
Abstract
Concussive traumatic brain injury (TBI) is the predominant type of brain injury in young adults and is a risk factor for the development of chronic traumatic encephalopathy and other neurodegenerative diseases late in life. Using a repetitive closed head injury mouse model, we found that treatment with PF04457845, a novel fatty acid amide hydrolase (FAAH) inhibitor that selectively elevated the brain levels of anandamide, improved locomotor function, learning, and memory in TBI mice examined by beam walk, Y-maze, and Morris water maze tests. The accumulation of microglia and astrocytes and the expression of proinflammatory cytokines, including interleukin (IL)-1β, IL-6, and tumor necrosis factor alpha (TNF-α), in the ipsilateral TBI mouse cortex and hippocampus were significantly reduced by drug treatment. The increased expression of amyloid precursor protein (APP), phosphorylated Tau (p-Tau), phosphorylated glycogen synthase kinase 3 beta (pGSK3β) and p35/p25 subunits and the decreased expression of the pre-synaptic proteins, synaptophysin, synaptosome-associated protein of 25 kDa (SNAP25), and cysteine string protein alpha (α-CSP), in TBI mouse brain were also normalized by PF04458745 treatment. The improved locomotor function and working memory were partially mediated by activation of both cannabinoid (CB)1 and CB2 receptors, whereas the improvement on spatial learning and memory seemed to be CB1 receptor dependent. Interestingly, the blockage of PF04457845 on the reduced expression of synaptophysin, but not SNAP25 and α-CSP, was reversed by coadministration of the CB1 receptor antagonist. These results suggest that the therapeutic effect of PF04457845 is mediated by both cannabinoid receptor dependent and independent mechanisms, and selective inhibition of FAAH possesses a great potential for the treatment of TBI.
Introduction
Traumatic brain injury (TBI) is the major cause of morbidity and mortality among young adults who are involved in accidents, contact sports, and military combats in the battle field. 1 –3 According to the Centers for Disease Control and Prevention, it is estimated that in the United States nearly 1.7 million people suffer from TBI each year and 5.3 million are living with TBI-related disabilities. 4,5 Although there is an increased understanding on the pathogenic mechanisms, the therapeutic agents for TBI are still unavailable.
Numerous studies have demonstrated that TBI can cause chronic neuroinflammation, memory deficits, and is a risk factor for the development of chronic traumatic encephalopathy (CTE) and other neurodegenerative diseases late in life. 6 Neuroinflammation is one of the principal mechanisms that contributes to the ongoing neurodegeneration and neurological impairments associated with TBI. 7 Activation of cannabinoid receptors by plant-derived or the synthetic cannabinoid agonists has been shown to elicit anti-inflammatory, antioxidative, and neuroprotective effects in many inflammatory and neurological diseases. However, the enthusiasm to use these compounds is still hampered by the potential psychotropic side effects caused by activation of cannabinoid type 1 receptor (CB1R) in neurons. 8,9
Modulation of the endocannabinoid system has emerged as an attractive strategy for the treatment of many neurological diseases, which include multiple sclerosis, neuropathic pain, stroke, Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis. 10,11 The endocannabinoid system is composed of CB1 and CB2 cannabinoid receptors, the endogenous cannabinoids, anandamide (AEA) and 2-arachidonoylglycerol (2-AG), and the enzymes for their biosynthesis and degradation. Unlike other neurotransmitters, the endocannabinoids are not stored in vesicles and are synthesized when/where they are needed. This on-demand synthesis enables the cannabinoid receptors to be activated locally, rather than globally, and therefore the psychotropic adverse effects caused by the exogenous cannabinoid ligands can be avoided. However, because of the rapid hydrolysis, the compensative protective effects of these endocannabinoids are transient and short-lived, and thus inhibition of their degradation can sustain the endogenous cannabinoid levels and prolong their protective effects. Studies from our group and others have shown that blockade of AEA and 2-AG hydrolysis is protective in several TBI animal models induced by controlled cortical impact and fluid percussion. 12 –17 Recently, inhibition of 2-AG hydrolysis has also been demonstrated in a mouse model of chronic repetitive closed head injury by suppressing neuroinflammation, preventing neuronal death, normalizing synaptic function, and improving learning, memory, and other neurological functions. 18 Despite that activation of both CB1 and CB2 receptors is known to have therapeutic effects in the TBI animal models, the contribution of the cannabinoid receptors in this model system has not been elucidated.
