
Abstract
Acute subdural hemorrhage (ASDH) is common and associated with severe morbidity and mortality. To date, the role of spontaneous cortical spreading depression (sCSD) in exaggerating secondary injury after ASDH, is poorly understood. The present study contains two experimental groups: First, we investigated and characterized the occurrence of sCSD after subdural blood infusion (300 μL) via tissue impedance (IMP) measurement in a rat model. Second, we compared the occurrence and influence of sCSD on lesion growth and neurological deficit in the presence and absence of whole blood constituents. In the first experimental group, three IMP traits could be distinguished after ASDH: no sCSD, recurrent sCSD, and constant elevated IMP (anoxic depolarization [AD]). In the second experimental group, sCSD occurred more often after autologous blood, compared with paraffin oil infusion. Lesion volume 7 days post-ASDH was 27.3 ± 6.8 mm3 after blood and 3.4 ± 2.1 mm3 after paraffin oil infusion. Subgroup analysis showed larger lesion size in animals with sCSD, than in those without. Further, occurrence of sCSD led to worse neurological outcomes in both groups. sCSD occurs early after ASDH and does not depend on the presence of whole blood constituents. However, numbers and degree of sCSD are more frequent and severe after autologous blood infusion, compared with an inert volume substance. The occurrence of sCSD leads to lesion growth and worse neurological outcome. Thus, our data advocate close monitoring and targeted treatment of sCSD after ASDH evacuation.
Introduction
In 1944, Leão discovered a cessation of electroencephalogram activity, which lasted for several minutes, while studying epilepsy in rabbits. 1 He later described the phenomenon as a loss of ion homeostasis spreading throughout the cortex, leading to spontaneous cortical spreading depression (sCSD) of electrical activity, after electrical or mechanical alteration. 1 –3 sCSD has since been studied in various animal models and humans and was found to represent cortical excitations followed by a spreading loss of functioning neurons and astrocytes. 4 The wave-like depolarization is associated with changes in cerebral blood flow (CBF), perfusion, swollen neurons, and alterations in dendritic spines. 5,6
Substances such as potassium, acetylcholine and glutamate, as well as conditions of hypoxia, hypoglycemia, ischemia, subarachnoid hemorrhage (SAH), traumatic brain injury (TBI), and seizures have been identified to cause sCSD. 7 –10 Changes of the ionic homeostasis, the underlying mechanism of sCSD, leads to cellular swelling after influx of cations, declining extracellular space, and consecutive loss of membrane potentials. 9 These changes can be detected using tissue impedance (IMP) measurement as an indicator for swelling. 11 It has been shown that each CSD, either spontaneous or triggered by potassium chloride (KCl), leads to a transient increase in tissue impedance (average of 2 kOhm). 12 The restoration of the ion homeostasis is energy dependent and under physiological conditions the brain copes with hyperemia and vasodilatation. 13,14 Increased energy demands are reflected in the form of decreased glucose, glycogen, and increased lactate concentrations. In sCSD under pathophysiological conditions (e.g., focal lesions), the surrounding microenvironment is acidic and high in potassium, which promotes vasoconstriction, leading to inverse hemodynamic reactions in regions with already compromised blood flow. 15 These intra-lesional conditions allow the persistent anoxic depolarization (AD) to become a transient depolarization spreading through the healthy, surrounding tissue.
Due to a poor neurovascular coupling, the raised energy demand in the wake of sCSD cannot be met; therefore, areas of watershed are at risk for becoming ischemic. 16 This undersupply may render more tissue susceptible to sCSD. 17,18 Consequently, ischemia and elevated energy demands lead to cell death in the peri-ischemic area and thus, secondary lesion growth. 18 The COSBID (Co-Operative Studies on Brain Injury Depolarizations) research group focuses on the causes and consequences of sCSD in lesion development under various pathological conditions (e.g., TBI, aneurysm/SAH and stroke). 19,20 Several COSBID studies demonstrate that electrocorticography (ECoG) depressions, identical to CSD in animal models, occur with a higher likelihood in patients with TBI and are estimated to be occur in as many as 56% of all patients with TBI. 21,22 Further, in clinical studies sCSD has repeatedly been proven by real-time monitoring of electrophysiology, blood flow, and cytotoxic edema to cause secondary damage. 23 It is reported that similar events might contribute to secondary lesion development after acute subdural hemorrhage (ASDH), an entity often associated with TBI, where extravasated blood causes elevated intracranial pressure (ICP) and consecutive ischemia. 24 It seems that factors other than merely volume mass lesions are important for cell death and lesion development, as subdural infusion of inert volume substances showed no substantial lesion formation despite similar ICP and CBF characteristics. 25 As ASDH pathophysiology comprises focal ischemia as well as the impact of blood constituents, sCSD may play a decisive role in secondary lesion progression and thus might be targeted by N-Methyl-D-aspartate (NMDA) antagonists (e.g., ketamine), anti-epileptic drugs (e.g., levetiracetam), NO donators, calcium channel blockers (e.g., nimodipine), or induced hypothermia. 7,26 –28
Our study was designed as a two-tailed approach to (1) characterize the occurrence of sCSD after ASDH and (2) to address whether sCSDs occurrence, lesion size, and neurological outcome depend on the presence of whole blood constituents rather than disturbances of ICP and CBF alone.
