
Abstract
Traumatic brain injury (TBI) is associated with high mortality and morbidity. Though the death rate of initial trauma has dramatically decreased, no drug has been developed to effectively limit the progression of the secondary injury caused by TBI. TBI appears to be a predisposing risk factor for Alzheimer's disease (AD), whereas the molecular mechanisms remain unknown.
In this study, we have conducted a research investigation of computational chemogenomics systems pharmacology (CSP) to identify potential drug targets for TBI treatment. TBI-induced transcriptional profiles were compared with those induced by genetic or chemical perturbations, including drugs in clinical trials for TBI treatment. The protein–protein interaction network of these predicted targets were then generated for further analyses.
Some protein targets when perturbed, exhibit inverse transcriptional profiles in comparison with the profiles induced by TBI, and they were recognized as potential therapeutic targets for TBI. Drugs acting on these targets are predicted to have the potential for TBI treatment if they can reverse the TBI-induced transcriptional profiles that lead to secondary injury. In particular, our results indicated that TRPV4, NEUROD1, and HPRT1 were among the top therapeutic target candidates for TBI, which are congruent with literature reports. Our analyses also suggested the strong associations between TBI and AD, as perturbations on AD-related genes, such as APOE, APP, PSEN1, and MAPT, can induce similar gene expression patterns as those of TBI.
To the best of our knowledge, this is the first CSP-based gene expression profile analyses for predicting TBI-related drug targets, and the findings could be used to guide the design of new drugs targeting the secondary injury caused by TBI.
Introduction
Traumatic brain injury (TBI) can cause significant mortality and morbidity. According to CDC (U.S. Centers for Disease Control and Prevention) reports, each year there are approximately 1.5 million new cases, 50,000 deaths caused by TBI, and more than 5.3 million people are living with permanent disabilities caused by TBI in the United States today. 1
TBI usually starts with a primary injury induced by mechanical forces followed by a secondary or delayed injury resulting from a complex set of cellular processes and biochemical cascades that occur in minutes to days after the initial trauma. Surviving patients then enter into a slow recovery stage or long-term disability. The most common neurological complications that accompany TBI include convulsions, stroke, and neurological infections.
Existing pharmacology research for improving outcome after TBI has investigated the following medications: mannitol, 2 dexamethasone, 3 progesterone, 4 xenon, 5 barbiturates, 6 NMDA antagonists, magnesium, 7 calcium channel blockers, 8 curcuminoids, 9 ethanol, 10 PPAR-γ agonists, 11,12 caffeine, 13 hyperbaric oxygen, 14,15 and hypothermia. 16 However, the progression from initial injury to secondary injury still cannot be stopped by any treatment, despite the tremendous efforts. 17 Clinical trials evaluating agents that were hoped would halt these subcellular mechanisms failed. In addition, TBI patients can be more sensitive to adverse effects and are unusually susceptible to some medications. 18
Gene expression patterns will change during disease progression and drug treatment. 19 Recent computational biology progress indicates that any perturbation to the transcriptional program of the cell can be summarized by a specific “signature,” a set of genes in a cell with a combined expression pattern that is uniquely characteristic to a biological condition or medical treatment. 19 Undiscovered drug–disease relationships can be unraveled by comparing gene expression signatures of drugs and disease perturbations. 19 It is also postulated that, if the effects of a medication on the transcriptional profile is contrary to the profile typically associated with a disease, the drug could have the potential to reverse the gene signature of the disease and hence the disease phenotype itself. With more and more gene expression profiles of TBI and drugs currently available, we can now employ in silico approaches to compare and align them. Such an analysis may provide valuable information as to which drugs would be either deleterious or beneficial to individuals suffering from TBI, as well as insights into the molecular pathogenesis of TBI-induced secondary injuries.
In addition, accumulating evidence implicates TBI as a probable predisposing factor in Alzheimer's disease (AD) development. 20 –22 AD is a neurodegenerative disorder characterized by the extracellular senile plaques made up of aggregates of amyloid beta (Aβ) peptides and intracellular neurofibrillary tangles (NFTs) formed by abnormally phosphorylated tau (p-Tau) microtubule-associated proteins. 23 TBI and AD share many common pathological features, including microgliosis, neurite degeneration, synapse loss, Aβ deposition, and tau phosphorylation. 24,25 The underlying molecular mechanism for TBI to trigger the neurodegenerative cascade in AD is still unresolved. 21
Chemical genomics is an interdisciplinary research field that derives from the chemoinformatics and bioinformatics disciplines, which produces useful genome-wide information provided by different in vitro and in silico assays. Computational chemogenomics applies chemogenomic data, predictive models, and ligand-/structure-based drug design approaches to systematically identify, analyze, and/or predict drug–protein interactions. 26 The advances in chemogenomics turn the traditional one-target one-drug paradigm to a novel multi-target, multi-drug development process. Systems pharmacology is the utilization of systems biology principles to pharmacology research. It considers how a drug works on a network of interactions in various biological systems of our bodies to achieve its therapeutic effect, instead of the effects from one, single, specific drug–target interaction. 27,28 Chemogenomics systems pharmacology (CSP) analyses of drugs in clinical use for TBI treatment will help to reveal the interactions of drug–target–disease at both molecular and systems levels.
