
Abstract
Ependymal cells (EpCs) are a kind of multi-potent stem cells in the central canal of adult spinal cord, which proliferate following spinal cord injury (SCI). Although they can differentiate into functional neurons in vitro, EpC progeny differentiate mainly into astrocytes after SCI, and the mechanism remains unclear. The present study aimed to explore whether neuroinflammation induced by classically activated macrophages (M1) or alternatively activated macrophages (M2) had an effect on EpC proliferation and/or differentiation.
EpCs were isolated from intact spinal cord of adult mice and co-cultured with M1 or M2, respectively, in vitro. EpC proliferation was detected using a Cell Counting Kit-8 (CCK8) assay and Ki67 staining. Expression of Sox2 (SRY-box 2) in EpCs derived from different groups was detected by immunofluorescence and western blotting. Also explored was whether the mitogen activated protein kinase (MAPK) signaling pathway was involved in EpC proliferation. Immunofluorescence staining of βIII-tubulin and MAP2 were performed to assess the differentiation direction of EpCs in different culture conditions.
Immunofluorescence and western blotting assays showed much more Sox2-positive EpCs in the group EpCs-M1 than the group EpCs-M2 in vitro (p < 0.01). The percentage of EpCs with positive Sox2 staining was decreased after tumor necrosis factor α (TNFα) antibody was added into the medium of EpCs-M1. Correspondingly, fewer Sox2-positive staining cells were observed in the central canal of TNFα-deficient mice with SCI. M1 co-culture promoted EpC proliferation significantly, which could be downregulated by Sox2 gene silencing (p < 0.01). Interestingly, M1 regulated the expression of Sox2 through the MAPK signaling pathway, especially the activation of ERK and p38 kinase. Co-culture in M2 conditioned medium obviously increased the proportion of βIII-tubulin-positive cells (p < 0.01). Small amounts of MAP2-positive neurons could be detected on day 7 in the M2 group and the control group.
M1 conditioned medium could promote EpC proliferation in response to SCI through the TNFα-MAPK-Sox2 signaling pathway; M2 conditioned medium favors EpCs differentiating toward neurons.
Introduction
Spinal cord injury (SCI) is a major health burden and currently there is no effective medical intervention. Stem cell transplantation, such as with bone marrow stem cells and neural stem cells, is considered a promising way to substitute necrotic tissue and reconstruct neural circuitry. 1,2 Several studies have found, however, that without manipulation, the transplanted stem cells predominantly give rise to astrocytes, limited numbers of oligodendrocytes, and neurons. 3,4 These results suggest that the beneficial effects of stem cells are mediated primarily through paracrine activities involving release of neuroprotective, angiogenic, 5 and/or immunomodulatory factors. 6 Moreover, transplantation through local parenchyma injection will even result in iatrogenic injury, which makes it far from a clinical treatment.
Research performed over the last decade revealed that ependymal cells (EpCs) are static multi-potent stem cells found in the adult tissue surrounding the central canal of spinal cord. EpCs proliferate in response to SCI in adult rodents, and their progeny cells are recruited toward the injury site. 7,8 Under suitable culture conditions, a certain proportion of EpCs obtained from mice suffering SCI could differentiate into functional spinal motor neurons with HB9-antibody-positive staining and electrophysiological properties, 9 which has raised hopes that new therapeutic strategies can be designed based on appropriate modulation to EpCs, and thus neural function can be restored without the need of transplantation.
However, the reactive EpCs differentiate in vivo mainly into scar-forming glial cells and, to a lesser degree, oligodendrocytes. 10,11 Very little is known regarding the differentiation mechanism of EpCs following SCI in vivo. Sox2 (SRY-box 2) is one of only a few key factors needed to regulate self-renewal and potency of embryonic neural stem cells. Some studies have observed that Sox2 expression was upregulated in EpCs following SCI in zebrafish and adult mice. 12,13 The mechanism of Sox2 upregulation and its impact on the fate of EpCs, however, remain ill-defined. Secondary inflammation is one of the most important events in the microenvironment of the injured spinal cord, which is characterized by the asymmetric polarization of macrophages, namely classically activated macrophages (M1) and alternatively activated macrophages (M2). 14,15 We hypothesized that neuroinflammation mediated by differently polarized macrophages at the injured site had an influence on EpC response to traumatic SCI and explored the possible involved mechanism.
Methods
Mice, macrophages isolation, culture, and polarization
Seven-to-nine-week-old male C57BL/6 mice were purchased from Hubei Provincial Center for Disease Control and Prevention (Wuhan, China). B6/tumor necrosis factor α (TNFα)-/- knockout (KO) mice were purchased from Shanghai Laboratory Animal Center (Shanghai, China).
Care, use, and treatment of mice in this study were in strict agreement with international guidelines for the care and use of laboratory animals. All animal procedures and care were approved by the Ethics Committee of Renmin Hospital of Wuhan University. This study was approved by the Animal Ethics Committee of the Medical School of Wuhan University.
M1 and M2 were generated using the following steps
16
: bone marrow cells were isolated from the femurs of C57/BL6 mice or B6/TNFα-/- mice. Cells (5 × 10
6
) were seeded onto 100-mm sterile non-tissue culture-treated polystyrene petri dishes (Falcon) with 12 mL R10 cell culture medium (RPMI 1640 [Corning] with 10% fetal bovine serum [FBS; Thermo Fisher], 1% penicillin/streptomycin [Thermo Fisher], and 1
EpC isolation and sorting
EpCs were obtained from healthy mice without SCI. After a laminectomy, spinal cord tissue was harvested and cut into 1-mm 3 pieces, enzymatically dissociated in a solution containing 0.01% papain and 0.01% deoxyribonuclease I (DNase I; Worthington Biochemicals) for 1–2 h at 37°C, and then mechanically dissociated into a cell suspension. Myelin debris was removed by centrifugation in 0.5–0.6 M sucrose/phosphate buffer saline (PBS). Erythrocytes were lysed with ammonium chloride (0.8% NH4Cl with 0.1 mM ethylenediaminetetraacetic acid (EDTA; Stemcell Technologies).
Fluorescence-activated cell sorting (FACS) was used to isolate EpCs as previously described by Pfenninger and colleaues. 17 In brief, spinal cord cells in suspension were incubated with an antibody mix, containing CD133-PE (eBioscience) and CD24-fluorescein isothiocyanate (BD Biosciences) for 30 min on ice. Isotype-matched control antibodies were used as negative control. 7-Amino-actinomycin D (final concentration 1 μg/mL, Sigma-Aldrich) was added to exclude dead cells. Cells were sorted with a FACS Divaflow cytometer (BD Biosciences).