Methods
Experimental design
Experimental group 1
To investigate the occurrence and influence of sCSDs on the outcome after subdural hematoma, 300 μL autologous blood was injected subdurally in 20 male Sprague-Dawley rats. Monitoring of physiological parameters, ICP, and tissue IMP was performed starting 10 min prior to ASDH induction and continued for 180 min. All animals were then returned to their housing for 7 days. Ultimately, all animals were sacrificed and the lesion size was histologically determined.
Experimental group 2
To analyze the influence of whole blood constituents on sCSD occurrence, consecutive lesion size, and neurological outcome, paraffin oil was used as inert volume substance to form a subdural mass lesion lacking blood constituents. Twenty-eight male Sprague-Dawley rats were randomly assigned to sham, autologous blood, or paraffin oil infusion. Monitoring of vital parameters, as well as extended neuro-monitoring of ICP and tissue IMP was performed, starting 10 min prior to ASDH induction and continued for 180 min. All animals were then returned to their housing for 7 days and ultimately sacrificed for determination of the lesion volume.
Animal care and anesthesia
A total of 48 male Sprague-Dawley rats (Charles River, Germany) were used in all experiments. All animals were housed at room temperature of 22 ± 2°C and humidity >50%. Animals were anesthetized by intraperitoneal (i.p.) injection of chloral hydrate. The initial dose was 36 mg/ 100 g body weight. To keep the animals under anesthesia, the same dose was given hourly through a permanently placed i.p. catheter. The animals were intubated and mechanically ventilated and their body temperature was closely monitored and kept at 37.5°C throughout the operation using a heating pad and a rectal thermometer (Homeothermic Blanket Unit, Harvard, Kent, UK). All experimental procedures were approved by the local ethics committee and were in accordance with the German guidelines for animal use and care.
Surgical preparation for ASDH
The tail artery was cannulated for blood pressure measurement and sampling of blood for blood gas analysis (BGA). The volume needed for each BGA was 210 μL (ABL615/EML105, Radiometer, Copenhagen, DK). A catheter was placed in the jugular vein to obtain blood for subdural infusion. Animals were harnessed in a stereotaxic frame; a craniotomy with a diameter of 3 mm was made between the bregma and lambda suture on the left side as previously described. 29 Briefly, the dura was opened and a bent, L-shaped, blunt cannula (23G) was inserted and secured with tissue glue (Histoacryl; B Braun, Melsungen, Germany) and dental cement. Two further burr holes were drilled anterior to the bregma suture (anterior-posterior: 2 mm, midline: 2.5–3 mm) to insert the IMP electrodes (two stainless-steel wires covered in polyvinyl chloride for electrical insulation except for the sharp-pointed tips). IMP was measured at 1 kHz (10 mV, bias-free). Changes in tissue IMP were accompanied by direct current shifts during sCSD occurrence. ICP was measured intraventricularly through a 26G needle on the contralateral side. All probes and needles were also fixed with tissue glue. A laser Doppler probe was placed on the ipsilateral hemisphere frontal to the bregma (Vasamedics Laserflo BPM2, Vasamedics Fiber-Optic Probe 8*200 mm; St Paul, MN). All parameters were recorded continuously every minute during a 10-min baseline, subdural infusion of 300 μL autologous non-heparinized blood or paraffin oil at a flow rate of 50 μL/min (6-min period) and continued for 180 min after ASDH induction. After injection, the catheter was removed and the scalp was sutured; the animal could recover and was returned to regular housing. The sham-operated groups received no infusion.
Evaluation of behavioral and neurological deficits
Then, a Euroscore was compiled to quantify sensory and motor integrity by evaluating motor activity, orientation, and reaction to tactile, visual, and auditory stimuli. 30 To determine the Neuroscore, 10 items were assessed and scored separately, and then totaled to derive the overall Neuroscore. Each item was scored in a relatively binary fashion (present/absent, normal/abnormal) and assigned a score of 0 or 10 with 10 denoting an abnormal finding, and totaled to a maximum total Neuroscore of 100, with a higher Neuroscore denoting a more severe neurological deficit.
The neuro-status was assessed on the day before the experiment and on day 7. The beam-walk and beam-balance test were conducted in a quiet room in dim light on day 7 after ASDH induction. Before the operation, all animals were trained to walk along a beam (diameter 1.8 cm, length 1 m, 2.5 cm between bars, 50 cm above ground) while the time to reach a black box was measured. 29
Neuro-histological evaluation
The animals were transcardial-perfusion fixed with 0.9% saline and 4% paraformaldehyde (pH 7.4). Brains were carefully removed, post-fixed for 24 h, and embedded in paraffin wax. Sequential 3-μm thick coronal sections were cut 250 μm apart throughout the visible lesion space and stained with hematoxylin and eosin. The lesion on each section was quantified using Optimas 6.1 software combined with a CCD Super Color CV950 camera (Denmark) mounted on a Zeiss Axiopod 2Plus. From the measured area (An [mm2]) on each section and the distance (d = 0.25 mm) between the consecutive sections the entire are of damage was calculated and expressed as the lesion volume (V, mm3) by the following equation:
Statistical analysis
Data are expressed as mean ± stand error of the mean (SEM). Comparison of the different groups was performed using a one-way analysis of variance (ANOVA) or standard t test (Sigma-Plot 11.0, Systat, CA). Differences were considered statistically significant at p < 0.05. Subgroup analysis was performed comparing animals with and without sCSDs.