Several studies have shown that sustained blockade of 2-AG hydrolysis can cause cannabinoid receptor desensitization and behavioral tolerance; therefore, the clinical application of this strategy remains under scrutiny. 19 –22 Given that augmentation of AEA does not have this adverse effect, and is able to suppress neuroinflammation and exert neuroprotection, in this study we aimed to examine the role of a recently developed fatty acid amide hydrolase (FAAH) inhibitor (PF04457845) in a mouse model of repetitive closed head injury and to explore the underlying therapeutic mechanisms.
Methods
Chemicals
The FAAH inhibitor, PF04457845, the CB1 receptor (CB1R) antagonist, AM281, the CB2R antagonist, AM630, and the deuterated AEA, 2-AG, and arachidonic acid (AEA-d4, 2-AG-d5, and AA-d8) were purchased from Cayman Chemicals (Ann Arbor, MI). All other chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO), unless stated otherwise.
Animals and drug treatment
Male, 8- to 10-week-old C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME). All animal procedures were carried out in accord with the guidelines established by the National institutes of Health (NIH) and approved by the Uniformed Services University Institutional Animal Care and Use Committee. Mice were intraperitoneally injected with vehicle or drugs at 30 min after each impact and then once a day for 5 consecutive days (eight injections in total). To determine the cannabinoid receptor dependency, PF04457845 (5 mg/kg) was coadministered with the CB1R antagonist, AM281 (3 mg/kg), or the CB2R antagonist, AM630 (3 mg/kg). The drugs were dissolved in dimethyl sulfoxide/cremophor/saline (1:1:18), which serves as a vehicle control.
Repetitive closed head injury
Repetitive closed head injury was performed in mice using an electromagnetic controlled stereotaxic impact device, as described previously, 18 with a minor modification. Briefly, mice were anesthetized with 3% isoflurane before stabilizing the head using ear bars in a mouse stereotaxic frame (Stoelting Co., Wood Dale, IL). A midline sagittal incision was made to expose the skull. The stereotaxic electromagnetic impactor with a 3.0-mm steel tip impounder was used to deliver a single controlled cortical impact, delivered at coordinates of 1.8 mm caudal to bregma and 2.0 mm left to midline with a controlled velocity at 3.0 m/s, an impact depth of 2.0 mm, and a dwell time at 100 ms. Mice with depressed skull fracture or visible hemorrhage were excluded from the study. Sham mice underwent the identical surgical procedures as the injured group, but no impact was delivered. After impact, the skin was sutured and the animals were allowed to recover from anesthesia and then returned to their home cages. A second and third identical closed head injury procedure was performed after removing the sutures on day 2 and day 3 after the initial injury. The skull fracture rate was less than 2.0% during the surgical procedures.
Beam walk test
The beam walk balance test was performed to assess fine motor movement as we previously described. 12 Briefly, the beam walk apparatus consists of a wooden beam measuring 6 mm in width, 120 cm in length, and suspended 30 cm above a table. Mice were trained to walk on the beam for 3 days before surgery, and at the end of the third day the baseline values (i.e., the number of foot faults per 50 steps) were collected. The beam walk balance test was performed at days 4, 6, and 10 after the initial impact, and the number of foot faults per 50 steps was recorded. The timeline for all the behavioral tests and molecular, biochemical, and immunohistochemistry assessments is illustrated in Figure 1A.