Transwell co-culture
A transwell system was used to evaluate the influence of polarized macrophages on EpCs according to our previous method. 18 Macrophages were cultured on 24-well plate inserts (1-mm size pore, Corning) with EpCs pre-plated on the bottom wells. In brief, the polarized macrophages (M1 or M2) were added into the inserts at an initial seeding density of 5 × 10 5 cells per insert. The inserts were placed on a 24-well plate that contained EpCs of 5 × 10 5 cells per well (bottom well). The bottom wells were pre-coated with poly-L-lysine (PLL; Sigma). The cells were cultured in medium (B27 [w/o vitamin A, 1 × ], L-glutamine [2 mM], penicillin [100 U/mL], streptomycin [100 μg/mL] [all from Invitrogen], partricin [0.5 μg/mL; Biochrom AG, UK]) for 3–7 days. In the control group, with empty inserts, EpCs were plated into the bottom well in attachment medium (B27 [w/o vitamin A, 1 × ], L-glutamine [2 mM], penicillin [100 U/mL], streptomycin [100 μg/mL] [all from Invitrogen], partricin [0.5 μg/mL, Biochrom AG, UK], rhEGF, and rhFGFbasic [both 20 ng/mL; PAN Biotech, Germany] in Neurobasal-A medium [NBA; Invitrogen]).
EpCs were co-cultured with M1 or M2 for 3–7 days. Macrophages in the inserts were changed every 3 days. Then EpCs were collected and used for cell proliferation assay, immunostaining, western blotting assays, or differentiation experiments.
Surgical procedure and animals group
Male B6 mice (n = 18, weight 18–20 g) were allocated into SCI group and sham operation group. After being deeply anesthetized with 40–60 mg/kg of pentobarbital i.p., WT mice in the sham operation group (n = 9) received a laminectomy. Animals in the SCI group, whether WT mice (n = 9) or TNFα-/- KO mice (n = 9), received a severe mid-thoracic (T8–T9) crush injury using Dumont-type forceps with a spacer of 0.2 mm.
Immunohistochemistry
On day 1, 3, and 7 post-injury respectively, 3 mice of each group were sacrificed by transcardial perfusion with 0.1 M PBS, followed by 4% paraformaldehyde in PBS. The entire spinal cord was isolated from the vertebral column, fixed for 2 h, soaked overnight in phosphate buffer, and cryoprotected in 30% sucrose. Tissue blocks centered on the SCI epicenter (3-mm blocks for transectional sections) were frozen on dry ice and sectioned in the transectional plane (10-μm thickness for transectional slices). The sections were incubated with anti-Sox2 monoclonal antibody (Cell Signaling Technology) according to the manufacturer's instructions at 4°C overnight, and then with anti-rabbit secondary antibody (Vector) followed as incubated in ABC (ABC Kit, Vector) at 37°C for 30 min. The blocking agent was bovine serum albumin (BSA) and visualized by reaction with diaminobenzidine (DAB; Vector).
Quantitative real-time polymerase chain reaction (qRT-PCR)
QRT-PCR was used to analyze messenger RNA (mRNA) expression. All RNA samples were DNA-free. The complementary DNA (cDNA) synthesis and quantitative PCR (qPCR) analyses were performed as follows. Total RNA was extracted from cells with Trizol (Invitrogen Life Technologies). The final RNA pellets were dissolved in 0.1 mM EDTA (2 μL/mg original wet weight). Reverse transcription reactions were carried out on 22 μL of sample using superscript II RNAse H-Reverse Transcriptase (Invitrogen Life Technologies) in a reaction volume of 40 μL. All samples were diluted in 160 μL nuclease-free water. RT-qPCR was carried out on a StepOnePlus Real-Time PCR System (Applied Biosystems, CA) using SYBR® Green Real-Time PCR Master Mix (Toyobo, Japan). The sequences of primers for PCR are listed in Table 1. PCR was conducted on an initial denaturing step of 3 min at 94°C followed by 35 cycles of 94°C for 10 sec, 58°C for 15 sec, and 72°C for 20 sec and then a final extension at 72°C for 7 min. The relative mRNA expression levels of each gene were normalized based on glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Relative fold differences were determined using the method of delta–delta CT, calculated as:
Primer Sequences for RT-PCR
GAPDH, glyceraldehyde 3-phosphate dehydrogenase; IL, interleukin; TNFα, tumor necrosis factor α.
Cell proliferation assay
A Cell Counting Kit (7 Sea Biotech, China) was used to assess cell proliferation. The experiment was performed strictly according to the manufacturer's manual. EpCs were cultured with or without M2/M1 cells as described in the method of transwell co-culture. After 72 h, the transwells were removed and Cell Counting Kit-8 (CCK-8) (Beyotime, China) assay was added to the medium of EpCs (50 μL/well). Following incubation for 4 h at 37°C the optical density (OD) value was measured at 450 nm by a SpectraMax® i3x Microplate Reader (Molecular Devices, CA). There were four replicate wells for each sample, and the result was expressed as a mean value ± standard deviation (SD).
For Ki67 staining, EpCs were co-cultured for 72 h as a CCK-8 test. Then EpCs in the bottom wells were digested with trypsin, and washed with PBS. EpCs were then fixed with 200 μL of fixation buffer and incubated at room temperature in the dark for 20 min. Cells were washed with BioLegend's FOXP3 Perm Buffer and then were placed in sterile conical tubes in aliquots of 500,000 cells in 100 μL of BioLegend's FOXP3 Perm Buffer each. EpCs were stained with fluorescein isothiocynate (FITC)-conjugated Ki-67 antibody (Invitrogen) and incubated at room temperature in the dark for 30 min. The cells were then washed with BioLegend Cell Staining Buffer and re-suspended in 0.5 mL cell staining buffer for flow cytometric analysis.
To determine the effect of Sox-2 on the proliferation of EpCs, EpCs cells were infected with Sox-2 shRNA (short hairpin RNA) Lentiviral Particles or control shRNA Lentiviral Particles (both from Santa Cruz, Biotechnology). After 6 h, the EpCs were then co-cultured with M1 as above.
Differentiation of EpCs into neurons
EpCs were co-cultured as described in the method of transwell co-culture. On day 3 and day 7, the phenotypic expression of βIII-tubulin (Covance) and MAP2 (Covance) in EpCs were examined by immunofluorescence as described below. Ten visual fields were selected randomly in the staining slices and the average positive cells were counted and regarded as the total cell number.