Results
ASDH elicits sCSDs (Experimental group 1)
To investigate the occurrence and influence of sCSDs on the outcome after ASHD, 300 μL autologous blood was subdurally injected and the physiological parameters, ICP, and tissue IMP were monitored starting 10 min prior to ASDH induction and were continued for 180 min. Arterial blood gases taken before and after infusion of blood remained stable with physiological boundaries over the course of the experiment.
Mean arterial blood pressure (MAP) showed peak values 10 min after ASDH induction (80.9 ± 3.44 mm Hg, p = 0.017) and recovered within 60 min to baseline levels of 61.08 ± 5.5 mm Hg (Fig. 1). No MAP increase was observed in sham operated animals. The ICP increased during ASDH induction with peak values of 47.14 ± 13.32 mm Hg at the end of the infusion (Fig. 1). Thereafter, it dropped to values twice as high as baseline and remained elevated during the entire monitoring period. As the ICP increased during ASDH induction, MAP showed a slightly shifted sinusoidal course as manifestation of the Cushing reflex (Fig. 1).

Measurment of ICP and MAP during ASDH induction. MAP and ICP were measured starting 10 min prior to the subdural injection of 300 μL autologous blood and continued for 180 min (n = 20). The MAP showed peak values 10 min after ASDH induction (80.9 ± 3.44 mm Hg, p = 0.017) and recovered within 60 min to baseline levels. No MAP increase was observed in sham operated animals (n = 3). ICP increased during ASDH induction with peak values at the end of the infusion (47.14 ± 13.32 mm Hg). ICP and MAP showed slightly shifted sinusoidal curves as manifestation of the Cushing reflex. ASDH, acute subdural hemorrhage; ICP, intracranial pressure; MAP, mean arterial blood pressure.
After ASDH induction, all animals displayed an initial increase of the tissue IMP, whereas no increased IMP was detectable in sham operated animals (Fig. 2A). The cerebral perfusion pressure (CPP) was inversely correlated to the IMP (Fig. 2B). Overall, three different traits of IMP were observed: (1) no sCSDs (n = 7), (2) detectable