Specifically, to systematically study current TBI drugs and also identify new molecular targets for TBI treatment, we analyzed the protein targets of current drugs in clinical trials for TBI treatment. We then compared TBI-induced gene expression profiles with these TBI drug- or genetic perturbation-induced gene profiles. Careful analyses of these gene expression signatures will help to elucidate the molecular mechanism of TBI and indicate potential proteins that may be targeted for better therapy.
Methods
Drugs and chemicals
We collected TBI drugs from the clinicaltrial.gov web site (
Differentially expressed genes (DEGs) induced by TBI, chemicals, and genetic perturbations
Differentially expressed genes (DEGs) of TBI were collected from commercial software Illumina BaseSpace (formerly Nextbio™, Santa Clara, CA;
Calculation of gene signature correlation
The calculations of correlation between DEGs were performed using a modified form of rank-based enrichment statistics implemented in BaseSpace software.
35
–37
BaseSpace pre-processed gene expression data with biomedical ontologies enables the comparison among heterogeneous datasets from different species. It also uses meta-analyses to provide consistent predictions from multiple instances of similar perturbations, such as the gene expression profiles from different cell lines induced by the same drug.
37
All analyses by BaseSpace were performed utilizing the default parameters. Figure 1 shows an example of a comparison between a TBI-induced gene signature and a dexamethasone-induced gene signature. (Detailed information for this example in Supplementary Tables 1–3; see online supplementary material at
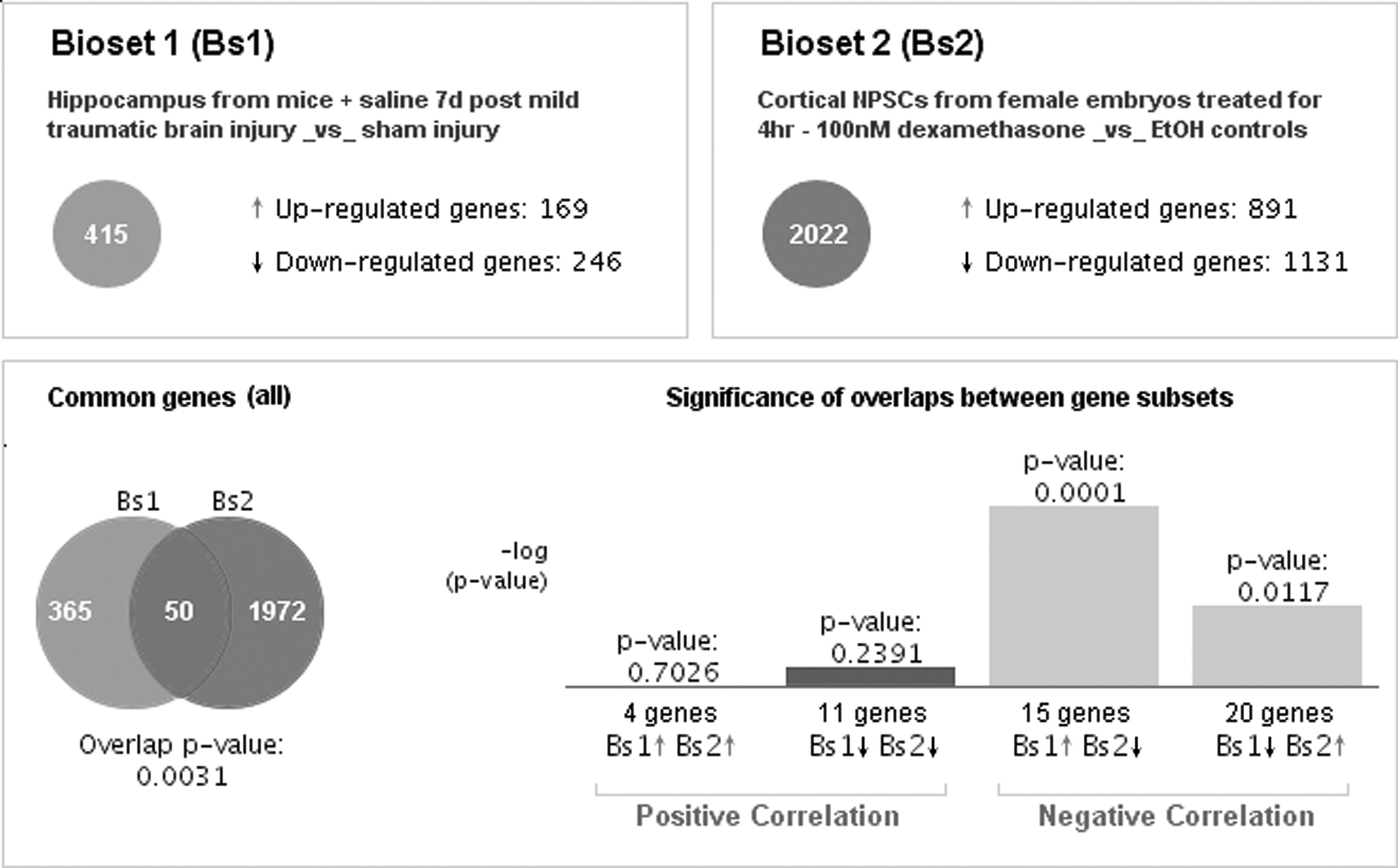
An example of comparison of gene expression profiles showing a negative association between the TBI-induced and dexamethasone-induced profiles. TBI, traumatic brain injury.