Immunofluorescence
The culture medium was discarded and the cover-slips from each group were rinsed with PBS (pH 7.4) twice. EpCs were fixed in 4% paraformaldehyde, and then washed in PBS 3 times, and permeabilized with 0.1% Triton X-100 in PBS for 30 min at room temperature. After being washed in PBS 3 times, EpCs were incubated in PBS Tween 20 (PBST) with 1% BSA for 60 min to block non-specific binding of antibodies. Then EpCs were incubated with primary antibody overnight at 4°C. The following primary antibodies were used: Sox2 (Cell Signaling Technology) for activated EpCs, and βIII-tubulin (Covance) and MAP2 (Covance) for neuronal progenitor cells. The nuclei were stained with DAPI. After being washed with PBS 3 times, cultures were incubated with fluorescent Alexa 488 or 568 secondary antibodies (1:500; Invitrogen) for 1 h. Immunofluorescent staining was examined using a Nikon Eclipse TE 300 microscope. Ten visual fields were selected randomly in the staining slices and the average positive cells were counted and regarded as the total cell number.
Western blotting
M1 and M2 were identified by expression of Arg-1 or iNos-1 after 7 days' culture. Sox2 expression in EpCs was detected using western blotting assays 24 h post-co-culture. Possible activation of mitogen activated protein kinase (MAPK) signaling pathway in EpCs co-cultured with macrophages were measured at 0, 15, and 30 min post-co-culture, respectively. In brief, cells were lysed with lysis buffer (Beyotime, China) containing phenylmethylsulfonyl fluoride (PMSF). Lysates was centrifuged at 12,000g at 4°C for 10 min, and the supernatant was collected and preserved at −80°C for later use. Protein concentrations were determined using a BCA Protein Assay Kit (Pierce). Proteins were separated by 15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to polyvinylidene difluoride (PVDF) membrane using a standard protocol, which was then blocked by incubation for 1 h at room temperature in 5% fat-free dry milk in tris-buffered saline containing 0.1% Tween 20 (TBS-T). The membrane was incubated with primary antibodies diluted 1:1000 in TBS-T containing 5% BSA and probed with the respective secondary antibodies. The membrane was exposed and scanned using an Odyssey® Infrared Imaging System.
The following antibodies were used: Sox2, Arg-1, iNos-1, p-p38MARK, p38MARK, and β-actin (all from Cell Signaling Technology). An image analytic system was used to detect the gray degree values of the positive signals.
To elucidate the signaling pathway involved in the Sox2-mediated EpC proliferation, EpCs were treated with specific inhibitors of MAPK (JNK inhibitor: SP600125, ERK inhibitor: PD98059, p38 inhibitor: SB203580; Cell Signaling Technology) for 2 h. Then EpCs were co-cultured with macrophages as above, and western blotting was performed.
Statistical analysis
Statistical comparisons were assessed by Student's t test. All p-values were derived from a two-tailed statistical test using SPSS 11.5 Software. A p-value of <0.05 was considered statistically significant. The results were always represented as a mean (at least three different assays) ± SD.
Results
M1 macrophages participate in regulating proliferation of EpCs
The mechanism regulating EpC proliferation and differentiation in vivo remains unclear. Microglia/macrophage activation and recruitment following SCI are associated with detrimental or reparative functions according to their polarized states. M1 and their associated cytokines inhibited the growth of the transplanted cells 19 and resulted in axonal dieback. 20 Few reports are available about the effect of an inflammatory microenvironment on EpC proliferation and/or differentiation. In the present study, isolated EpCs were co-cultured with M1 or M2 in vitro to explore whether M1/M2 had an effect on EpC properties. Polarized M1 or M2 showed different morphology under inverted microscope. M1 showed a small circular shape; M2 showed a spindle-like shape with an obvious longitudinal axis (Fig. 1A). Arg-1 and iNos-1 expression were detected by western blotting analysis and confirmed a successful polarization (Fig. 1B,C). RT-PCR was used to detect the cytokines secreted by polarized macrophages (Fig. 1D). The mRNA levels of iNOS, TNFα, and IL-1β upregulated significantly in M1, whereas arginase and IL-10 mRNA increased markedly in M2.
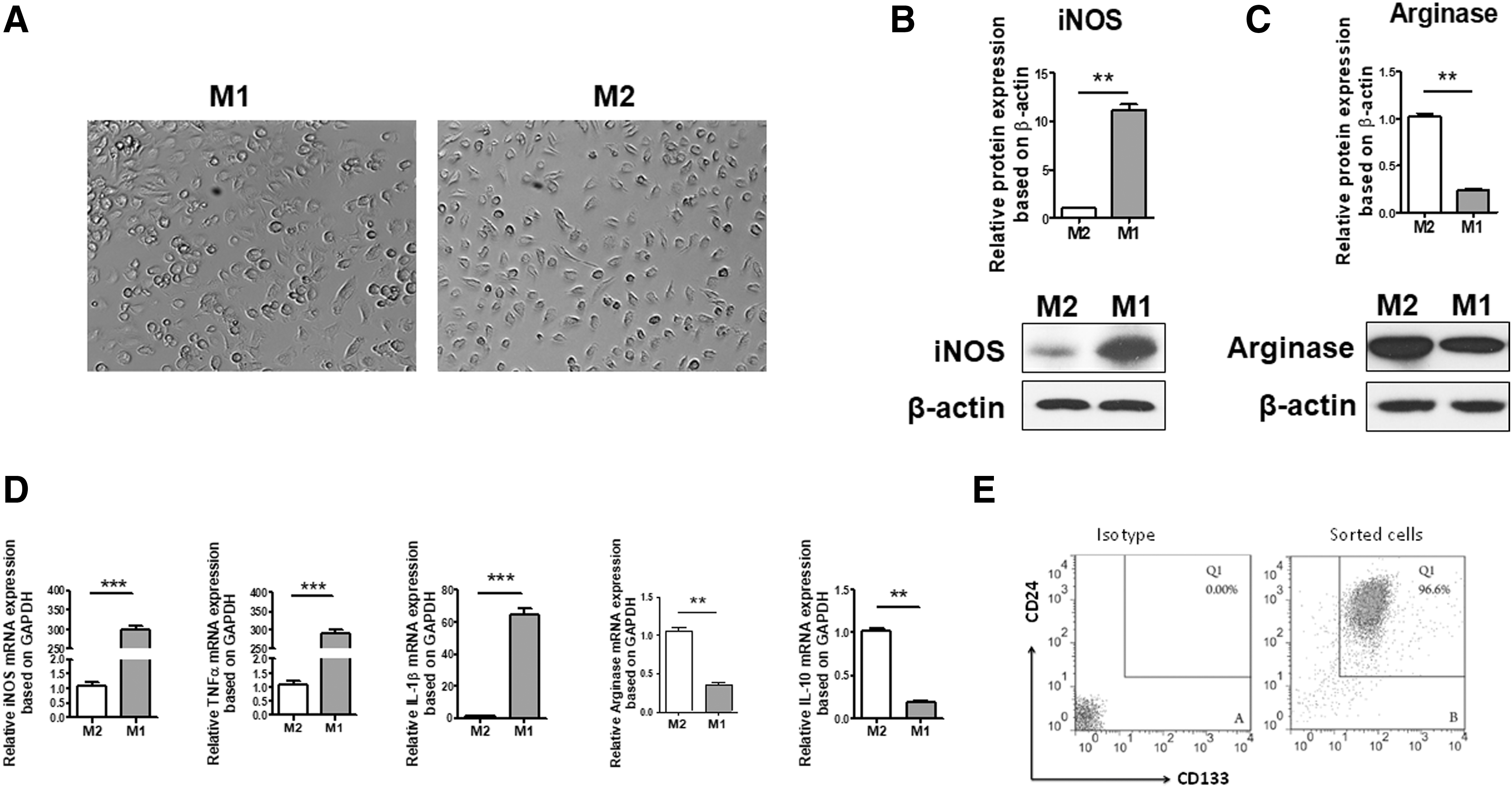
M1, M2, and EpC preparation. (
