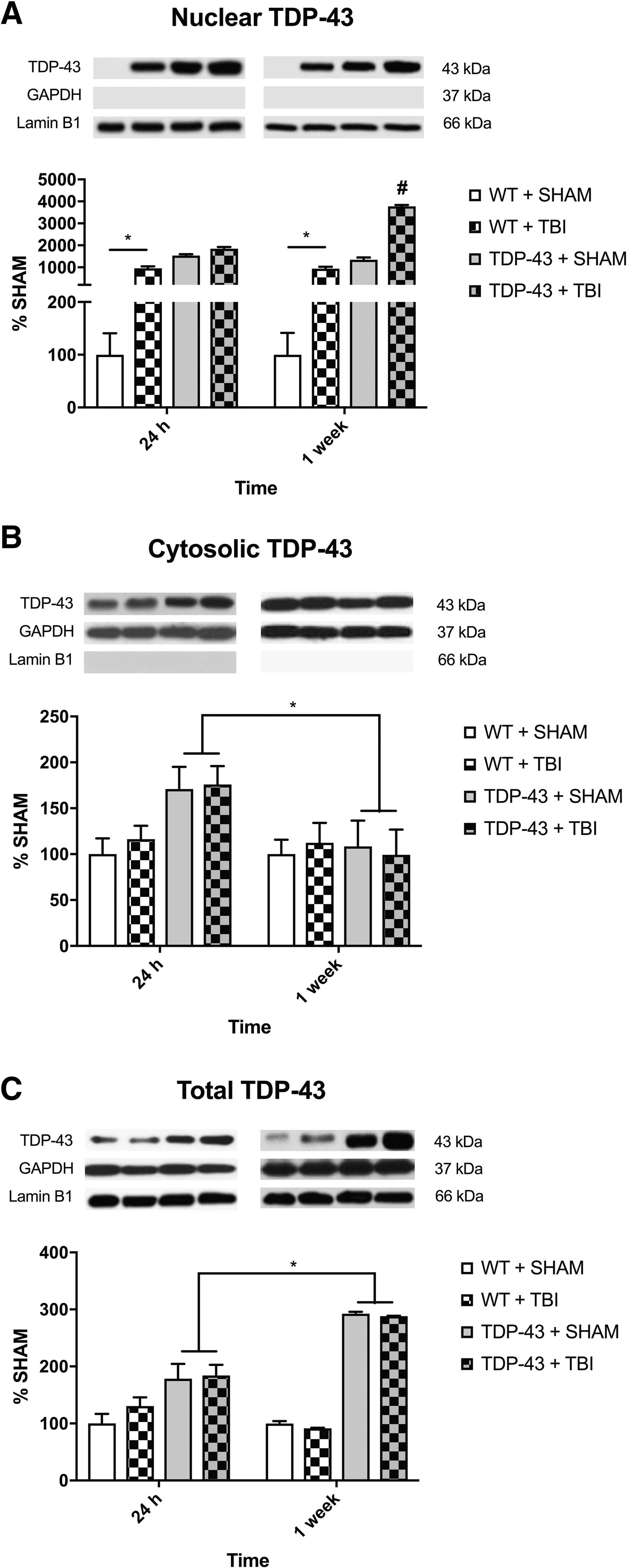
Representative western blots of nuclear, cytosolic, and total full-length TDP-43 (43kDa).
Cytosolic fractions
There was a significant strain × time interaction (F1,24 = 4.893, p < 0.05; Fig. 3B) for cytosolic TDP-43. Post hoc analyses showed that the TDP-43A315T mice had increased expression of cytosolic TDP-43 at 24-h compared with the TDP-43A315T mice at 1-week post-injury (p < 0.05).
Total samples
There was a significant strain × time interaction (F1,24 = 39.290, p < 0.001; Fig. 3C). Post hoc analyses showed that the 1-week recovery TDP-43A315T mice had increased expression compared with their 24-h TDP-43A315T counterparts (p < 0.01).