Protein–protein interaction networks
To investigate the interactions between the predicted TBI targets, we used STRING software developed by the European Molecular Biology Laboratory (EMBL) to plot the protein–protein interaction networks. 40 The STRING store literature reported protein–protein interactions and predicted potential interactions. 40 We assume that TBI-related protein targets may be closely connected or clustered together because they are all involved in signaling pathways of TBI progression, so that modulating one protein may have similar biological effects as regulating closely connected proteins.
Results
TBI-related gene expression datasets
Table 1 shows the full list of 26 TBI-related gene expression datasets in the BaseSpace database after processing. These datasets were from either mice or rats with time-points ranging from 3 h to 7 days after injury, which covered the initial stage of TBI secondary injury.
The List of TBI-Related Biosets, Features, and Associated Gene Expression Omnibus (GEO) IDs
Positively and negatively correlated genetic perturbations
Table 2 lists the top most positively and negatively correlated genetic perturbations, for example, knockdown or knockout (detailed information in Supplementary Tables 4 and 5; see online supplementary material at
The Associations between TBI-Induced and Gene Knockdown/Knockout-Induced Gene Profiles
DEGs, differentially expressed genes; TBI, traumatic brain injury.
Table 3 lists the top most positive and negative associations between TBI-induced DEGs and gene mutation-induced DEGs. We can see that DEGs induced by mutations on AD-related genes such as PSEN1 (Presenilin-1), MAPT (gene encodes tau protein), and APP (amyloid protein precursor) were positively correlated with TBI-induced DEGs (18, 16, and 16 among 26, respectively). In addition, superoxide dismutase1 (SOD1) is the top positively related gene and mutations in SOD1 have been implicated in amyotrophic lateral sclerosis (ALS; a neurodegenerative disease),
48,49
which may link TBI with ALS,. In addition, SOD1 plays an important role in protecting cardiac cells against reactive oxygen species (ROS) bursting induced by cardiac ischemia injury under oxidative stress.
50
Further, the results also suggest that HPRT1 (hypoxanthine phosphoribosyltransferase 1) could be a new potential drug target, as DEGs of HPRT1 mutation (usually loss of function) are negatively correlated with 21 of the 26 TBI-induced profiles. HPRT1 catalyzes the conversion from hypoxanthine to inosine monophosphate and guanine to guanosine monophosphate through transferring of the 5-phosphoribosyl group from 5-phosphoribosyl 1-pyrophosphate. It plays a critical role in the generation of purine nucleotides through the purine salvage pathway.
51
HPRT1 is also the target of FDA-approved immunosuppressive drugs, such as azathioprine and mercaptopurine. These results imply that these two drugs may be repurposed for TBI treatment (detailed information in Supplementary Table 6; see online supplementary material at
The Associations between TBI-Induced and Gene Mutation-Induced DEGs
DEGs, differentially expressed genes; TBI, traumatic brain injury.
Gene set enrichment analysis
Table 4 shows the top enriched gene sets in the 26 TBI-induced gene expression profiles calculated by the BaseSpace meta-analysis. All these gene sets are upregulated and most of them are related to inflammation and immune signaling pathways, including response to wounding, innate immune response, response to bacterium, response to other organism, and regulation of immune response, as well as genes involved in cytokine signaling in the immune system. It is well known that neuroinflammation is the key process involved in the secondary injury.
52
Such information further confirms that some TBI drugs improve TBI symptoms via modulation of the neuroinflammation and immune signaling, as shown in Table 5 (detailed information in Supplementary Table 7; see online supplementary material at
Results of Top 20 Gene Sets by Enrichment Analysis on 26 TBI Datasets
DEGs, differentially expressed genes; TBI, traumatic brain injury.