Cell purity of the sorted EpCs derived from mice undergoing sham operation was determined by flow cytometry analysis. The purity of EpCs was 98% (Fig. 1E), which confirmed an effective sorting with anti-CD133 and CD24. After a co-culture with M1 or M2 for 3 days, the CCK-8 method was used to assess whether M1 or M2 was involved in modulating EpCs proliferation in vitro. EpCs in the control group were cultured in NBA medium supplemented with recombinant human epidermal growth factor (rhEGF) and recombinant human fibroblast growth factor (rhFGF) basic, which is known to favor the growth and differentiation of neural stem cells. Consistent with this, EpCs in the control group showed the most obvious cell proliferation in the present study. There was no significant difference between M1 EpCs and control EpCs (p > 0.05) (Fig. 2A). In contrast, the proliferation of EpCs co-cultured with M2 was significantly decreased (p < 0.05 vs. EpCs control).

EpC proliferation under different conditions by CCK8 method and Ki67 FACS.
At the same time, Ki67 expression in EpCs under different culture conditions was detected by FACS and the percentage of positive cells was counted. The number of positive cells in the M1 co-culture group was higher significantly (p < 0.01) than that in the M2 group, and the mean ratios of positive cells were 33%, 27%, and 11%, respectively (control vs. M1 vs. M2) (Fig. 2B,C). These results suggested that M1-mediated inflammatory response could stimulate the proliferation of EpCs in vitro.
M1 promoted EpC proliferation by upregulating Sox2 expression via TNFα
The experiments described above showed that M1 could promote the proliferation of EpCs co-cultured. However, the exact mechanism was poorly understood. Transcription factor Sox2 has been shown to play a key role in regulating embryonic stem cell proliferation, and studies have found that the expression of Sox2 is upregulated in the ependymal region after SCI. 12,13
Could M1 directly or indirectly affect the expression of Sox2 through the factors they secreted, and thus regulate EpC proliferation? In the present study, EpCs were co-cultured with M1 or M2, respectively, in vitro to evaluate Sox2 expression in EpCs. A certain amount of EpCs showed positive Sox2 staining in the control group, where they were cultured in NBA medium supplemented with EGF and FGF basic. Following co-culture with M1 for 3 days, the percentage of Sox2-positive cells in EpCs co-cultured with M1 was similar to that of the control group according to immunofluorescent assays, which was significantly higher than that of the M2 co-culture group (EpCs-M2 vs. control, EpCs-M2 vs. EpCs-M1, p < 0.05; Fig. 3A,B). As a result, when co-cultured with M1, 36% of EpCs stained positively for Sox2 and the percentage in the control group was 43%. By contrast, only 12% of EpCs showed Sox2 expression when co-cultured with M2 (Fig. 3A,B). These results were in accordance with western blotting assays. EpCs-M1 had similar Sox2 expression to EpCs in the control group, whereas EpCs-M2 had markedly decreased Sox2 expression (Fig. 3C,D).

M1 promoted EpCs proliferation by upregulating Sox2 expression via TNFα.
M1 medium is more conducive to triggering the expression of Sox2 compared with M2 medium, which may be a protective response to spinal cord trauma. The locally aggregated M1 after SCI releases a large number of inflammatory mediators, triggering the proliferation of EpCs in an attempt to fill the cavities and limit the damage. M1 macrophages may induce Sox2 expression through secreting some cytokines. TNF-α is one of the important cytokines secreted by M1. 21 So TNFα antibody was added into M1 medium of the transwell system to explore whether it participated in Sox2 activation. Interestingly, the percentage of EpCs with Sox2-positive staining decreased to 26% in the presence of TNFα antibody (Fig.3A,B). This suggested that TNFα may be one of the cytokines through which M1 medium plays its role in activating Sox2 expression and promoting EpC proliferation.
Western blot assay provided consistent results (Fig. 3C,D). Similar results were also obtained from experiments in vivo. Sox2-positive staining was found mainly in the ependymal cells of the central canal in WT mice following SCI, which was determined based on cellular location and morphology, although no immunohistochemical identification was performed. By contrast, EpCs from animals with sham operation showed negative Sox2 staining. After SCI, protein levels of Sox2 were significantly increased at and near the injury site. By 1 day, Sox2 was upregulated in the ependymal zone, which peaked at day 3 post-SCI. The high expression of Sox2 declined at 1 week after SCI in the present models. In TNFα KO mice, weakly positive staining to Sox2 was detected at the ependymal zone 3 days post-trauma (Fig. 4A). Semi-quantitative analysis with western blotting analysis indicated that the Sox2 expression of TNFα KO group was significantly lower than that of the WT group (p < 0.05, Fig. 4B,C).