α-Fodrin
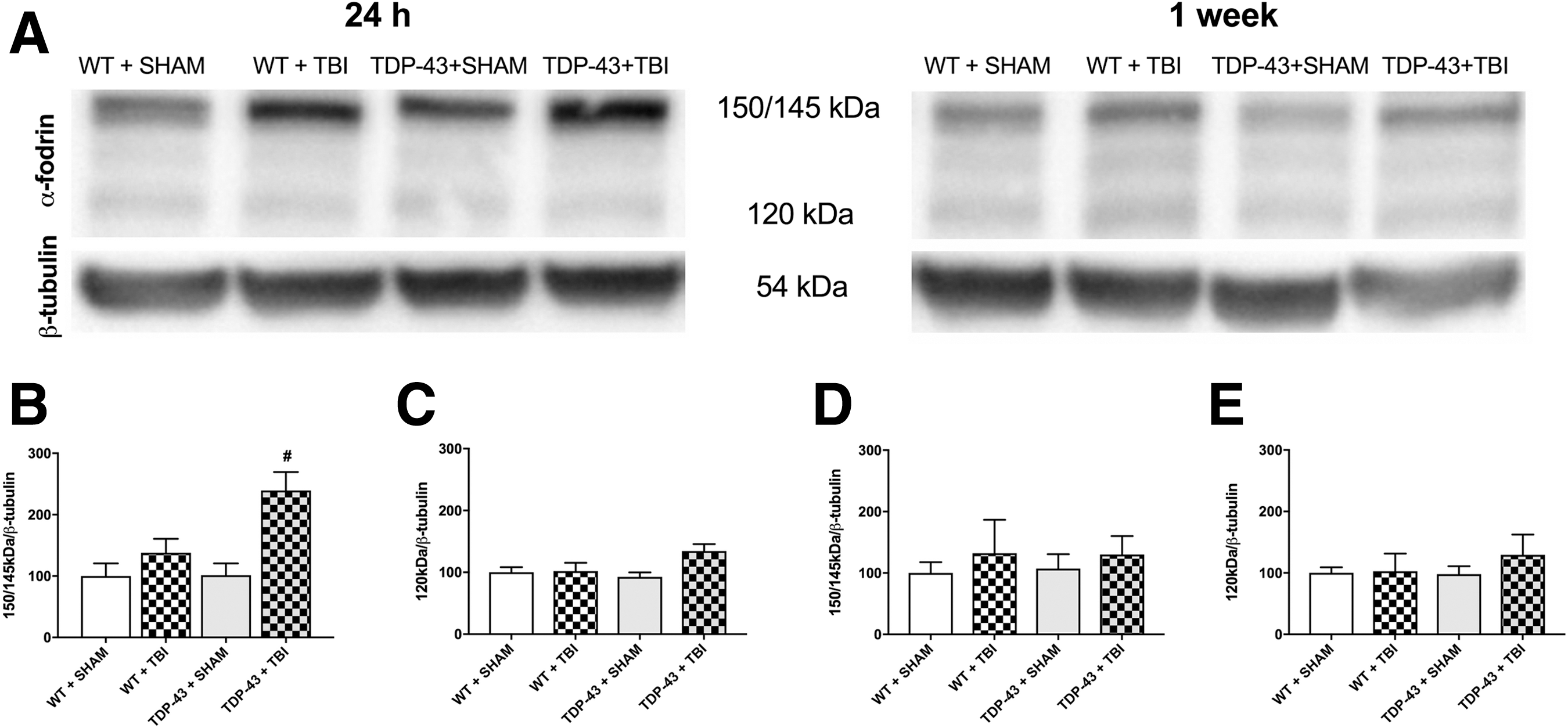
α-Fodrin cleavage was used as a marker for cell death (i.e., calpain-dependent = 150/145kDa, caspase-dependent = 120kDa). There was a significant strain × injury interaction for the 150/145kDa fragment (F1,16 = 4.519, p < 0.05; Fig. 4B) at 24-h post-injury, with post hoc analyses indicating that the TDP-43A315T + TBI mice had increased α-fodrin cleavage compared with all other groups. There was no statistically significant finding at 1-week post-injury. There were no statistically significant findings for the 120kDa fragment.
Behavior
There was a significant strain × injury interaction (F1,53 = 4.146, p < 0.05; Fig. 5A) for the measure of time in the novel arm of the Y-maze at 6 days post-injury. Post hoc analyses indicated that the TDP-43A315T + TBI mice spent significantly less time in the novel arm of the Y-maze than the other three groups (p < 0.05). There were no statistically significant findings on the measure of old arm entries (p > 0.05; Fig. 5B).
