
Abstract
Plasminogen activator inhibitor-2 (PAI-2/SerpinB2) inhibits extracellular urokinase plasminogen activator (uPA). Under physiological conditions, PAI-2 is expressed at low levels but is rapidly induced by inflammatory triggers. It is a negative regulator of fibrinolysis and serves to stabilize clots. In the present study, PAI-2 expression is upregulated 25-fold in pericontusional brain tissue at 6 h after traumatic brain injury (TBI), with a maximum increase of 87-fold at 12 h. To investigate a potentially detrimental influence of PAI-2 on secondary post-traumatic processes, male PAI-2–deficient (PAI-2-KO) and wild-type mice (WT) were subjected to TBI by controlled cortical impact injury. Brain lesion volume and cerebral inflammation were not different. Total brain volume was significantly smaller in PAI-2-KO, indicating reduced brain swelling. The brain water content at 24 h post-insult was significantly smaller in PAI-2-KO mice. Markers of vasogenic brain edema showed no difference in blood–brain barrier integrity and expression of blood–brain barrier proteins (claudin-5, zonula occludens-1). In contrast to plasminogen activator inhibitor-1 (PAI-1), PAI-2 plays a limited role for brain lesion formation and does not influence blood–brain barrier integrity. PAI-2 contributes to brain edema formation and could therefore be a promising new target to treat post-traumatic brain edema.
Introduction
Traumatic brain injury (TBI) is the most common cause of trauma-related death and long-term disability in industrialized countries. It affects all ages, with a high prevalence in children and young adults, and remains a serious social and health care burden. 1 –3 The initial trauma triggers multiple pathophysiological cascades and puts the healthy brain parenchyma at risk for secondary brain injury within the first hours after insult. 4 –6 Among the most prominent events are neuroinflammation, apoptosis, and cerebral malperfusion due to hypotension or microclot formation. 7
Vascular occlusion is caused by a pro-coagulant state early after trauma, which leads to increased clot formation. 8 The coagulation system is affected substantially by tissue-type and urokinase plasminogen activators (tPA/ uPA). 9 Besides the impact on early coagulation, it was shown that tPA and uPA are central mediators of coagulopathy following TBI by enhancing delayed cerebral hemorrhage. 10 An effective inhibitor of tPA and uPA is plasminogen activator inhibitor type 2 (PAI-2). 11 PAI-2 is over-expressed in astrocytes, microglia, and endothelial cells around the lesion zone of severely injured brain tissue. 12 Recent studies credited PAI-2 the ability to influence the innate immune response and act in a neuroprotective manner. 13,14 However, the role of PAI-2, especially the tPA:PAI-2 complex formation after TBI is still far from being well understood. 9
Therefore, the present study was designed to determine the post-traumatic cerebral regulation of PAI-2 and to investigate the influence of PAI-2 in a genetic model on brain lesion formation, cerebral inflammation, and brain edema formation after experimental traumatic brain injury in mice.
Methods
Animals
In the time-course experiments, male C57Bl/6N (Charles River Laboratory, Sulzfeld, Germany) were studied after traumatic brain injury. Male PAI-2–deficient mice (Stock #007234, Jackson Laboratory, Bar Harbor, Maine) 15 and corresponding wild-type C57Bl/6J mice (Charles River Laboratory) were investigated 24 h after brain injury. The animals were 2 months old and weighed from 27 to 30 g at the time of injury. The Federal Animal Ethics Committee of the Landesuntersuchungsamt Rheinland-Pfalz approved all experiments (protocol number 23177-07/G10-1-024). The presentation of the experiments is in accordance with the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines.
Experimental groups
Experimental group A. The time-course of PAI-2 messenger RNA (mRNA) expression was determined in brain tissue from healthy C57Bl/6N animals (naïve) and in contused brain tissue after controlled cortical impact injury (CCI; 15 min, 3 h, 6 h, 12 h, and 24 h after brain injury; n = 6 per group).