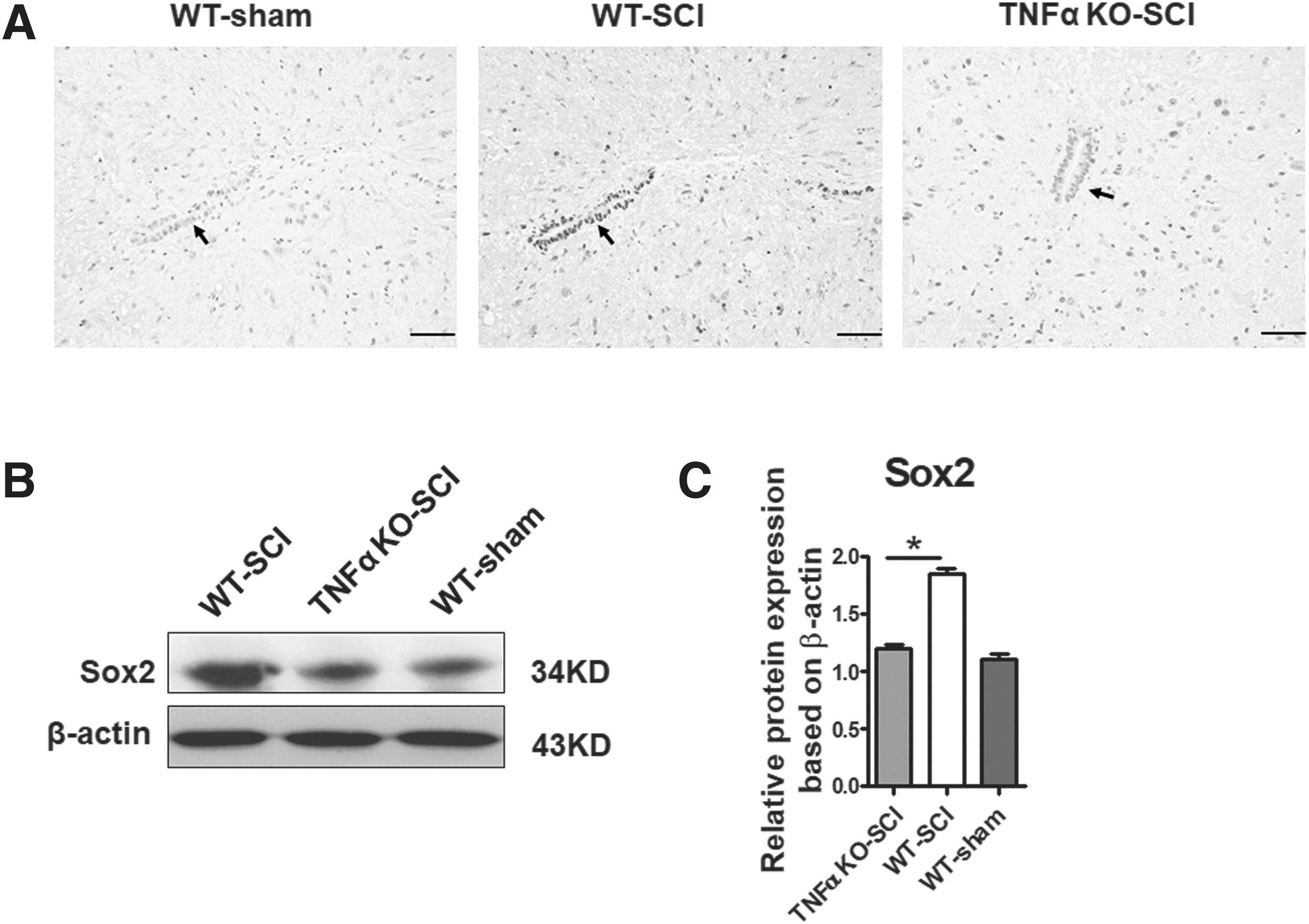
Sox2 expression in the central canal of spinal cord on day 3 post-SCI.
To determine the effect of Sox2 on proliferation of EpCs, EpCs were infected by Sox2 shRNA Lentiviral Particles. Six hours later, the EpCs were further co-cultured with M1 according to the methods before. Proliferation of EpCs decreased dramatically following Sox2 blockage (p < 0.01) (Fig. 2A–C). Just as they acted in the development of embryonic neural stem cells, Sox2 played a key role in regulating proliferation of EpCs.
Taken together, M1-mediated inflammatory response can promote EpC proliferation, and the effect was achieved at least partly by activating Sox2 expression.
TNFα upregulated Sox2 expression in EpCs through MAPK signaling pathway
M1 conditioned medium could activate the expression of Sox2 in EpCs. The signaling pathway involved in Sox2 upregulation needs to be elucidated. The MAPK pathway is one of the most important signaling pathway that mediates development of inflammation. 22,23 In the present study, EpCs were co-cultured with M1 from WT mice or TNFα KO mice for 12 h, then MAPK (ERK, JNK, and p-38) expression was detected by western blot.
First, compared with EpCs co-cultured with M1 derived from TNFα KO mice, EpCs co-cultured with M1 from WT mice showed significantly increased activity of the three kinases noted above. There was no significant difference between EpCs co-cultured with M1 of WT mice and that of the control group (p > 0.05). It verified that M1 could activate the phosphorylation of the MAPK signaling pathway and the activation was dependent at least partly on TNFα (Fig. 5A).
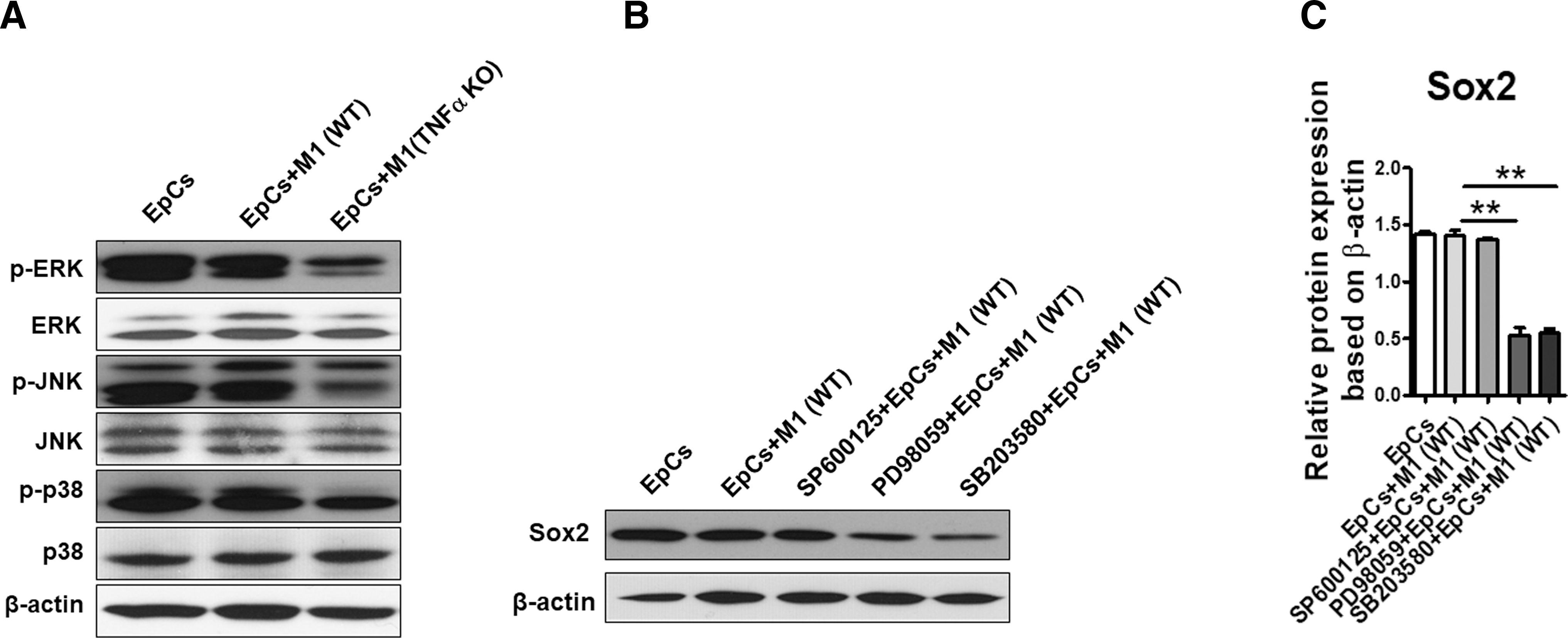
The MAPK signaling pathway was activated in Sox2-expressing cells.