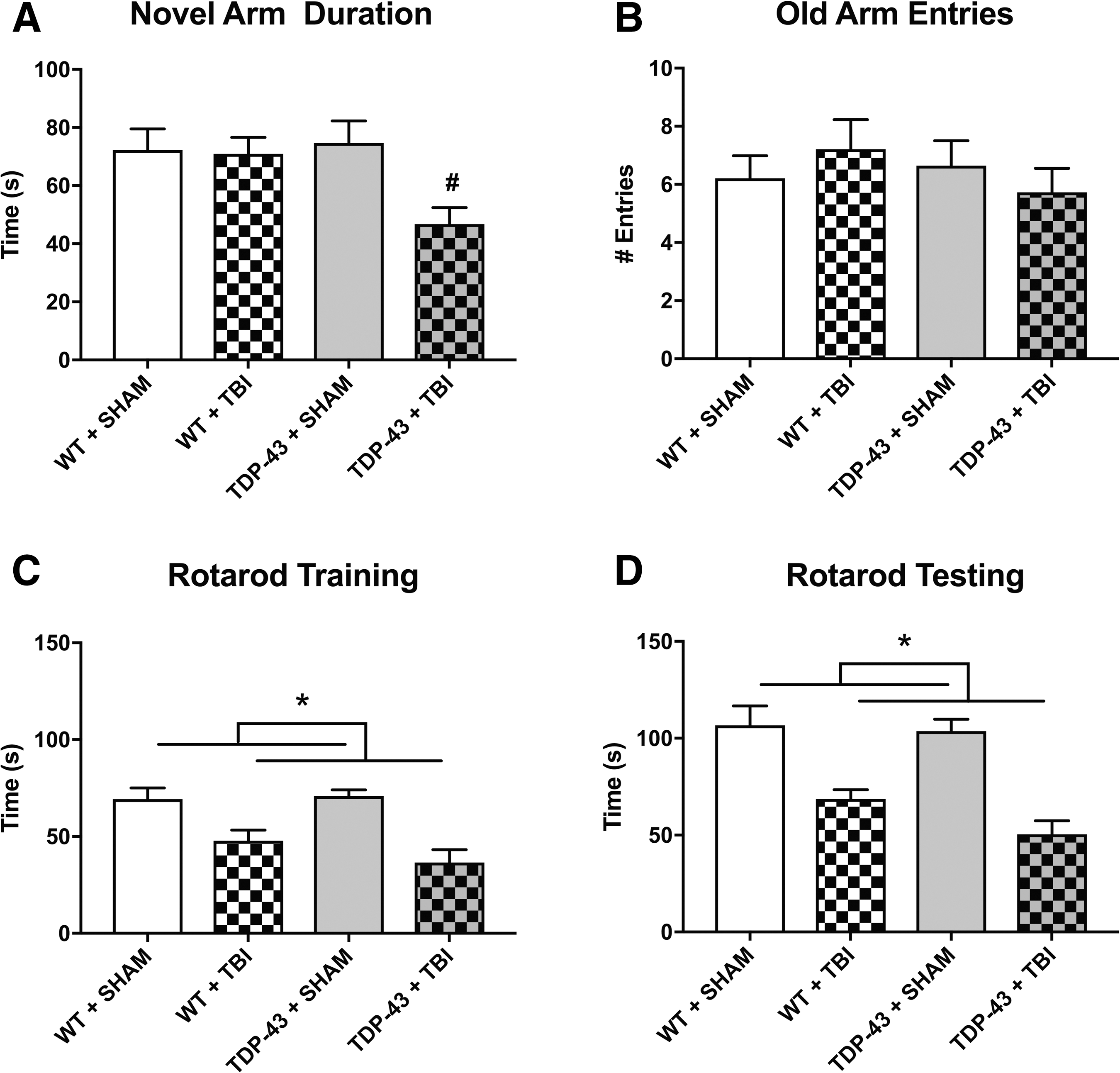
There was also a significant main effect for injury during both rotarod training (F1,53 = 25.897, p < 0.001; Fig. 5C) and testing (F1,53 = 40.194, p < 0.001; Fig. 5D) at 5 and 6 days post-injury, respectively, with TBI mice spending significantly less time on the rotarod compared with sham-injured mice, regardless of strain. There were no significant findings on the elevated plus maze (data not shown).
TDP-43 findings in TBI patients
The TBI patients had increased nuclear accumulation of phosphorylated TDP-43 25kDa fragment compared with the control group for both S403/404 (t13 = 2.782, p < 0.05; Fig. 6A) and S409/410 (t13 = 4.137, p < 0.01; Fig. 7A) phosphorylation sites. A similar result was found for the nuclear levels of phosphorylated TDP-43 35kDa fragment, with the TBI group accumulating more than the control group for both the S403/404 (t13 = 2.441, p < 0.05; Fig. 6B) and S409/410 (t13 = 2.622, p < 0.05; Fig. 7B) phosphorylation sites. There were no other statistically significant differences found on any of the other human western blot measures (Figs. 6 –8).