Associations between TBI-Induced and TBI Drug-Induced Gene Profiles
DEGs, differentially expressed genes; TBI, traumatic brain injury.
Associations between TBI-induced and TBI drug-induced gene expression profiles
We then analyzed the association between TBI-induced DEGs and those TBI drugs evaluated in clinical trials. As shown in Table 5 (detailed information in Supplementary Table 9, see online supplementary material at
TBI drugs and their associated targets
We summarized the drugs being applied to treat TBI and their targets (Fig. 2; detailed information in Supplementary Table 8; see online supplementary material at
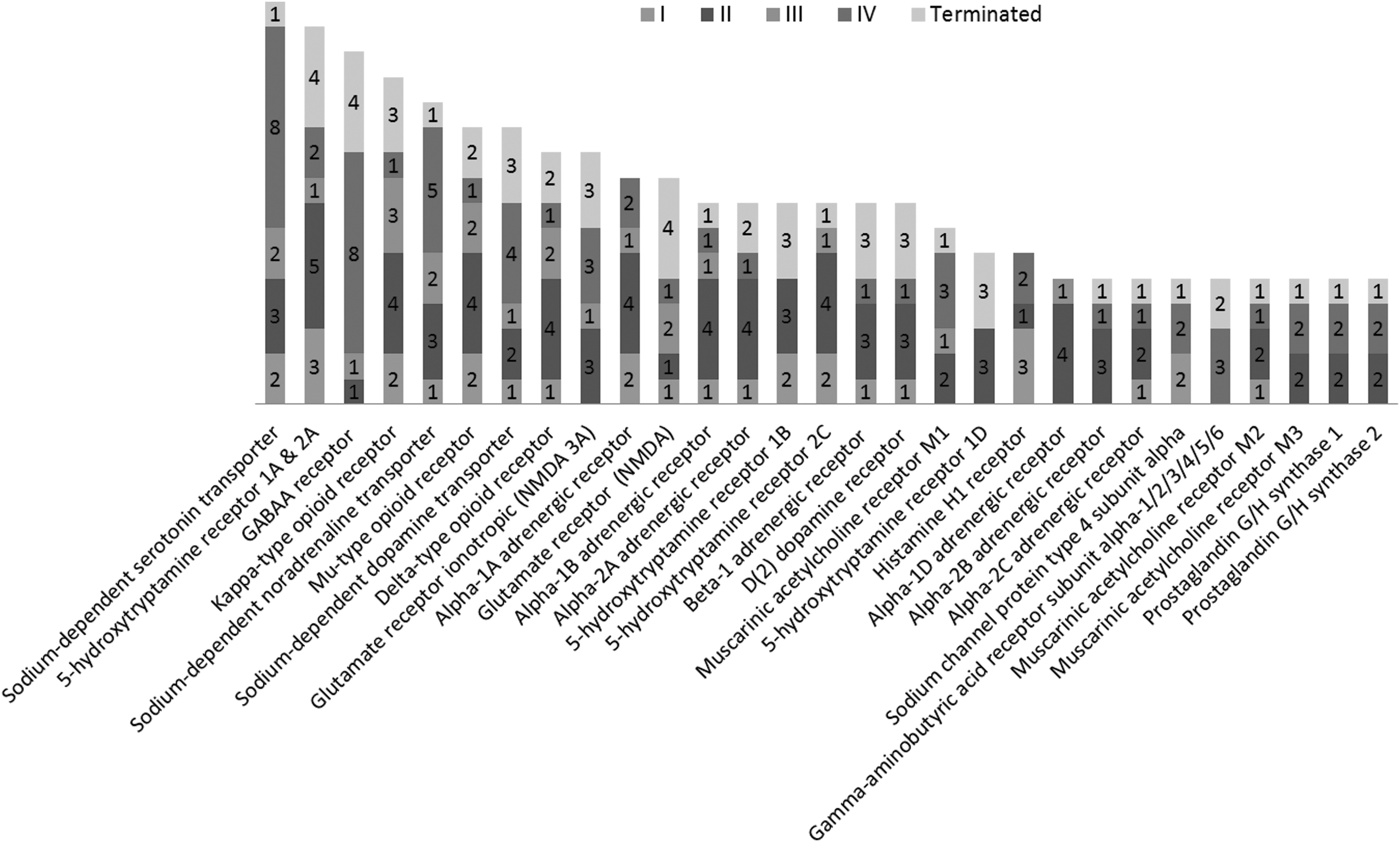
Summary of known TBI drugs and the associated protein targets. TBI, traumatic brain injury.