Experimental group B. The effect of PAI-2–deficiency on secondary brain damage, brain edema formation, markers of blood–brain barrier, and cerebral inflammation was investigated. Inflammatory marker genes tumor necrosis factor α (TNFα) and interleukin 1β (IL-1β) as well as blood–brain barrier markers claudin-5 (CLDN5) and tight junction protein 1 (TJP1) were measured in pericontusional brain tissue 24 h post-injury. The secondary brain damage was determined after 24 h survival time (n = 9) and the primary lesion was set 15 min after injury (n = 7). The contusion volume was analyzed in cresyl violet–stained cryosections. To analyze the blood–brain barrier integrity, CLDN5 and TJP1 as blood–brain barrier markers were stained in cryosectioned brain slices after 24 h survival time. Brain edema formation was quantified in non-operated (sham; n = 6) and brain-traumatized mice (n = 10) after a 24-h survival time.
Neurological function
Before trauma and 23 h post-trauma, the body weight of the mice was measured and the mice underwent neurological testing as described previously. 16 To be included in the study, animals were required to be of normal weight pre-injury and to achieve 0 or 1 point in the modified 15-point neuroscore that verifies the adequate neurological status of a healthy mouse. 17
Traumatic brain injury
Traumatic brain injury was induced by CCI) injury as described previously. 18 Mice were anesthetized with isoflurane (Abbott, Wiesbaden, Germany) mixed with 40% O2 and 60% N2 (induction in a bell jar filled with 4% isoflurane, maintenance via face mask with 1.6% isoflurane) and placed in a stereotaxic frame (Kopf Instruments, Tujunga, CA). Rectal-measured body temperature was maintained at 36.5°C with a feedback-controlled heating pad (Hugo Sachs, March-Hugstetten, Germany). After craniotomy at the right parietal cortex with a high-speed drill (Paggen, Starnberg, Germany), a pneumatic brain trauma was induced on the intact dura with a custom fabricated impactor (L. Kopacz, Mainz, Germany) with the following parameters: 3 mm diameter, impact velocity 8 m/sec, impact duration 150 msec and 1.0 mm brain penetration depth. After injury, the craniotomy was closed and skin wounds were sutured. After discontinuation of anesthesia, mice were placed for 2 h in an incubator heated to 33°C and at a humidity of 35% (IC8000; Draeger, Lübeck, Germany) to recover in their individual cages.
Sham operation
Sham-operated mice underwent the same procedures as described above except for trauma induction.
Histological evaluation
The brains were carefully removed from isoflurane-anesthetized animals, immediately frozen in powdered dry ice and stored at −20°C. Brains were cut in coronal plane with a cryostat (HM 560 Cryo-Star, Thermo Fisher Scientific, Walldorf, Germany) as described previously. 19 Sections with 10 μm thickness were collected every 500 μm, placed on Superfrost plus slides (Thermo Fisher Scientific) and stained with cresyl violet (Nissl). The area of both hemispheres and contused brain tissue, defined as region with lack of cresyl violet staining, were analyzed by an investigator blinded to experimental groups using a computerized image system (Delta Pix Insight; Delta Pix, Maalov, Denmark). Contusion volume was calculated by multiplying contusion areas obtained from 16 consecutive sections with a 500 μm distance between histological sections based on following formula: 0.5 mm*(A1[mm 2 ] + A2[mm 2 ] + … + A16[mm 2 ]).
Brain water content
After a 24-h observation period, the brains were carefully removed from isoflurane-anesthetized animals, longitudinally cut, and separated in contused ipsilateral and non-injured contralateral hemispheres correspondent to previously described protocol. 20 The weight difference of wet and dry brain tissue was determined after 48 h speed-vacuum-drying. The hemispheric brain water content [%] was calculated according to following formula: (wet weight - dry weight)/wet weight * 100. Afterwards, delta-sham was calculated for each post-traumatic value minus mean of sham wild-type respectively PAI-2–deficient mice [%].
Gene expression analysis by quantitative polymerase chain reaction (qPCR)
To determine mRNA regulation, brain tissue samples of the right upper quadrant were collected between the histologic slice intervals as described previously. 16 Samples were transferred into a pre-cooled tube and frozen in liquid nitrogen prior to long-term storage at −80°C. As previously described in detail, RNA extraction, complementary DNA (cDNA) synthesis, and real-time PCR were performed following standard protocols and RNA concentration was photometrically calculated using the NanoVue system (GE Healthcare Europe, München, Germany). 21 An amount of 1 μg extracted RNA was reverse-transcribed afterwards into cDNA by QuantiTect Reverse Transcription Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions.