sCSDs (n = 8), and (3) continuous IMP elevation as equivalent of AD (n = 5) (Fig. 2C). IMP recordings were calibrated to 2254.44 ± 61.50 Ω as baseline readings and normalized to 100%. Subdural blood infusion initiated a collinear increase of IMP and ICP. No statistically significant difference in the initial IMP increment (p = 0.685) or duration (p = 0.084) was detected between animals with and without further sCSDs. Peak value of 172.55 ± 17.60% of baseline was reached at the end of infusion and IMP decreased within 30 min after ASDH induction. First sCSDs occurred 53.86 ± 1.73 min after ASDH in eight animals, whereas no sCSDs were observed in seven animals. On average 6 ± 0.58 sCSDs were observed. In five animals a continuous IMP elevation with peak values of 166.67 ± 21.01% of baseline was observed. Three of these animals died within 24 h after ASDH. All recorded sCSDs revealed similar traits. Each lasted 2.66 ± 0.31 min, whereas the first one lasted longest (3.43 ± 0.34 min) and the last one shortest (2.47 ± 0.28 min). The mean elevation of baseline was 15 ± 0.02 % and was consistent for each animal.
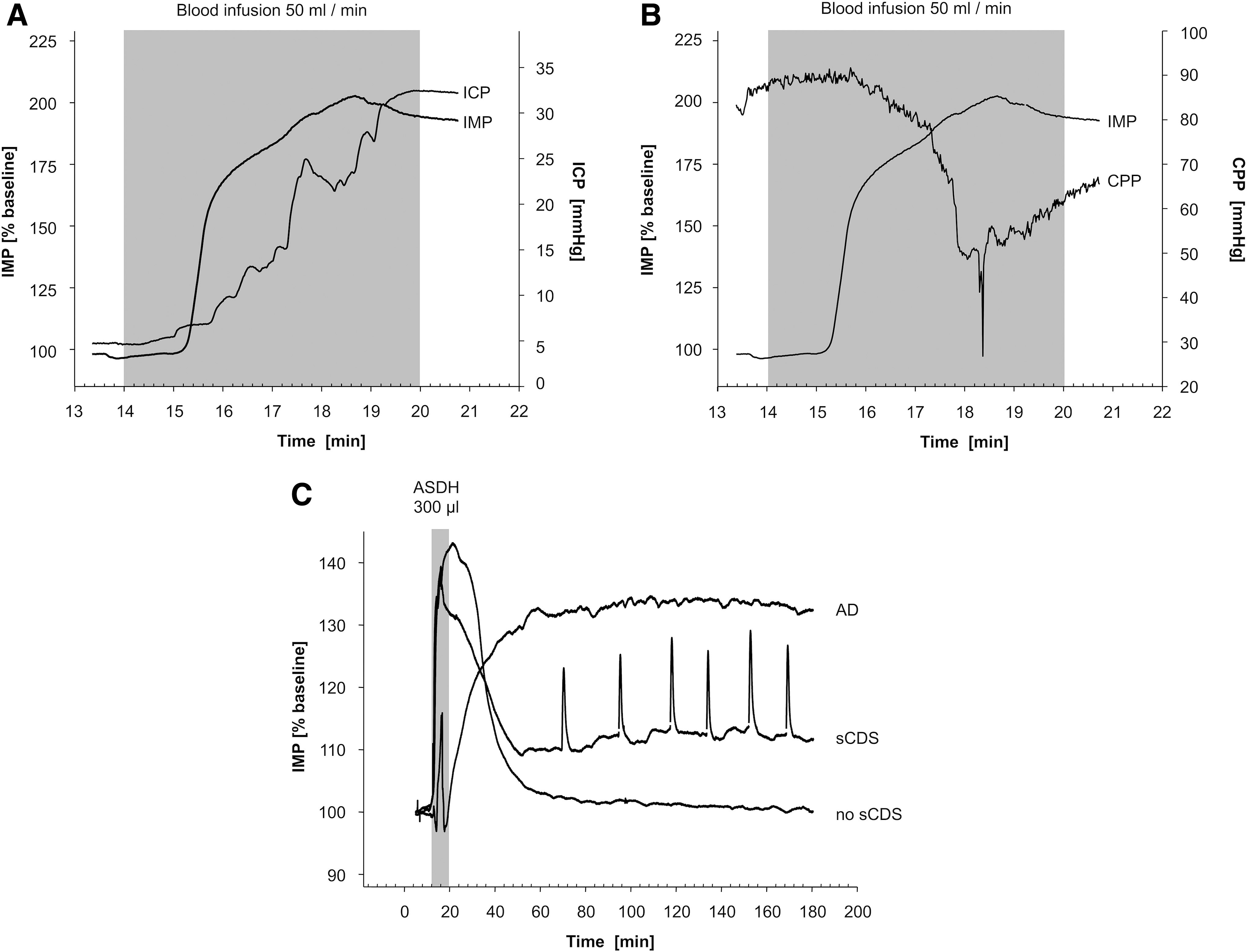
IMP and ICP changes during and after subdural blood infusion. Depicted are the tissue IMP measurements in % of baseline.
To evaluate the overall lesion volume, sequential coronal sections were cut throughout the visible lesion and stained with hematoxylin and eosin. The overall lesion volume for all animals was 46.2 ± 17.1 mm3. If sCSDs had been detected, the lesion size was found to be larger by a statistical significance (56.7 ± 10.7 mm3), compared with those without initial sCSDs (27.7 ± 10.4 mm3; p = 0.01) (Fig. 3). Two animals classified as AD had a mean lesion size of 55.8 ± 3.7 mm3.
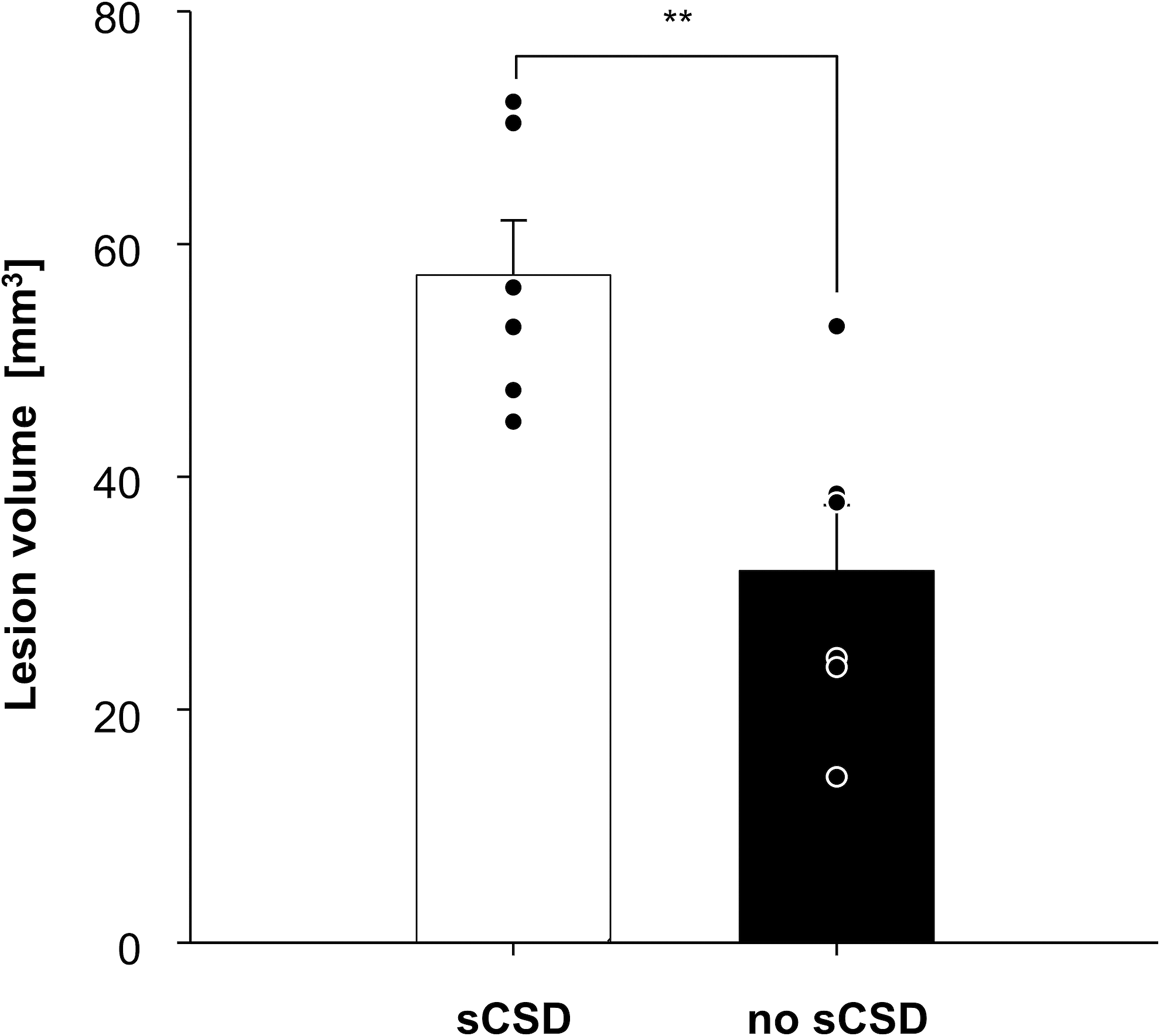
Lesion size differs with the occurrence of sCSD. Lesion size of animals with sCSD showed significantly (p = 0.01) larger cortical lesions 7 days after ASDH induction compared with animals without sCSD. ASDH, acute subdural hemorrhage; sCSD, spontaneous cortical spreading depression.