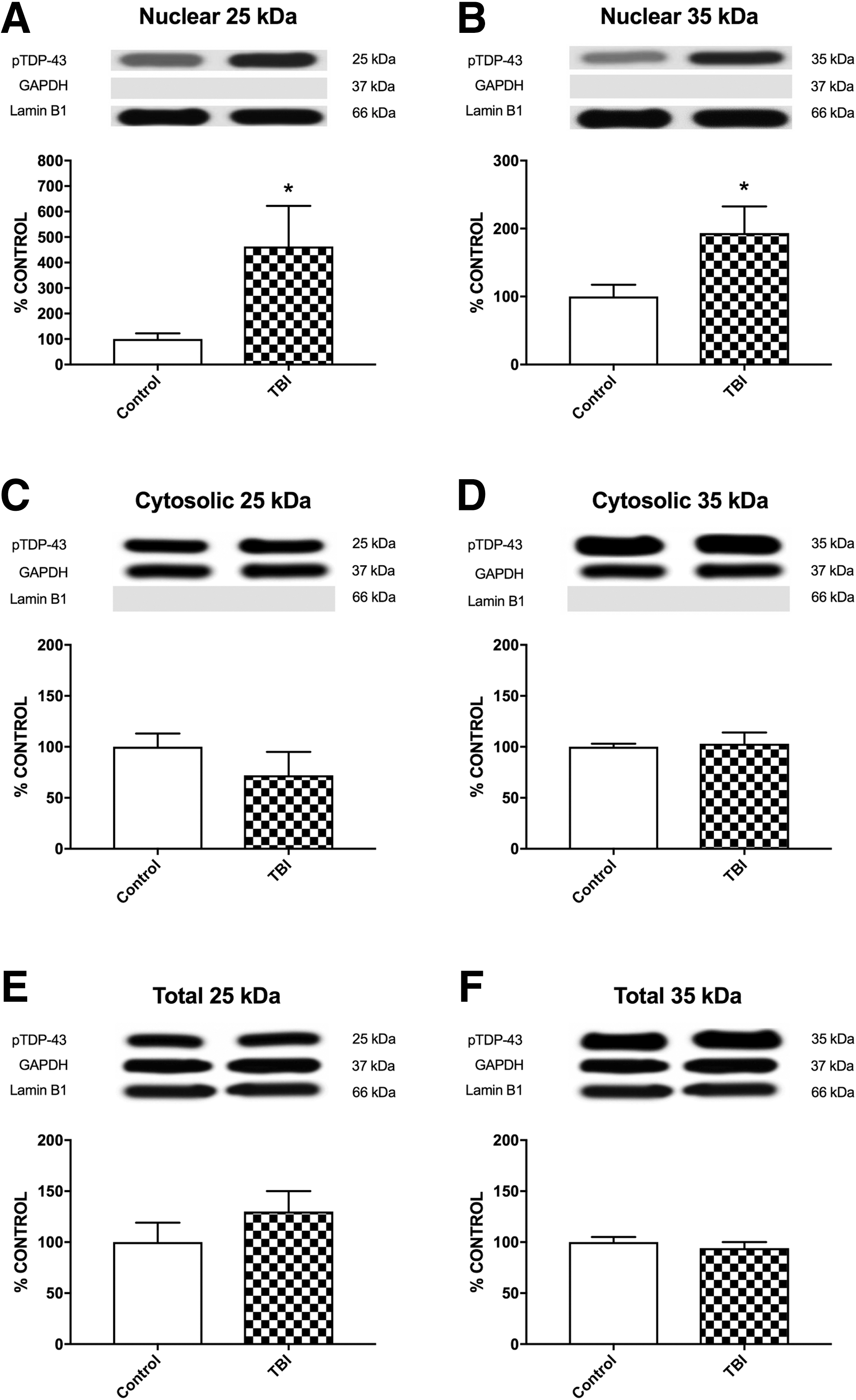
Human TBI patient western blots of nuclear, cytosolic, and total phosphorylated TDP-43 (S403/404) for 25kDa and 35kDa fragments. There were significant differences between the control and TBI groups for the nuclear 25kDa fragment
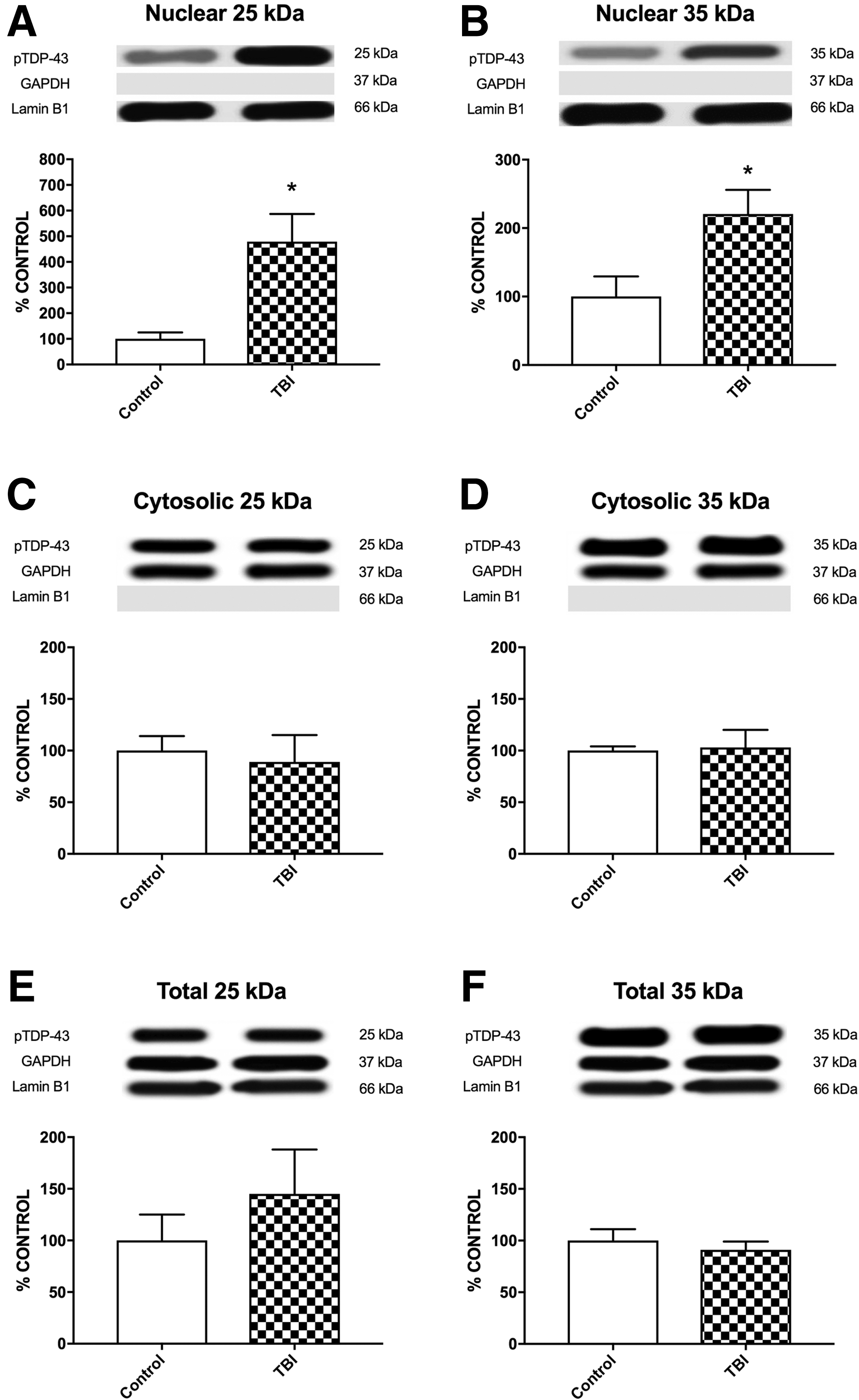
Human TBI patient western blots of nuclear, cytosolic, and total phosphorylated TDP-43 (S409/410) for 25kDa and 35kDa fragments. There were significant differences between the control and TBI groups for the nuclear 25kDa fragment
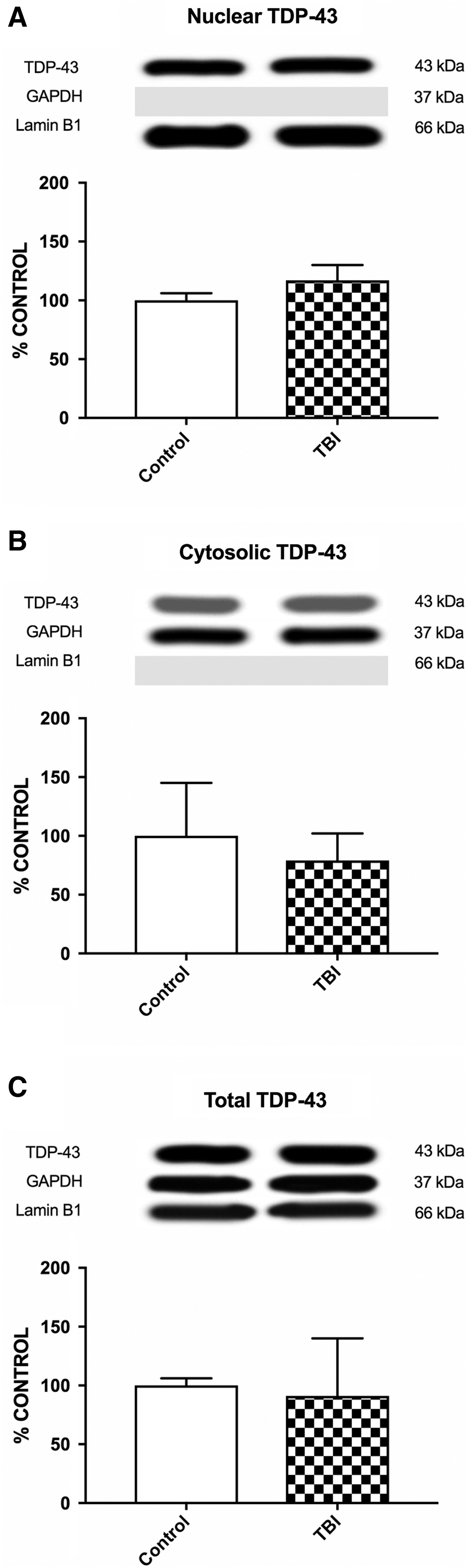
Human TBI patient western blots of nuclear, cytosolic, and total full-length TDP-43 (43kDA). There were no significant differences between the control and TBI groups. TBI, traumatic brain injury; TDP-43, transactive response DNA-binding protein 43.
Discussion
Here we examined abnormalities in TDP-43 and behaviors in WT and TDP-43A315T mice given a TBI. For both of the phosphorylated TDP-43 sites (i.e., S403/404 and S409/410), the WT + TBI mice revealed increased total, nuclear, and cytosolic levels of the 25kDa and 35kDa fragments relative to the WT + SHAM. The WT + TBI mice also had increased nuclear accumulation of full-length TDP-43 at 24-h and 1-week post-injury compared with the WT + SHAM, and mice given a TBI had motor deficits on the rotarod. The effect of TBI was exacerbated in the TDP-43A315T mice on many of the outcomes, as theTDP-43A315T + TBI group had increased TDP-43 abnormalities and expression of 150/145kDa α-fodrin (i.e., an indication of calpain-mediated brain cell death), and spent significantly less time in the novel arm during Y-maze testing (i.e., an indication of more severe cognitive deficits). We also examined post-mortem cortical tissue from humans who had suffered a TBI. Whereas there was increased nuclear accumulation of phosphorylated 25kDa and 35kDa TDP-43 fragments for both S403/404 and S409/410 sites in the TBI patients, no other significant findings were found.
The role of TDP-43 in TBI?
This study found that TBI induced the phosphorylation, fragmentation, and mislocalization of TDP-43, and that the group with the worst TDP-43 abnormalities (i.e., the TDP-43A315T + TBI mice) also had exacerbated cell death and functional deficits. Under normal physiological conditions, TDP-43 primarily resides in the nucleus, 33 although it is capable of shuttling between the cytoplasm and nucleus. 34,35 Under pathological stress, TDP-43 might undergo pathological modifications such as C-terminal truncation, phosphorylation, and aggregation, causing toxic gain or loss of functions. 15,36,37 Of note, oxidative stress, which is common in TBI, 1,38,39 has previously been shown to induce pathological modifications of TDP-43. 40
Studies have found 35kDa and 25kDa TDP-43 fragments in the brains of ALS and FTLD patients, 15 –17 and there is some evidence that fragmentation has neurotoxic effects. Due to its intrinsic caspase cleavage sites in its primary sequence, TDP-43 cleavage by caspases has been reported to generate C-terminal 35kDa and 25kDafragments. 41 –43 The 25kDa C-terminal TDP-43 fragment has been reported to be cleaved by caspase 4, which then activates caspase 3/7 to accelerate the TDP-43 fragmentation, and ultimately leads to cell death. 17 Calpain has also been reported to be involved in the generation of the 35kDa and 25kDa TDP-43 fragments. 16,44,45 For example, calpain-1 is involved in the generation and cleavage of the 25kDa fragments. 16 Although this study only found evidence for calpain-mediated cell death in rats given TBI, it is well established that TBI triggers numerous apoptotic processes, 16,44,45 including caspase-mediated cell death. 46,47 Therefore, it is possible that both calpains and caspases contributed to the generation of the TDP-43 fragments found in this study.