Identical amounts of cDNA were utilized in duplicates and amplified with Roche (Grenzach-Wyhlen, Germany) Lightcycler 480 Probes Master for IL-1β and PAI-2, Thermo Scientific ABsolute Fast QPCR Mix for cyclophilin A, Thermo Scientific ABsolute Fast SYBR Green Mix for TJP1, or Thermo Scientific ABsolute Blue QPCR SYBR Green Mix for CLDN5 and TNFα. The absolute copy numbers of the target genes were normalized against the absolute copy numbers of cyclophilin A (PPIA) as a housekeeping gene. Applied primers and probes with optimized temperature conditions were: cyclophilin A (PPIA) Forward: 5′-GCG TCT SCT TCG AGC TGT T-3′, Reverse: 5′-RAA GTC ACC ACC CTG GCA-3′, Cyanine 5: Cy5-TTG GCT ATA AGG GTT CCT CCT TTC ACA G-Phos, Fluorescein: 5′-GCT CTG AGC ACT GGR GAG AAA GGA-FL [146 base pairs; 55°C annealing temperature; 10 sec annealing time, 15 sec extension time; gene bank numbers NM_008907 and NM_01710-1]; PAI-2 Forward: 5′-GAA ATC CCA AAC CTG CTA CC-3′, Reverse: 5′-ACC ACA ACA TCA TCT TCA TCC-3′, Fluorescein: 5′-CTC CTG TTG CTT CCC GAT GAG A-3′, Cyanine 5: 5′-TTG AGG ACG CAT CCA CTG GCT T-3′ [383 base pairs; 55°C annealing temperature; 30 sec annealing time, 30 sec extension time; gene bank numbers NM_011111 and NM_001174170]; TNFα Forward: 5′-TCT CAT CAG TTC TAT GGC CC-3′, Reverse: 5′-GGG AGT AGA CAA GGT ACA AC-3′ [212 base pairs; 62°C annealing temperature; 10 sec annealing time, 10 sec extension time; gene bank number NM_013693]; IL-1β Forward: 5′-GTG CTG TCG GAC CCA TAT GAG-3′, Reverse: 5′-CAG GAA GAC AGG CTT GTG CTC-3′, Cyanine 5: Cy5-CAG CTG GAG AGT GTG GAT CCC AAG C-Phos, Fluorescein: 5′-TAA TGA AAG ACG GCA CAC CCA CCC-Fl [348 base pairs; 55°C annealing temperature; 10 sec annealing time, 15 sec extension time; gene bank number NM_013693]; CLDN5 Forward: 5′-CGT TGG AAA TTC TGG GTC TG-3′, Reverse: 5′-AGA TTC ATA CAC CTT GCA CTG-3′ [194 base pairs; 58°C annealing temperature; 10 sec annealing time, 15 sec extension time; gene bank number NM_013805]; TJP1 Forward: 5′-TGT CCC TGT GAG TCC TTC AG-3′, Reverse: 5′-CCA GGT TTT AGG GTC ACA GT-3′, Light Cycler Labeled: L640-ATG CCA CGA GCT GTA GCC ACT ACA-Phos, Fluorescein: 5′-CTC AAC ACA CCA CCA TTG CTG TT-FL [331 base pairs; 55°C annealing temperature; 20 sec annealing time, 30 sec extension time, gene bank number NM_009386].