sCSDs are more frequent and severe after autologous blood infusion leading to increased lesions and worse neurological outcomes (Experimental group 2)
In the second experimental group, paraffin oil was used as an inert volume substance to form a subdural mass lesion lacking blood constituents. Within this group the influence of whole blood constituents on sCSD occurrence, consecutive lesion size, and neurological outcome was analyzed. Twenty-eight animals were randomly assigned for autologous blood (n = 13), paraffin oil (n = 12), or sham (n = 3) operation. All peri-procedural parameters were measured comparable to the first experimental group.
MAP rose from 64.8 ± 3.1 mm Hg to peak values of 80.1 ±7.3 mm Hg 6 min after the end of the infusion in the blood-infused animals. During paraffin oil infusion, MAP increased from 76.4 ± 2 to 80.6 ± 4.7 mm Hg showing no differences between both groups. ICP increased similarly during blood and paraffin injection and peaked at the end of the infusion (sham: 9 ± 2 mm Hg; blood: 52 ± 6 mm Hg; paraffin: 41 ± 6 mm Hg). CBF dropped in both treatment groups, reaching minimum values at the end of the infusion (blood: 26.9 ± 7.7% of baseline; paraffin: 35.4 ± 6.8% of baseline) (Fig. 4). During the observational period CBF temporarily exceed the baseline level after 15 min, before flattening of below baseline (blood: 74.1 ± 9.3 %; paraffin: 62.9 ± 4.3% after 180 min post ASDH induction). All CBF changes were of statistical significance within each group compared with baseline (p < 0.001), whereas there was no significant difference between blood and paraffin oil infusion. Further, subgroup analysis revealed no significant difference in CBF between animals with and without sCSD.
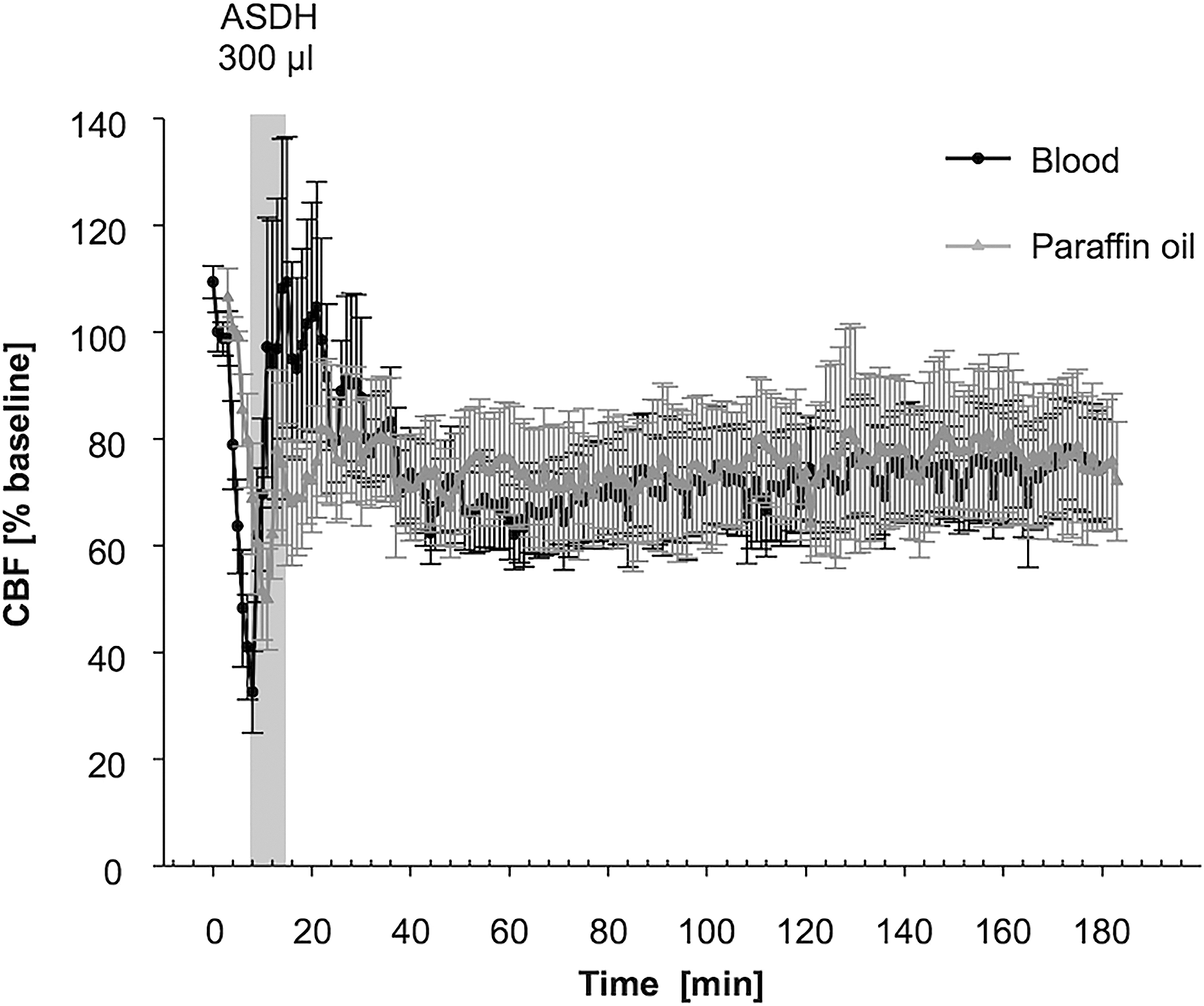
Comparison of CBF in blood and paraffin oil treatment groups. The CBF was measured starting 5 min prior to the subdural injection of 300 μL autologous blood (n = 13) or paraffin oil (n = 12) and continued for 180 min. Measurment of CBF shows no significant variations between blood and paraffin oil treatment groups. CBF, cerebral blood flow.