The 35kDa TDP-43 fragment has also been associated with the recruitment of TDP-43 to the cytoplasm. 15 TDP-43 has a nuclear localization signal and a nuclear export signal. 34,35 The absence of the nuclear localization signal, together with the presence of the nuclear export signal, results in the 35kDa fragment becoming a cytoplasmic resident protein and subsequently recruits more TDP-43 protein and may form aggregation. 15 Of relevance, our laboratory previously found that rats given an experimental TBI had increased cytoplasmic levels of TDP-43 in neurons, which was associated with progressive neurodegeneration and motor dysfunction. 20 Another study found increased levels of cytosolic 25kDa fragment in mice after TBI that was associated with the formation of TDP-43 aggregates. 48
Previous studies have reported that TDP-43 phosphorylation occurs in all sporadic and familial forms of TDP-43. 49 –51 TDP-43 has been recognized to have up to 29 phosphorylation sites, including the well-documented S403/404 and S409/410 sites that were examined in the current study. 49 –52 These sites are mainly phosphorylated by casein kinase-1, 52 in particular the hyperactive truncated form of casein kinase, 53 although the actual mechanism of phosphorylation and the specific function of the different phosphorylation sites are unclear. Although the pattern of abnormalities in this study were for the most part consistent between the S403/404 and S409/410 sites, there were also some differences. For example, nuclear expression of 25kDa S409/410 fragments in the WT mice rose from 24-h to 1-week post-TBI, and this was much more pronounced than the nuclear expression of the 25kDa S409/410 fragments that remained relatively stable at the 24-h and 1-week recovery times. On the other hand, increased expression of cytosolic 35kDa S403/404 fragments at 24 h was the first indication of mislocalization post-TBI, and this continued to increase at the 1-week recovery time, whereas cytosolic expression of 35kDa S409/410 fragments in WT mice was unaffected by TBI at 24-h post-injury before increasing at 1 week. The significance of these findings remains to be determined, but suggest that there may be mechanistic differences to the phosphorylation sites.
Although the overall literature does suggest that TBI induces TDP-43 abnormalities, there are also inconsistencies within these findings that should be considered. For example, whereas a single, severe TBI in humans has been found to result in increased phosphorylation-independent cytoplasmic TDP-43, 19 the only human evidence for phosphorylated TDP-43 inclusions in the cytoplasm (i.e., pathological TDP-43) has been found in cases of repetitive mild brain trauma. 18 On the other hand, the current study found evidence for both TDP-43 phosphorylation and cytosolic mislocalization in mice given a single TBI. There are a number of possible explanations for these findings, all of which will require further examination to confirm. First, the study by Johnson and colleagues in patients with single, severe TBI relied on immunohistochemical analysis of sections from various brain regions, 19 whereas the current study relied on western blot analyses of the mouse parietal cortex directly at the injury site. The severity of TBI might be another factor that could account for the different findings, as the Johnson study examined severe TBI, whereas the mice in this studied received a relatively mild TBI (i.e., no evidence of cognitive impairment in the WT + TBI mice). 19 The antibodies and phosphorylation sites that were examined in the studies also differed. 19 For these reasons, it is difficult to make any concrete conclusions regarding the inconsistencies between the findings, and future studies should attempt to use similar methodologies so that comparisons across the studies are possible. For example, the current study would have been strengthened by the inclusion of immunohistochemical analysis to determine whether the TDP-43 abnormalities identified by western blot are accompanied by classical TDP-43 pathology hallmarks.
The human findings from the current study also differed from our mouse findings. Specifically, whereas the finding that the human TBI samples had increased nuclear accumulation of phosphorylated TDP-43 fragments was consistent with the mouse findings, no other significant findings were found in the human samples (e.g., no evidence of mislocalization). There are a number of important considerations when interpreting our human data. Perhaps most importantly is that our group sizes were very low, especially considering the heterogeneity of human TBI. To date, all human studies investigating TDP-43 in TBI have consisted of relatively low group sizes, and larger scale studies are required to comprehensively characterize TDP-43 abnormalities in human TBI patients. Further, our mouse data suggest there are temporal complexities involved in TDP-43 abnormalities that could account for the discrepancy between our mouse and human findings. For example, cytoplasmic mislocalization of TDP-43 was most evident at 1-week post-injury in the mice, whereas the average survival time for the TBI patients was ∼18 h (range, 1–76 h), which may have been too early to detect cytoplasmic changes. It should also be noted that the mean age of the TBI and control patients in our study was high in comparison with previous pre-clinical and clinical studies, which could impact TDP-43 abnormalities.