Immunohistochemistry
Frozen brains were cryosectioned (10 μm) and fixed in ice-cold (-20°C) acetone for 10 min. Brain sections were washed with phosphate-buffered saline (PBS) 0.01 M and blocked and permeabilized with 7% normal donkey serum (Jackson ImmunoResearch via Dianova, Hamburg, Germany) and 1.0% triton (Sigma-Aldrich, Steinheim, Germany) in PBS 0.01 M for 2 h at room temperature. Endogenous mouse immunoglobulin G (IgG) was blocked by Fab Fragment (donkey) anti-mouse IgG 1:20 in PBS 0.01 M for 2 h at room temperature. For immunohistochemical staining slices were incubated with the primary antibodies (1:100 rabbit anti-zonula occludens-1; 61–7300; Life Technologies, Carlsbad, CA) or 1:50 mouse anti-CLDN5 (35–2500; Life Technologies) in 2% bovine serum albumin (Dianova, Hamburg, Germany) with 0.05% azide and 0.5% triton in PBS 0.01 M overnight at room temperature. Incubation with secondary antibodies was as follows: Antibody DyLight 488 conjugated donkey anti-rabbit IgG (A120-208 D2, Biomol, Hamburg, Germany) or Cy3-conjugated donkey anti-mouse IgG (715-165-151, Dianova) diluted in 2% bovine serum albumin with 0.05% acid for 2 h at room temperature. After washing in PBS, 0.01 M slices were embedded in Fluoromount (Southern Biotech, Birmingham, AL).
Confocal microscopy
For imaging a Leica SP5 confocal laser-scanning microscope with 40 × oil immersion objective was used. Used excitation for DyLight 488 was 488 nm with emission 499–540 nm and for Cy3 an excitation at 561 nm and emission 571–698 nm was used. Images were analyzed and processed with and ImageJ (National Institutes of Health software; Bethesda, MD).
Western blot
Dried brain tissue samples of right hemisphere were dissolved in ice-cold lysis buffer (Thermo Fisher Scientific) containing 1 × protease inhibitor cocktail (Roche). After 30 min incubation on ice, probes were sonicated for 3 × 5 sec at 4°C. Samples were centrifuged (14,000 rpm, 20,817 × g, 15 min) at 4°C and the supernatant was used for protein determination; 250 μL supernatants included 2 mg protein, which was incubated overnight with 1:50 anti-mouse IgG antibodies (#7076S, Cell Signalling, Cambridge, UK). Equal amounts of protein (16 μg) were put on a nitrocellulose membrane. After drying at room temperature, the membrane was blocked with 5% milk for 1 h. Samples were detected with IRDye 800 goat anti-mouse (#926-32210, LI-COR, Lincoln, NE). The detection was made by LI-COR Odyssey.
Statistical analysis
All animals were randomized (computer-based randomization) and experimental measurements performed by blinded investigators. To determine the required sample size, an a priori power analysis using G∗Power was performed using lesion volume data from previously published studies. 22 The a priori power analysis was performed to determine an effect size of 0.7, standard statistical power (1-β) of p = 0.95, and a significance level (α) of 0.05. Statistical analysis was performed using GraphPad Prism 7 statistical software (GraphPad Software Inc., La Jolla, CA). For comparison of multiple groups, Kruskal-Wallis test with post hoc Dunn's multiple comparison test was employed. For pairwise comparison, significance was determined by exact Wilcoxon Mann Whitney tests. Values of p < 0.05 were considered significant. Data are presented as mean ± standard error of the mean.
Results
Traumatic brain injury induces PAI-2 upregulation in naïve brain tissue
To specify the time-dependent regulation of PAI-2, mRNA expression was determined in naïve animals and in pericontusional tissue 15 min, 3 h, 6 h, 12 h, and 24 h post-CCI (n = 6 each; Fig. 1A). The expression increased 6 h after CCI (25-fold; p = 0.0255) with 87-fold peak 12 hours post-injury (p = 0.0002). The expression remained elevated at 24 h (73-fold; p = 0.0094).