Second, EpCs were treated with specific inhibitors of MPAK (JNK inhibitor: SP600125, ERK inhibitor: PD98059, p38 inhibitor: SB203580, respectively) for 2 h, then the cells were co-cultured with M1 for another 24 h. Western blot was employed to determine the upstream signaling pathway of Sox2 expression. According to the experimental results, JNK inhibitors did not change the expression of Sox2 markedly, whereas ERK or p38 inhibitors significantly attenuated Sox2 expression (Fig. 5B,C). In general, M1 regulated the expression of Sox2 through the MAPK signaling pathway, especially the activation of ERK and p38 kinase.
M2 conditioned medium favored EpC differentiation toward mature neurons
Polarized macrophages, mainly M1, can activate the expression of Sox2 and promote the proliferation of EpCs in vitro and in vivo. It was also studied in the present research whether macrophages participated in regulating EpC differentiation, especially toward neurons. EpCs underwent immunofluorescence assay following co-culture with M1 or M2 for 3 or 7 days to observe their possible differentiation. βIII-tubulin and MAP2 were identified as specific markers of early neurons and mature neurons, respectively. 24,25
Immunofluorescence staining to βIII-tubulin but not MAP2 was observed in all three groups on day 3 (Fig. 6A). About 45% of cells with positive βIII-tubulin staining and longer neurites were observed in the EpCs-M2 group according to cell counting analysis, which was a little more than that of the control group (41%) (p > 0.05). βIII-tubulin-positive cells could also be observed in EpCs co-cultured with M1; however, the mean percentage was merely 9%. On day 7 post–co-culture, MAP2 staining remained negative in the M1 co-culture group, whereas some EpCs demonstrated positive MAP2 staining in the M2 co-culture group and control group (Fig.6B). This indicated that macrophage response, especially M2 type, facilitated the differentiation toward not only early neurons marked with βIII-tubulin staining, but also mature neurons with MAP2-positive staining.
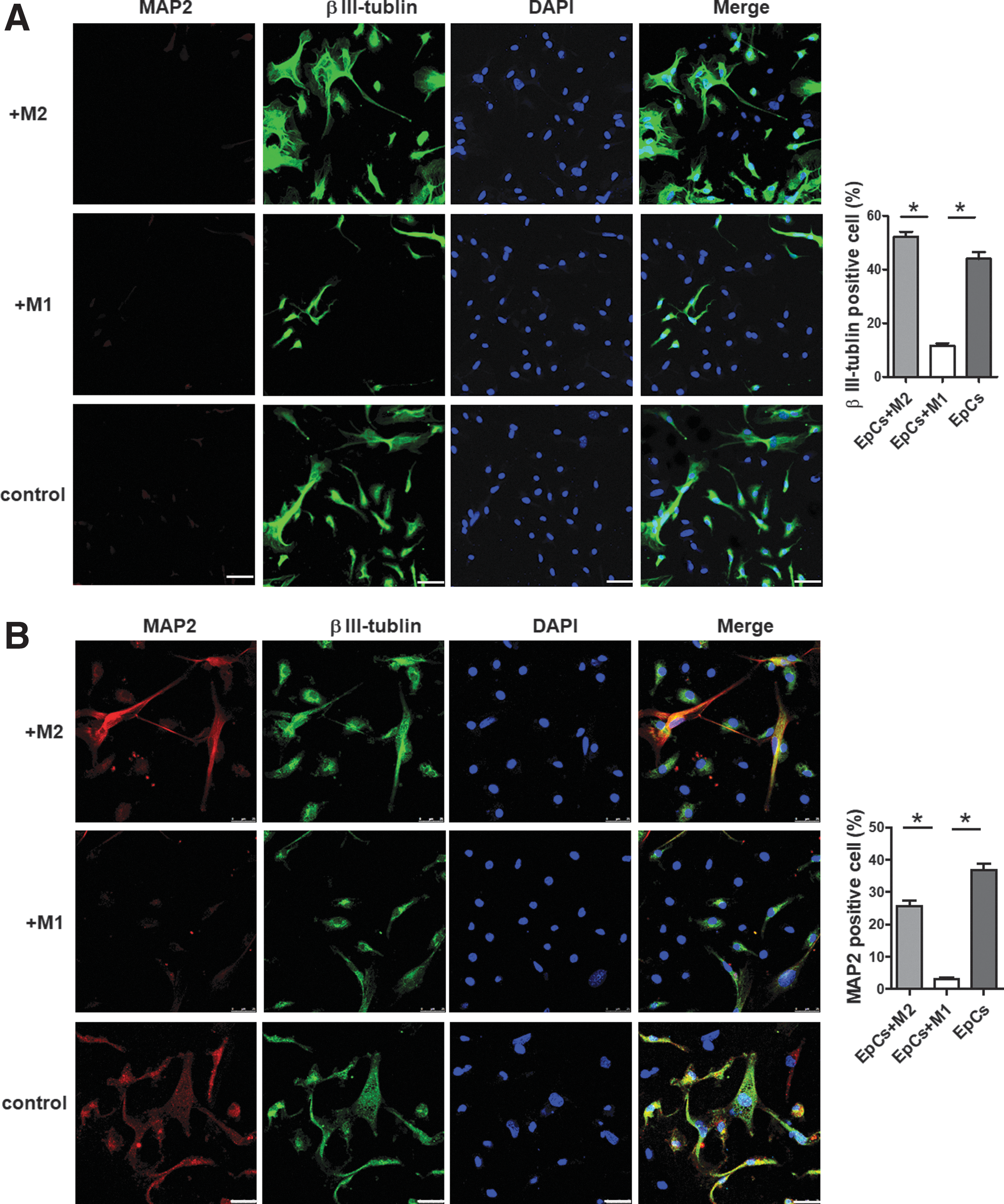
Differentiation potential of the EpCs co-cultured with differently polarized macrophages. The differentiation potential of EpCs toward neurons in the presence of M1 or M2 conditioned medium was assessed.