Measurement of tissue IMP confirmed the classification of different IMP traits as observed in the first experimental group. Animals displaying no sCSDs, sCSDs, or AD were observed equally after autologous blood (no sCSDs, n = 4; sCSDs, n = 4; AD, n = 5) or paraffin oil infusion (no sCSDs, n = 4; sCSDs, n = 3; AD, n = 5). IMP recordings ranged from 1487 to 3023 Ω (2229 ± 358 Ω). All values were normalized to each individual baseline (100%). In those animals receiving autologous blood, an initial IMP increase of 149.8 ± 12.8% (p = 0.008) compared with baseline levels was registered. Peak values were recorded 8.8 ± 1.6 min after the start of the infusion. In those animals receiving paraffin oil, peak values were registered after 5 min and reached 128.5 ± 10.6% of baseline levels. IMP remained elevated during the time of monitoring (blood: 106.09 ± 1.8 % [p = 0.015]; paraffin: 108.0 ± 2.2% [p = 0.008]). sCSDs were detected in both groups (blood, n = 4; paraffin oil, n = 3). After paraffin oil infusion, the first sCSD occurred after 14.9 ± 6.9 min (peak 118.2 ± 5.9%). An average number of 1.7 sCSD, which lasted for 11.2 ± 1.9 min, were detected in this group. In contrast, after blood infusion the first sCSD occurred after 91.11 ± 26.36 min (peak 114.8 ± 2.7%). In this group, an average of 6.8 sCSD, with a mean duration of 5.84 ± 0.69 min were detected. No sCSD occurred in sham operated controls. AD occurred after blood (n = 5) and paraffin infusion (n = 5). All AD animals died either intraoperatively or before the primary end-point of our study, and were therefore excluded.
Lesion volumes were significantly larger after blood infusion (27.3 ± 6.8 mm3) compared with paraffin infusion (3.4 ± 2.1 mm3) (p = 0.004) (Fig. 5A). A subgroup analysis showed that the lesion volumes largely differed within both groups between animals with sCSD (blood: 43.2 ± 5.8 mm3; paraffin: 4.4 ± 2.4 mm3) and those with no sCSD (blood: 11.5 ± 3.5 mm3; paraffin: 2.4 ± 0.8 mm3). Although the differences within the blood group reached statistical significance (p = 0.003), the differences within the paraffin infusion group did not (Fig. 5B).