TBI and TDP-43 have both been implicated in the neurodegenerative conditions CTE and ALS. 6 –9,18 CTE is postulated to be a neurodegenerative condition caused by repetitive, mild brain injuries, and some studies have reported that TDP-43 pathologies can occur in CTE. 7,18 As noted above, the TBI severity given to the mice in this study was relatively mild and our findings that WT mice given a TBI had TDP-43 abnormalities, and that TDP-43 + TBI mice (i.e., mice with TDP-43 abnormalities at the time of injury) had worse outcomes, may bear relevance to a scenario where an individual sustains repetitive brain injuries and suffers cumulative/exacerbated effects. Similarly, TBI has also been linked to the later onset of ALS, and the TDP-43A315T + TBI mice (i.e., mice predisposed to TDP-43 pathologies/ALS) findings may also be relevant to this scenario and warrant further investigation. Overall, the relationships between TBI and neurodegenerative conditions such as CTE and ALS remain controversial; however, TDP-43 may provide a mechanistic link between the conditions that should be further explored. Unfortunately, due to the eventual onset of motor deficits and short life span of the TDP-43A315T mice, this study was limited to studying the acute and sub-acute effect of TBI on TDP-43. Future studies must also be done in the chronic post-TBI setting to continue to characterize how TDP-43 abnormalities evolve and contribute to progressive neurodegeneration.
If future studies can provide a better understanding of the role of specific TDP-43 abnormalities in the neurological consequences of TBI, it may be possible to target TDP-43 to improve TBI outcomes. Compounds such as rapamycine and treholose have been found to reduce TDP-43 levels and aggregation. 54,55 Targeting the unfolded protein response might also be a treatment strategy to combat the neurotoxic effects of TDP-43 abnormalities. 56 Importantly, our novel findings of acute and sub-acute TDP-43 abnormalities provide initial insight into the potential therapeutic window of drugs that may be used to target TDP-43 pathologies after TBI. In particular, whereas nuclear TDP-43 abnormalities (i.e., fragmented phosphorylated TDP-43 and increased expression of full-length TDP-43) in the WT + TBI mice were present by 24-h post-injury and remained relatively stable at 1-week post-injury, cytosolic abnormalities were much more pronounced 1 week after TBI. Another interesting finding related to the timing of the TDP-43 abnormalities was that although there was increased nuclear expression of full-length TDP-43 and phosphorylated fragments at both 24-h and 1-week post-TBI, there was no evidence for increased expression of cytosolic full-length TDP-43 at either 24-h and 1-week post-TBI. This suggests that fragmentation may have occurred prior to transportation to the cytoplasm, although future studies with additional antibodies, time-points, and methods are required to further investigate if this is the case.
Conclusion
This study found that WT mice given TBI had increased TDP-43 phosphorylation, mislocalization, and fragmentation compared with sham-injured WT mice, and this was exacerbated in the TDP-43A315T mice that are predisposed to TDP-43 pathologies. Further, the TDP-43A315T mice given a TBI had worse cell death and cognitive deficits compared with all other groups. Findings from western blot analysis of brain tissue from human TBI patients showed increased nuclear accumulation of phosphorylated TDP-43 fragments, but were otherwise unremarkable and inconsistent with the robust mouse findings. Factors including heterogeneity in clinical TBI, the low group sizes in the patient study, and temporal complexities with TDP43 abnormalities could account for this discrepancy. Although there were limitations with this study and future research is required, the findings of this study indicate that TBI can induce TDP-43 abnormalities, and that these changes may contribute to the neurological consequences of TBI.
Footnotes
Acknowledgments
This study was funded by grants and fellowships awarded to SRS from NHMRC and the Motor Neuron Disease Research Institute of Australia (Mick Rodger & Benalla Act to d'feet MND Grant), a Stafford Fox Medical Research Foundation awarded to BJT, and a grant to MS from the Alzheimer's Australia Dementia Research Fund. Human tissues were received from the Victorian Brain Bank, supported by The Florey Institute of Neuroscience and Mental Health, The Alfred and the Victorian Institute of Forensic Medicine and funded in part by Parkinson's Victoria, MND Victoria, and Cure for MND Foundation. The Florey Institute of Neuroscience and Mental Health acknowledges the strong support from the Victorian Government and in particular the funding from the Operational Infrastructure Support Grant.
Author Disclosure Statement
No competing financial interests exist.