Discussion
Spinal cord injury can be divided into two stages pathophysiologically. In the acute phase, usually within the first week after injury, primary mechanical trauma and secondary injury cause tissue edema and damage to microvessels, resulting in metabolic disorders and microenvironment disorders, including generation of free radicals, release of excitatory amino acids, lipid peroxidation, and release of inflammatory mediators. In the chronic phase, usually several weeks after injury, the inflammatory response cascade amplifies, and demyelination and neural cell apoptosis occur. Finally, this leads to neural cell loss and disruption of neural circuitry, which is the pathological basis of the irreversible damage and permanent paralysis.
It is a pre-condition for the recovery of neural function to fill the cavities formed following SCI with neural cells and to reconstruct the neural pathway. Currently, there are two main types of cell sources used to replace the necrotic neurons. One is transplanted exogenous stem cells and the other is proliferated endogenous stem cells. As representatives of the former, embryonic stem cells and neural stem cells have been proven to be able to differentiate into neurons in appropriate conditions in vitro. 26,27
Studies have found, however, that they usually maintain an undifferentiated state after transplantation, and even are cleared in the short term. EpCs, representative of the latter, are a kind of multi-potent stem cells in the central canal of adult spinal cord. They proliferate rapidly following SCI and have been thought of as an ideal candidate for cell transplantation. 28,29 Although they can differentiate into functional neurons in vitro, EpC progency differentiate mainly into astrocytes after SCI. It has been widely accepted that the differentiation direction is due partly to the microenvironment in the injured spinal cord, although the exact mechanism remains unclear. 30
Secondary inflammation, usually induced by activated macrophages, is one of the most important events in the microenvironment of the injured spinal cord, where more and more attention has been paid recently. Activated microglia/macrophages have either detrimental or beneficial effects on neural regeneration based on differently polarized M1/M2 subsets. Pro-inflammatory M1 cell response, which is rapidly induced and sustained after SCI, contributes to protracted cell and tissue loss, whereas the alternative, anti-inflammatory M2 type, which is induced transiently, is believed to promote cell protection, regeneration, and plasticity. 31,32
Few reports are available about the effect of the inflammatory microenvironment on EpC proliferation and/or differentiation. In the present study, isolated EpCs were co-cultured with M1 or M2 in vitro to explore whether inflammation induced by M1/M2 had an impact on EpC properties, and if so, how they did. M1 conditioned medium was used to mimic the inflammatory microenvironment dominated by M1 macrophages that were formed following SCI in vivo. NBA medium supplemented with EGF and bFGF was used as a positive control, which has been well-known to promote stem cell growth and differentiation toward neurons. As a result, in the M1 co-culture group, EpC vitality was improved significantly, and the number of Ki67-positive cells increased markedly. The possible mechanism was also explored preliminarily, especially whether transcription factor Sox2 played a role in it. A primary answer was obtained in the present study.
First, much more Sox2-positive EpCs were observed when co-cultured with M1, which reduced significantly following TNFα blockage. These results suggested that M1 type neuroinflammation activated Sox2 expression in EpCs through the cytokines they secreted, such as TNFα. A well-designed experiment in vivo obtained similar results regarding M1 facilitating EpC proliferation.
Second, when co-cultured with M1, cellular viability and number of EpCs with Ki67-positive staining decreased dramatically if the Sox2 gene was silenced previously. It revealed the critical role that Sox2 played in EpC proliferation. Third, it has not yet been unveiled how TNFα modulates the expression of Sox2, in particular, whether the MAPK signaling pathway participates. Evidence in the present study suggested that the MAPK signaling pathway, especially the activation of ERK and P38 kinase, was associated with Sox2 activation. Chen and colleagues reported TNFα could activate the p38 MAPK signaling pathway and thus regulate neural stem cell proliferation, which was consistent with the present study. 33
Taken together, neuroinflammation induced by M1 could promote EpC proliferation, and the effect was achieved at least partly by activating Sox2 expression. In combination with other research findings, 34,35 the present study indicated that Sox2 high expression post-SCI was a spontaneous protection against injury. At the early stage following traumatic SCI, usually within the first week, M1 inflammation is activated rapidly and dominates the spinal cord environment. The body “perceives” the harmful cytokines released by M1 and thereby triggers the self-rescue program, such as activating Sox2 expression. Just as it acts in embryonic stem cells, Sox2 activation promotes EpC proliferation and generates a significant number of new neural cells, attempting to remedy neural loss.
It had been elucidated in the present study that M1 participated in promoting EpC proliferation in vitro, whereas M2 had little effect in this respect. So what is the role of M2, appearing almost simultaneously with M1 in another polarization state post-SCI? Many studies have shown that EpCs differentiate into functional neurons in suitable culture conditions. In the present study, EpCs with βIII-tubulin and MAP2-positive staining were observed on day 7 in the control group, which was consistent with other reports. At the early stage of co-culture, a small number of βIII-tubulin-positive neurons appeared in the M1 group, whereas the number of positive cells increased markedly in the presence of M2. More significantly, after a long period of incubation (7 days in the present study) some mature neurons with MAP2 staining could be detected in the M2 co-culture group.
These results seem to support that M2 conditioned medium was in favor of the differentiation of EpCs into neurons, although detailed mechanisms, such as the cytokines involved, have not been explored yet. EpCs can differentiate into mature MAP2-positive neurons in appropriate conditions in vitro; however, the local neuroinflammation after SCI is predominant with M1 reaction, and the M2 reaction is very short-lived. Although a small number of early neurons with βIII-tubulin-positive staining may spontaneously arise after SCI, this unfavorable microenvironment does not support their long-term survival, let alone further differentiation into mature neurons with MAP2 staining.