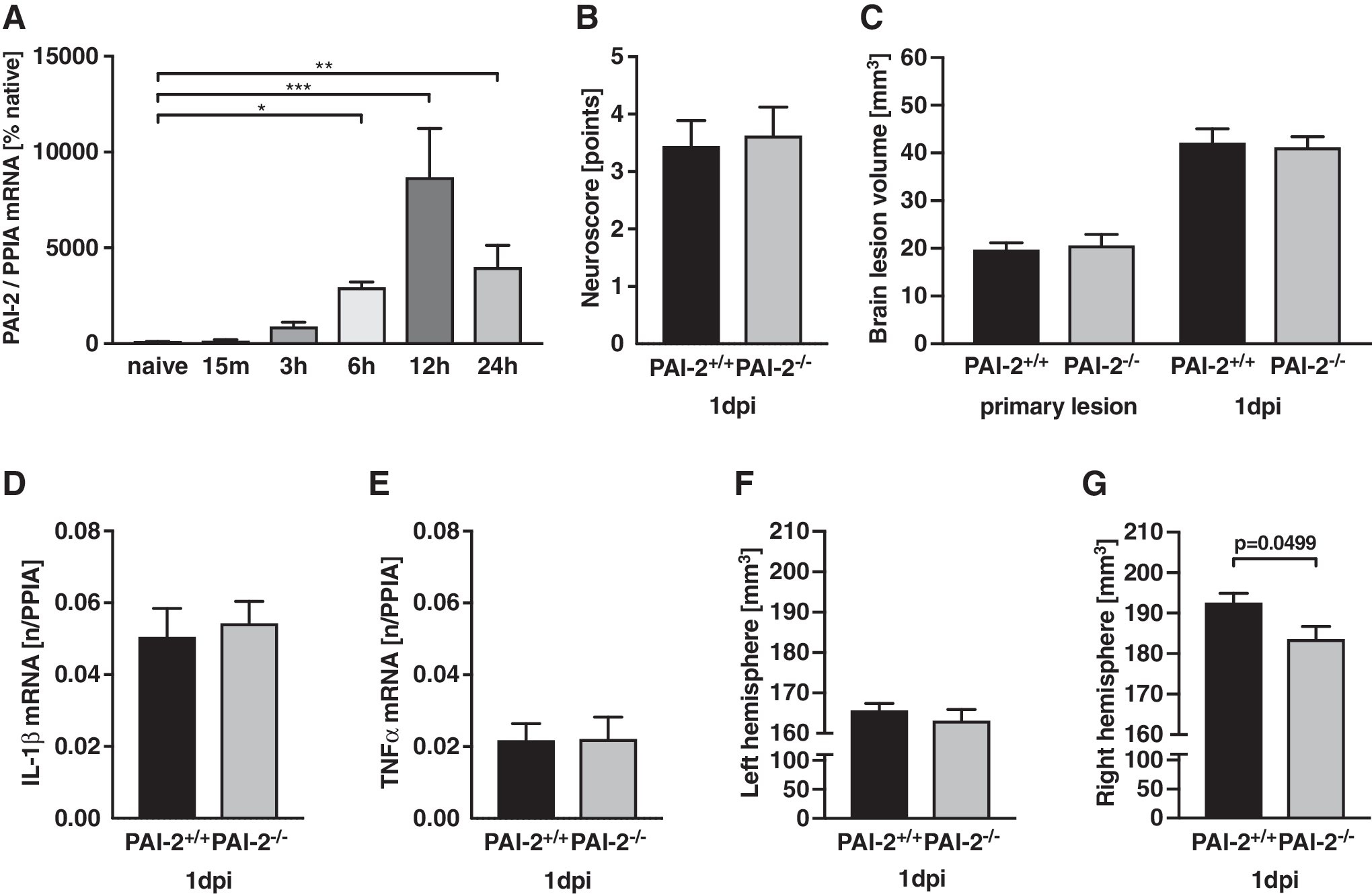
Neurocognitive function and brain lesion volume is not influenced by PAI-2 deficiency
After brain trauma, neurological impairment was determined with a neurofunctional score at 23 h after insult, which demonstrated post-injury deficits without differences between groups (wild-type [WT]: 3.44 ± 0.44 points, PAI-2-/- : 3.63 ± 0.50 points, n = 9, per group; Fig. 1B). The influence of PAI-2 on secondary brain damage was determined in cresyl violet–stained sections at 15 min (primary lesion) and 24 h after trauma. Brain lesion volume increased 2.1-fold between primary lesion (19.72 ± 1.44 mm 3 ) to 24 h after insult in both wild-type (42.16 ± 2.94 mm 3 ) and PAI-2-/- mice (15 min: 20.62 ± 2.32 mm 3 ; 24 h: 41.18 ± 2.26 mm 3 ). Brain lesion volume at 24 h did not vary between groups (Fig. 1C).