Most of the drugs used to ameliorate symptoms of TBI modulate neurotransmission in the central nervous system (CNS). Among them, we found that monoamine transporter inhibitors comprise a large class of TBI drugs. In the CNS, monoamines such as serotonin, dopamine, and norepinephrine are important neurotransmitters that are involved in both physiological functions as well as pathological conditions. Monoamine transporters, including dopamine transporter (DAT), serotonin transporter (SERT), and norepinephrine transporter (NET), are important drug targets for mental disorders. Their inhibitors, such as bupropion, amoxapine, mazindol, fluoxetine, paroxetine, GBR-12909, imipramine, and nisoxetine, have been developed to treat various CNS-related disorders, including but not limited to depression, AD, anxiety disorders, epilepsy, mood disorders, personality disorders, psychosexual disorders, addiction and abuse, Parkinson's disease, bipolar disorder, chronic pain, stroke, and trauma. Here, some of these inhibitors are used to treat post-trauma mood disorders and to calm down patients to prevent secondary brain injury after trauma. Among these drugs, fluoxetine, which has much more negative association (17 of 26) than positive association (9 of 26), is a first-line drug approved for TBI treatment. Serotonin receptor (5HTR) antagonists are another class of TBI drugs that are mainly used to treat post-trauma mood disorders, especially depression (Fig. 3).
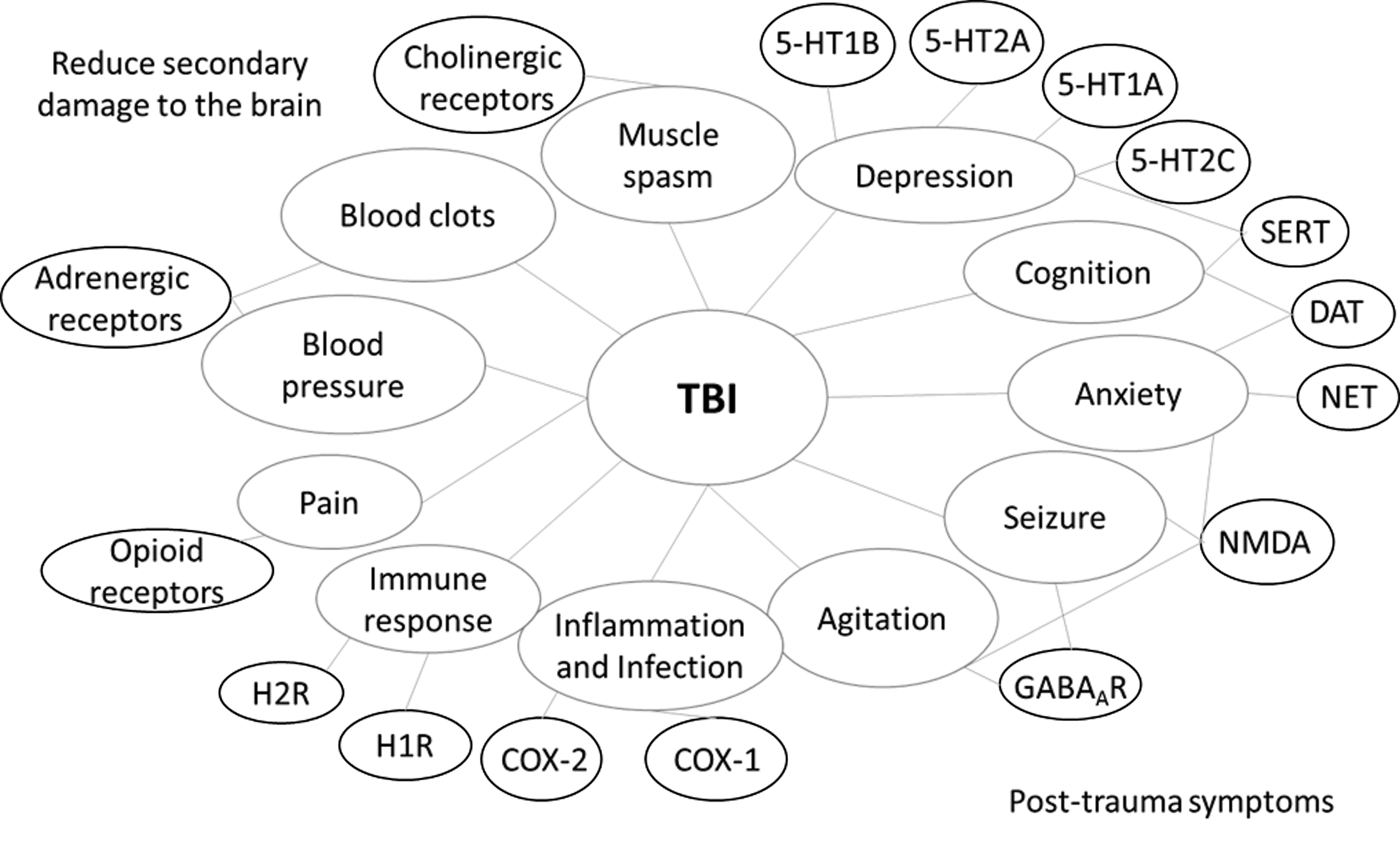
The overview of post-trauma symptoms of TBI and drug targets for intervention. TBI, traumatic brain injury.