Combined with the results of other studies, 30,36 at the early stage, usually immediately after traumatic SCI, local and recruited macrophages in the injured spinal cord are activated, in which the M1 response is dominant and persistent, whereas the M2 response is transient. M1 activate Sox2 expression through releasing some cytokines such as TNFα, and thus promote EpC proliferation, attempting to remedy neural loss. Due to the lack of M2-related neurotrophic factors in the local microenvironment, EpC progeny mainly differentiate into astrocytes and participate in the formation of glial scar, which, on the one hand, restricts the diffusion of injury, and on the other hand, hinders axonal elongation. Asymmetric polarization of macrophages is the cause of differentiation of EpCs mainly to astrocytes rather than neurons and oligodendrocytes. Enhancement of M2-type neuroinflammation locally after SCI may provide a permissive microenvironment and pre-condition for neural restoration. 37,38
EpCs could differentiate into neurons in vitro, which provides a promising strategy to promote spinal cord recovery without the need of transplantation. Neuroinflammation mediated by differently polarized macrophages following SCI had an influence on the biological characters of EpCs. EpCs proliferated in response to SCI, which owed mainly to M1 macrophages, and the signaling pathway TNFα-MAPK-Sox2 was involved. In addition, M2 facilitated EpC differentiation toward mature neurons (Fig. 7). Although it was just a preliminary study and more work should be done, the present study provides a strategy to obtain better neurorecovery through regulating EpCs following SCI.
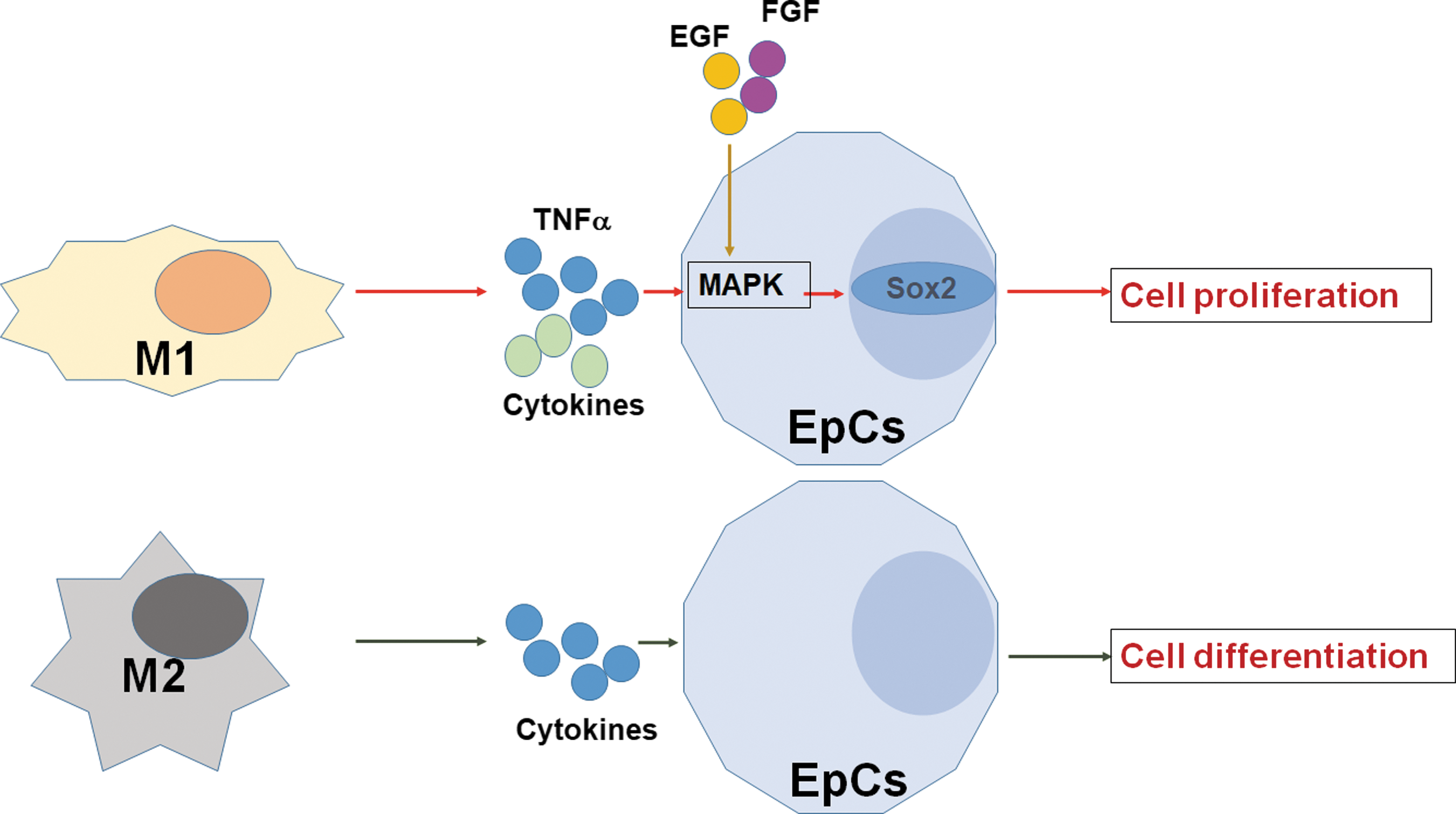
Schematic diagram of the effect of polarized macrophages on EpCs. According to the findings of the present research, M1 mainly promote EpC proliferation through the TNFα-MAPK-Sox2 signaling pathway; M2 facilitated the differentiation of EpCs into mature neurons, which may be achieved through some cytokines they secreted. EpCs, ependymal cells; M1, classically activated macrophages; M2, alternatively activated macrophages; MAPK, mitogen activated protein kinase; Sox2, SRY-box 2; TNFα, tumor necrosis factor α.
Conclusion
EpCs are a kind of static endogenous stem cells in the central canal of adult spinal cord, which proliferate rapidly and recruit into the injured site following SCI. Although they can differentiate into functional neurons in vitro, EpC progeny differentiate mainly into astrocytes after SCI. The mechanisms regulating EpC proliferation and differentiation in vivo remain unclear. In the present study we confirmed that neuroinflammation at the local microenvironment mediated by M1 or M2 had different effects on the fate of EpCs. M1 conditioned medium could promote EpC proliferation through the TNFα-MAPK-Sox2 signaling pathway, whereas M2 conditioned medium favors EpC differentiation toward mature neurons.
Footnotes
Acknowledgments
This study was funded by: WJ2015MB087, Health and Family Planning Commission of Hubei Province, design of the study; 2014CFB205, Natural Science and Technology Foundation of Hubei Province, design of the study; and 81501377, National Natural Science Foundation of China, analysis and interpretation of the data.
Author Disclosure Statement
No competing financial interests exist.