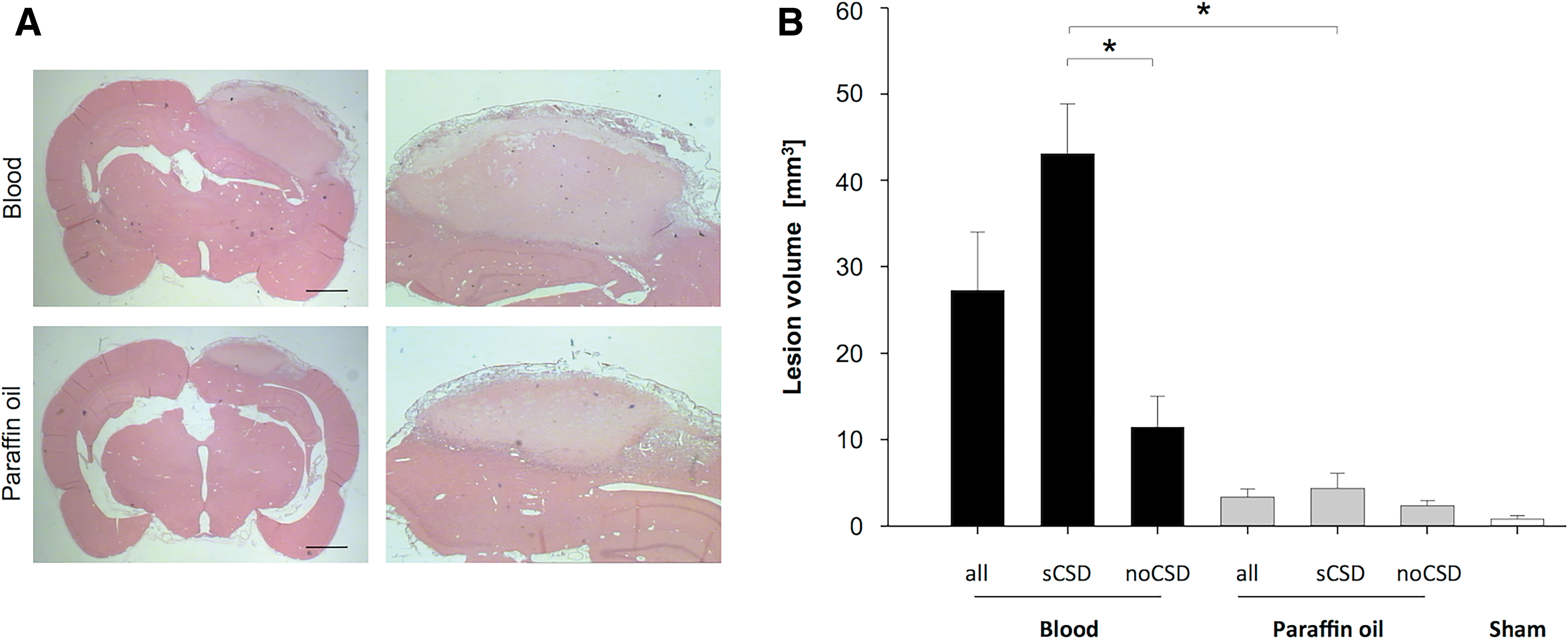
Lesion size is larger after the occurrence of sCSDs. Lesion volume differed between autologous blood and paraffin oil injection and within the subgroups sCSD and no sCSD.
A Neuroscore assessment was used to evaluate motor and behavioral deficits in relation to the subsequent analyzed lesion size. In both groups, animals with large lesions scored higher 7 days after ASDH, compared with those with smaller lesions (blood: p < 0.001, r2 = 0.95; paraffin: p = 0.014, r2 = 0.59), representing more severe deficits (Fig. 6A). Subgroup analysis revealed a linear association of sCSD and poorer scoring values (p < 0.001, r2 = 0.96), however not in the no sCSD group (p = 0.12, r2 = 0.35) (Fig. 6B). Animals needed significantly longer to solve the beam-walk test after autologous blood infusion (53.4 ± 2.6 sec), compared with those with paraffin lesions (30.4 ± 3.0 sec) (p < 0.001). Additionally, all subgroups showed a significant correlation between a shorter beam-walk time and a larger lesion size (blood: p = 0.0068, r2 = 0.73, paraffin: p = 0.025, r2 = 0.66; sCSD: p = 0.0034, r2 = 0.85; no sCSD: p = 0.0019, r2 = 0.82). Finally, the beam-balance test revealed significant deficits in correlation to the lesion size in both treatment groups and the subgroup analysis (blood: p = 0.0003, r2 = 0.91; paraffin: p = 0.0013, r2 = 0.89; sCSD: p = 0.006, r2 = 0.88; no sCSD: p = 0.045, r2 = 0.52). A further subgroup analysis revealed that animals with sCSD balanced for a shorter time, compared with those without sCSDs (sCSD: 17.6 ± 3.6 sec; no sCSD 44.4 ± 4.4 sec; p = 0.005) confirming that animals with sCSD have more severe neurological defects than these animals without sCSD.

Linear correlation between Neuroscore, beam-walk, and beam-balance test with the lesion volume 7 days post-operation. The association was analyzed between autologous blood and paraffin oil injection
Discussion
Our experiments demonstrate that sCSD occur in the wake of ASDH in rats. These sCSDs are more often, but not exclusively, observed in the presence of whole blood constituents. Although the appearance of sCSDs was in all cases associated with a larger lesion volume and more severe neurological deficits, statistical significance was only reached in those animals receiving subdural infusion of autologous blood. AD was associated with pre-term death of the animal.
sCSDs is an electrophysiological phenomenon with little impact on healthy tissue. However, cerebral ischemia seems to trigger sCSDs under various pathological conditions, leading to a supply-demand mismatch that creates a vicious circle propagating the delayed cerebral ischemia syndrome. 31 In compromised microvasculature, the attempted vasodilatation often fails, leading to a vasoconstriction and depolarization in this region instead. 20 If the spreading depolarization-induced perfusion deficit is so severe that it delays the energy-dependent recovery from sCSD, it leads to a prolonged change of the cortical slow potential indicating prolonged neuronal calcium overload. 13,20,32 This inverse hemodynamic response to spreading depolarization locally converts a harmless short-lasting depolarization into a harmful intermediate or even terminal depolarization. 20 The resulting perfusion deficit aggravates the need for energy substrates and increases the tissue at risk. 21 Under pathological conditions such as cerebral ischemia, TBI, or SAH, sCSD are known to increase O2 consumption, resulting in neuronal swelling and contributing to neuronal injury. 5,7,33 The swollen neurons become acidic and lose their hyperpolarization, leading to the release of glutamate among other neurotransmitters and thus causing excitotoxicity of the surrounding tissue while propagating the spread. 10 sCSDs can last from several minutes to hours and cause vasoconstriction and consecutive lesion growth. 34 –36 Additionally, the accumulation of hemolysis products hemoglobin and potassium after SAH can induce spreading ischemia, an inverse hemodynamic response to sCSD in tissue at risk, thus expanding cortical infarction. 37 In our experiments, the largest lesion size in the no sCSDs group was comparable to the smallest lesion in the sCSD group. Thus, the initial lesion size is not solely responsible for the occurrence of sCSDs and other contributory factors, such as blood constituents, are necessary to trigger their occurrence. Artificial induction of sCSDs after SAH, using cortical KCl, lead to significantly larger lesions in rats. 3 In 13 patients with SAH, hypoperfusion was coupled to the clustered occurrence of sCSD, resulting in local development of ischemic stroke. 38 These observations advocate a driving role of sCSD in lesion growth after TBI and SAH, rather than being an epiphenomenon of more severe brain damage. This observation is reinforced by another COSBID study, where spreading depolarization was found to cycle around an ischemic lesion and contribute to its perpetual enlargement. 39 In our experimental groups, increased numbers of sCSDs likewise coincided with larger lesions and more severe neurological deficits.