PAI-2 deficiency does not influence inflammatory marker gene expression
To determine the effect of PAI-2 on cerebral inflammation, the inflammatory marker genes IL-1β and TNFα were quantified in pericontusional brain tissue after trauma. There were no differences between wild type and PAI-2-/- mice at 24 h post-injury (IL-1β, Fig. 1D; TNFα, Fig. 1E).
PAI-2–deficient mice show lower hemispheric brain volume after injury
To our surprise, analysis of total hemispheric volumes in histological slides showed similar values in the contralateral hemisphere (Fig. 1F), whereas the total volume of the lesioned hemisphere was significantly smaller in PAI-2-/- animals compared with wild-type (Fig. 1G). These data suggest that the size of damage tissue is not influenced by the genetic PAI-2 deficiency. Instead, PAI-2 deficiency could reduce swelling of the perilesional brain parenchyma.
Brain edema formation is ameliorated in PAI-2–deficient mice
To confirm the histological findings suggesting reduced brain swelling, brain water content was determined in sham-operated and traumatized mice at 24 h after injury. The brain edema was significantly smaller in PAI-2-/- compared with WT (p = 0.0089; Fig. 2A). Brain water content was not lower in sham-operated PAI-2–deficient mice. To rule out a baseline effect of PAI-2 on brain water content in sham-operated animals, we calculated the relative increase of brain water content to confirm the previous analysis. The increase in post-traumatic brain water content of the contused ipsilateral hemisphere was calculated as the difference between post-traumatic value minus the mean of sham of the corresponding genotype. In this analysis, PAI-2-/- mice also demonstrated a significantly smaller increase in brain water content (WT [%], 5.78 ± 0.47; PAI-2-/- [%], 4.53 ± 0.37; p = 0.0288; Fig. 2B).

Blood–brain barrier integrity is not changed in PAI-2–deficient mice
To investigate if reduction in brain water content is a result of blood–brain barrier (BBB) stabilization (vasogenic brain edema), post-traumatic IgG extravasation from the blood stream into brain parenchyma was analyzed using Western blot technique and the data were analyzed as x-fold increase of sham. Despite clear effect on brain water content, IgG extravasation was not different between lesioned hemispheres of PAI-2-/- and wild type mice (Fig. 2C).
To determine the post-traumatic modulation of BBB proteins, mRNA expressions of CLDN5 and tight junction protein 1 (TJP1; also known as zonula occludens-1, [ZO-1]) were measured in pericontusional brain tissue 24 h after trauma in wild-type and PAI-2–deficient mice. Both CLDN5 and ZO-1 expression were not different between both groups (Fig. 2D, 2E). To identify a possible post-transcriptional regulation, the tight junction proteins were immunohistochemically stained. No difference of the immunostaining signal was visually detectable (Fig. 2F). As expected, the stained sections showed mechanically ruptured vessels in PAI-2-/- and wild-type mice.
Discussion
Although cerebral mRNA expression of PAI-2 was highly upregulated 12 h after TBI, indicating a shift towards inhibited fibrinolysis, brain lesion volume and expression of inflammatory marker genes were unchanged in PAI-2–deficient mice. Brain water content was significantly attenuated by PAI-2 deficiency. The lack of effect on the BBB integrity and BBB protein expression suggests that vasogenic brain edema is not modulated by PAI-2 deficiency.
PAI-2 is predominantly present in a 47 kDa non-glycosylated intracellular form and in minor amounts as a 60 kDa glycosylated extracellular protein. 23 Under normal conditions, expression of PAI-2 is detectable in keratinocytes, macrophages, activated monocytes, placenta, and in cells of neuronal origin. 24,25 Basal PAI-2 expression levels are usually low or barely detectable, but can be markedly upregulated upon stimulation. In endothelial cells, PAI-2 expression is modulated by lipopolysaccharide, phorbol ester, TNFα, IL-1, and angiotensin II. 24 PAI-2 signals also are seen in microglia and vascular endothelial cells of human brains and increased levels of PAI-1 and PAI-2 were detected in injured human brains. 12,26 In our experimental model, PAI-2 mRNA shows an 87-fold up-regulation 12 h post-injury in pericontusional brain tissue, suggesting a pivotal role of PAI-2 in secondary processes. Upstream modulators of PAI-2 (e.g., proinflammatory protein IL-1β and TNFα) show increased levels within 3 h after brain tissue damage. 27 Genetic deficiency of PAI-2 did not influence expression of these proteins, suggesting the lack of a feedback-loop from PAI-2 to these inflammatory genes and confirming the downstream characteristic of PAI-2.