Drugs of opioid receptors (kappa, delta, and mu types) agonists are used as painkillers in emergency and post-trauma conditions to relieve pain and calm down patients. In addition, control of the body's response to stimulus is important to stabilize the patient and promote survival after severe TBI.
53
Therefore, the adrenergic and cholinergic systems are also therapeutic targets for TBI emergency treatment. Some findings also indicate that enhancing the norepinephrine system will help improve the recovery of neurons from trauma injury in animal studies.
54
–57
ADRB1 (adrenoceptor beta), a receptor of norepinephrine is also shown in Table 3 above with 10 positive correlations in the gene expression level with the TBI condition, which makes it more reasonable to be a therapeutic target for TBI. Whereas hyperfunction in cholinergic systems is observed immediately following TBI, inhibition of muscarinic acetylcholine receptor has exhibited an enhanced recovery of brain function after TBI.
58
In the late stage, it has been reported that cholinergic agonists (e.g., galantamine, an anti-AD drug, currently in TBI clinical trials) were able to improve cognition after TBI (clinical trials #NCT00645190 and #NCT01734395 from
Suppressing the inflammation and immune response is critical to prevent further damage to the brain as well as relieve post-trauma symptoms. Here, we can find prostaglandin G/H synthase 1/2 (COX1/2; with two drugs [ibuprofen and aspirin] in Phase II and two approved drugs [acetaminophen and diclofenac]) and histamine receptor 1 (H1R; with one approved drug [citalopram], two drugs [escitalopram and amitriptyline] in Phase II, and three drugs [trazodone, diphenhydramine and mirtazapine] in Phase I) are frequently used drug targets for TBI treatment. COX2 expression will be induced by the increased prostaglandin production and cytokine activation in response to the pathological condition initiated by TBI.
61,62
Both the overexpression of COX2 and increased prostaglandin and ROS production may exacerbate neuroinflammation and cause secondary insults as well as delayed neuron death in the brain.
63
Thus, COX inhibitors may benefit and protect injured neurons after TBI (Supplementary Table 8; see online supplementary material at
Protein–protein interaction networks
It is a surprise that there is no overlap between lists of knockdown/knockout and gene mutation. This may indicate that only a limited number of gene perturbations have been studied. Another possibility is that although they are not the same targets, these targets are highly related with each other with some underlying mechanisms. Thus, to investigate the interactions between proteins encoded by those genes listed in Tables 2 and 3, we further built protein–protein interaction networks using the STRING service provided by EMBL. 40 As shown in Figure 4, the most connected nodes include SOD1, APOE, APP, PSEN1, MAPT, PTEN, ABL1, SMAD3, PDGFRB, HRAS, and FGFR. AD-related proteins such as APOE, APP, PSEN1, and MAPT are enriched and highly connected in this