Similar predisposing factors as described for the occurrence of sCSD after SAH, can also be observed after ASDH. We could demonstrate that ASDH elicits sCSDs, which occur more often in the presence of blood constituents, rather than in the presence of paraffin oil. Further, sCSD occurred independently of changes in CBF or ICP. In our study, lesion size and neurological deficit correlate more closely with the occurrence of sCSD in the presence of autologous blood and, subsequently, blood hemolysis products, than in the presence of paraffin oil. Number, frequency, and duration of sCSD were higher in animals injected with whole blood, underlining a possible interrelation of hemolysis products and sCSD pathogenesis. Nevertheless, neither the occurrence nor the number of sCSDs was a sole predictor of larger lesion volumes and neurological deficits in the absence of blood constituents. We demonstrated that larger lesion size does not necessarily lead to sCSD occurrence but rather is shaped by those spreading depressions. These results fall in line with the recent literature while broadening the scope by introducing ASDH as disease influenced by the occurrence of sCSD. 3,39
It has long been thought that increased ICP was the main cause for the damage development underneath the blood clot. Recently, it became more evident, that this expansion is not solely due to elevated ICP, generated by a subdural volume, but a result of the interplay of ICP, CBF, and blood-derived products in the acute phase of hemorrhage. 40 MAP and ICP has been previously shown to follow similar traits after subdural infusion of blood or paraffin oil, although difference in lesion size is eveident. 41 Lesion volume, development of neurological deficits, and the occurrence of sCSD seem dependent on the presence of whole blood constituents such as thrombin, complement, and other blood-derived products. In contrast, direct intra-parenchymal thrombin injection, subdural infusion of lysed blood, or cortex superfusion with a mixture of hemoglobin and potassium caused a slow CBF decrease and sCSD. 36,42,43 sCSD elicitation by KCl injection causes a similar expansion of focal venous ischemic injury. 12 In our study, animals receiving paraffin oil instead of autologous blood as subdural volume substance, not only had smaller lesion volumes, but also performed better in neurological outcome assessments. sCSD occurred more seldom in the absence of blood constituents but still correlated with a poorer neurological outcome. It was recently shown that a combination of acute ICP increase, local CBF reduction, and the presence of blood constituents are critical for the damage caused by ASDH. 41 We detected no dependency of sCSD occurrence from CBF or ICP. On the contrary, it has recently been shown that each sCSDs increases the peri-infarct area where the CBF is reduced by 19%. 30,34 Pharmacological prevention of these periodical CBF drops leads to decreased lesion volumes 24 h after onset. 34 Thus, the occurrence of sCSDs represents an influencing factor, which might also determine the lesion size and neurological outcome after ASDH. It is noteworthy that no interrelation of sCSD and lesion size has been detected after controlled cortical impact, where no subdural blood collection was present. 44
Results from a recent multi-center approach by the COSBID research group demonstrated that sCSD is a critical factor for outcome in patients with various traumatic and spontaneous pathologies. 19 Our presented data argue in a likewise fashion and emphasize that the largely excepted paradigm of sCSD contribution to secondary lesion development also applies to ASDH. In the light of these findings, clinical decision-making should consider a combination of blood removal to prevent the negative influence of blood constituents released by its degradation and monitoring of sCSD using ECoG. The invasive nature of monitoring sCSD over a prolonged period of time does currently hamper a broader usage. 19 Nevertheless, recognition and monitoring of sCSD might lead to a paradigm shift in our understanding of the pathophysiology of several brain injuries and thus pave the way for targeted anti-sCSD therapies including weak NMDA antagonists such as ketamine, NO donors, or nimodipine. 19
In conclusion, we provide evidence that the accumulation of autologous blood increases the likelihood of sCSD occurrence in a rat ASDH model. sCSDs correlate with compromised neurological outcome in the pre- and absence of whole blood. Lesion growth was increased by sCSDs in animals receiving subdural autologous blood, whereas it only remained a trend in the animals receiving paraffin oil. We postulate that sCSDs are a contributing factor, but not the sole driver of lesion development and neurological deficit after ASDH.
Footnotes
Acknowledgments
We thank A. Ehlert, F. Kafai, L. Kopacz, and M. Malzahn for their technical and secretarial support during the experiments.
Author Disclosure Statement
No competing financial interests exist.