As a member of the serine protease inhibitor (serpin) superfamily and as key regulator of fibrinolysis, PAI-2 may influence two major processes of secondary brain damage, such as post-traumatic microclot formation and BBB impairment. Therefore, PAI-2 could in theory increase lesion formation, cerebral inflammation, and vasogenic brain edema formation.
In the first hours after TBI, pericontusional clot formation is required to limit post-traumatic bleeding. 28 –30 PAI-2 may support this reaction by reducing the thrombolytic effect of tPA at the expense of increased risk for tissue ischemia. 31 –33 In the present study, PAI-2 deficiency did not influence the extend of the lesion size. A possible explanation for the limited role of PAI-2 deficiency could be that PAI-2 acts on uPA rather than tPA 13 and that the intravascular effects are fully compensated by PAI-1. In PAI-2-/-, more uPA is bound by PAI-1 and forms a uPA:PAI-1 complex, which, for example, inhibits activation of astrocytes, blocks synaptic recovery after brain ischemia, and modulates various signaling pathways. 34,35
The mechanical disruption of brain tissue after TBI causes an opening of the BBB, which is followed by extravasation of plasma proteins and vasogenic brain edema formation. 36 –38 The tPA:PAI-1 complex increases the BBB permeability, 9 whereas no information is present to the best of our knowledge on the role of PAI-2 on the BBB integrity. PAI-2 was shown to promote endothelial apoptosis in inflammatory environments. 39,40 We therefore expected an effect of PAI-2 on BBB integrity and brain edema formation. In the present study, brain water content was significantly lower in PAI-2–deficient mice. Unexpectedly, markers of BBB integrity were not different between wild-type and PAI-2–deficient mice. Taken together, a tPA:PAI-1 complex–mediated BBB opening appears to play a subordinated role in PAI-2–deficient mice, as brain edema as well as BBB integrity was not negatively affected. In general, it is believed that after mechanical (TBI/CCI) 18 and ischemic brain injury, 41 vasogenic brain edema plays a prominent role, whereas other work shows that not vasogenic edema alone, but rather swelling (e.g., of astrocytes and cytotoxic brain edema formation by NF-κB activation 42 or purinergic 2Y1 receptor stimulation). is important in the early phase after insult. 43 The present data support this latter work and underline the role of cytotoxic brain edema after TBI.
Finally, we want to address a limitation. In the present study, a global PAI-2 knockout strain was investigated. We therefore cannot rule out that compensatory regulations may have influenced the results. PAI-2–deficient mice show normal development, survival, and fertility. The animals demonstrate no alteration of the plasminogen-activating system or compensation by PAI-1 regulation. 15,44 We recently showed that PAI-2–deficient mice show no PAI-1 mRNA expression differences compared with wild-type mice. 45 We are therefore confident that PAI-2 expression is not compensated by upregulation of PAI-1.
In summary, the present study shows, to the best of our knowledge, for the first time, an important role of PAI-2 in cerebral injury. PAI-2 does not influence the extent of secondary brain damage formation and BBB integrity, but contributes to brain edema formation. Due to lack of effect on the blood–brain barrier, we propose that PAI-2 does not have a prominent role in vasogenic brain edema formation. PAI-2 could therefore be a promising new target to manage post-traumatic brain swelling.
Footnotes
Acknowledgments
The authors thank Frida Kornes and Dana Pieter for their excellent technical and organizational assistance, and Dr. Robert Dickinson (Imperial College London, UK) for critically reading the manuscript. The support by the Microscopy Core Facility of the Institute of Molecular Biology (IMB), Mainz, is gratefully acknowledged.
This work was supported by a grant from the Focus Program Translational Neurosciences (FTN) of the Johannes Gutenberg-University Mainz to SCT and by a “Stufe 1 grant” of the University Medical Center of the Johannes Gutenberg-University Mainz to JH. No additional external funding sources were used for this study.
Author Disclosure Statement
No competing financial interests exist.