network. Interestingly, ABL1 has been reported as a biomarker for TBI by Feala and colleagues, 64 and our analysis further indicated that mutation in ABL1 can induce a similar gene expression pattern as patterns induced by TBI. Thus, modulation of ABL, or its closely related proteins, could potentially serve for TBI treatment. Murakami and associates reported that impaired SOD1 expression and hypoactivity were associated with Aβ protein aggregation and cognition loss in AD mouse models. 65 In addition, SOD1 is a critical antioxidant enzyme and it is one of the major targets in response to oxidative stress damage in the brain of patients suffering from AD and TBI. It has been reported that overexpression of SOD1 will reduce the oxidative stress and neuron injury after brain injury and cerebral ischemia. 66 SOD1 was shown to have a neuroprotective effect and reduce neurotoxic inflammatory signaling, consequently preventing secondary damage to the brain after injury. 67
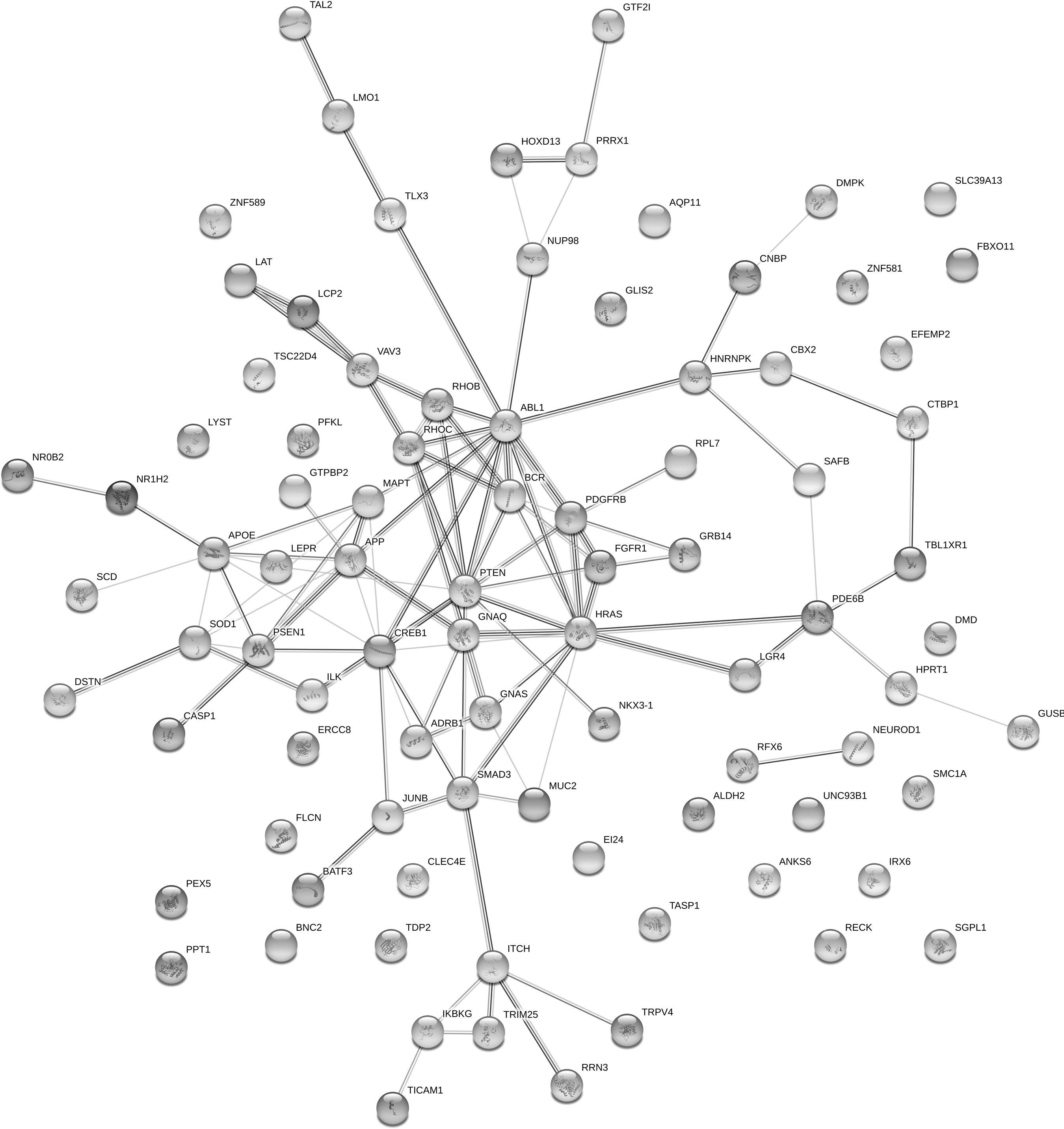
The protein association networks of potential TBI drug targets generated by STRING software. TBI, traumatic brain injury.
Further, our analysis indicated that mutation of SOD1 would induce a gene expression signature similar to TBI-induced. Therefore, SOD1 could be a potential therapeutic target for TBI. Those nodes not connected with other ones mean that they do not have any literature-reported interactions. These probably suggest that not linked proteins are not related with other targets, and cannot be a signaling hub for genetic perturbations.
Discussion
The greatest number of TBI deaths occurring in hospitals is caused by secondary injury, a complicated set of biochemical cascades and cellular processes that happen in minutes to days after the trauma. In this study, we conducted a chemogenomics analysis on potential drug targets for TBI treatment, focusing on those that can reverse the gene expression signatures of secondary injury during TBI. The analysis included comparing the TBI-induced gene signatures with those of chemical and genetic perturbations. Our analysis not only confirmed previously reported potential TBI targets such as TRPV4 and NEUROD1, but also predicted novel protein targets such as HPRT1. As HPRT1 is the target of a few FDA-approved drugs, it is likely they can be repurposed for TBI treatment. This analysis provides a comprehensive target map for TBI, which will also help to identify characteristics and patterns of TBI.
We systematically explored the associations between the gene signatures induced by TBI and those induced by TBI drugs. This association study validated the mechanisms of some TBI drugs on modulating neuroinflammation, which aligns well with the pathway meta-analysis result. Our results also indicated the limitations of these TBI drugs: None of them are negatively associated with all 26 TBI-induced gene signatures. This analysis will facilitate pharmaceutical scientists to discover novel therapies, such as drug combinations for TBI treatment to fulfill the unmet clinical needs.
Although a few databases for AD and TBI have been built, for example, Alzforum (
Because a medical history of TBI is one of the risk factors for AD, 20 –22 there is a strong association between TBI and AD, 68,69 laying the foundation for further TBI-AD studies. Yin and co-workers suggested that oxidative stress could be a link that connects TBI and AD. Oxidative stress initiated by TBI would induce the aggregation of Aβ and phosphorylated tau, two well-known biomarkers for AD. 70,71 Sivanandam and Thakur reviewed several factors that could form a bridge between TBI and AD. 22 First, TBI and AD share a common denominator, neuroinflammatory response. 72 The upregulated APP expression and increased neuroinflammatory response initiated by acute brain injury may contribute to a cycle of Aβ deposition and microglial activation that ultimately leads to chronic neuropathology for AD. Moreover, the axonal/pre-synaptic trafficking of APP was impaired in pathological conditions of injury, because there is a deficiency of APP processing enzyme following TBI. 73
Further, TBI deregulated the gene expression patterns of α-synuclein, APP, BACE1 (beta-secretase 1), tau and ApoE4, which are closely related to Aβ and tau pathological signaling, frequently implicated in neurodegenerative disorders. In addition, TBI induces caspase 3 activity, which contributes to APP cleavage and apoptosis signaling in AD. Injury to axons following TBI will cause abnormal formation of axonal spheroids and block neuron transport. 25,74 Blocked transport will increase the accumulation of APP, BACE1, and PSEN1 in axons, thus leading to enhanced APP processing and Aβ formation. 75 This increased Aβ generation, together with other factors initiated by TBI, including the neuroinflammatory response, triggers a cascade of events that might increase tau phosphorylation and NFT formation for AD development. These studies are attractive, but further evaluations and molecular mechanism studies are still needed. It is necessary to better understand the molecular physiological responses to TBI and its relationship with the pathological mechanism of AD to prevent further brain injury and subsequent AD development after TBI.
Our protein–protein interaction plot indicated the overlap between TBI and AD-related protein targets. The link between early post-injury changes in tau and Aβ peptides and the future risk of developing AD remains unclear. Our research will also help in understanding the pathology of tau. As the pathology of tau has been well studied in AD, the information collected from AD will help us unravel the basic pathological mechanisms of tau associated with TBI and AD. As shown in the protein–protein interaction networks (Fig. 2), MAPT (the gene encoding Tau protein) is directly connected with other important AD-related proteins such as APOE, APP, and PSEN1. TBI induces similar gene profiles as MAPT mutation (Table 3). Other non-neuronal cells can also be involved in this progression. The roles of non-neuronal cells in TBI/AD pathogenesis can be better studied in their molecular context as shown in the examples of NeuroD1 (neurogenic differentiation factor 1) and PPT1 (palmitoyl-protein thioesterase 1) (Table 2), where modulating these proteins in glial cells may have an influence on TBI.
Although the above approaches are intuitive, we need to be careful in using them for prediction: Different cells in different biological contexts, such as pathological conditions, genomic features, protein expressions, and species diversity, might respond differently to the same pharmacological agent treatment. Thus, our approaches might not produce optimal results, because they may differentiate gene expression profiles resulting from consistent experimental settings that were manipulated by researchers not under natural condition. In this study, we used multiple pieces of evidence, including both chemical and genetic perturbations as well as meta-analysis, to provide solid support for our analysis. Another problem is that although proteins listed in Tables 2 and 3 could be potential therapeutic targets, their druggability may be a problem, because not all of them can be manipulated by small molecular drugs or chemical probes. Nonetheless, we believe that advances in new biotechnologies may afford development of selective agents that can be used to manipulate these proteins. Additionally, whereas our analyses focus on gene signature profile alterations, more in-depth studies are still necessary to investigate other aspects of these potential targets. Also, the number of available bioassays may have a negative influence on our results, but with more transcriptional data for TBI becoming available in the near future, we will be able to refine our study. Finally, we point out again that the predictions need to be further validated by experiments.
Conclusion
In this study, we performed a CSP study on potential TBI drugs and targets by meta-analysis of the TBI-induced gene transcriptional profiles against those induced by chemical and genetic perturbations. We predicted new drug targets and also revealed the association between TBI and AD at gene signature and protein levels. Our research will have a significant impact toward identifying new therapeutic targets, and repurposing drugs for TBI intervention and AD preventionto reduce the burden on affected individuals and caregivers, especially in military and veteran communities.
Footnotes
Acknowledgments
The authors would like to acknowledge the funding support to the Xie laboratory from the Department of Defense (DOD) (W81XWH-16-1-0490), and the National Institutes of Health (NIH) NIDA (P30 DA035778A1).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.