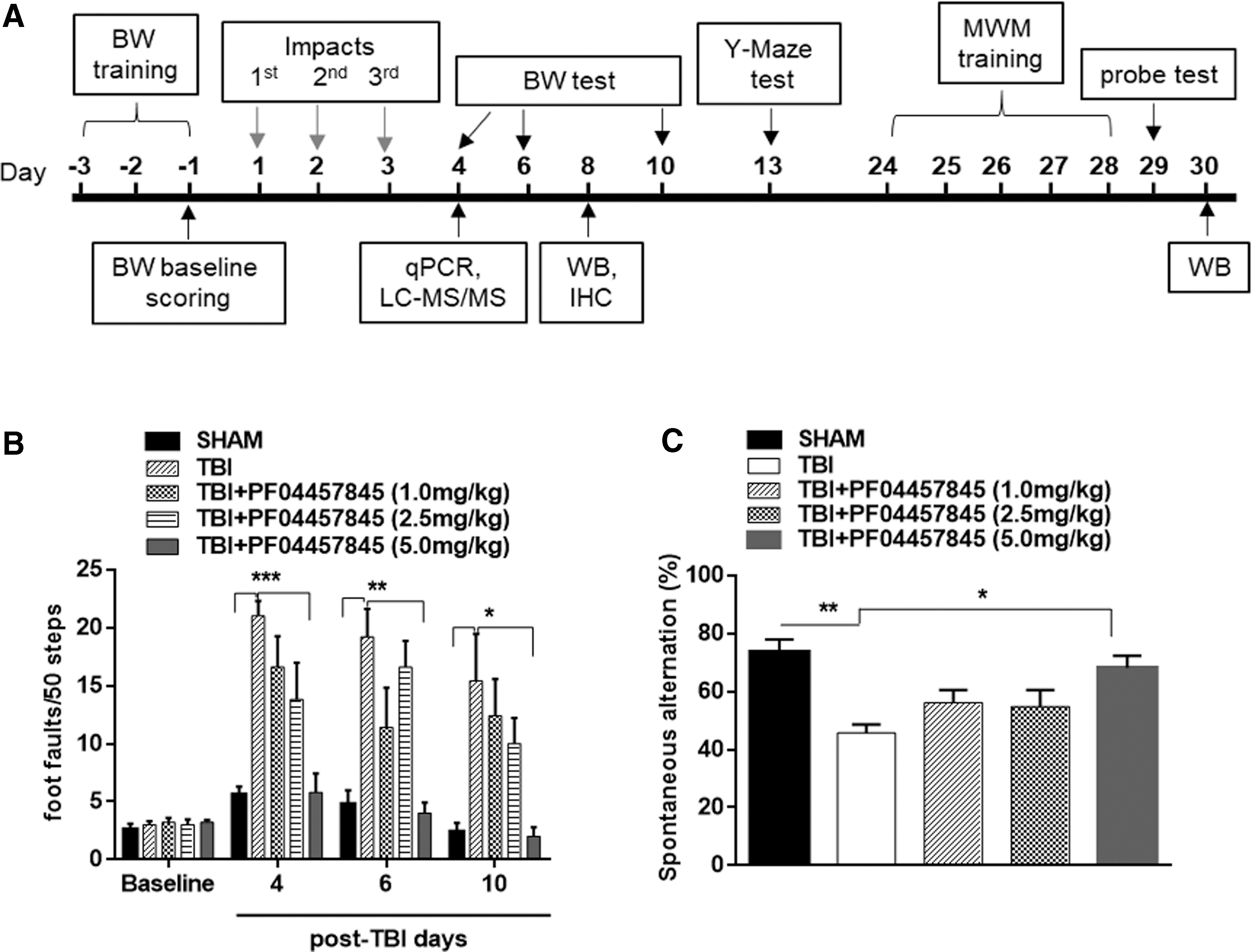
PF04457845 treatment attenuated TBI-induced deficits in motor coordination and working memory performance. (
Spontaneous alternation Y-maze test
At 13 days after repetitive closed head injury, mice were tested in a Y-maze apparatus. The Y-maze device (Stoelting Co.) has a symmetrical plastic Y shape with arms measuring 25 cm long, 8 cm wide, and 15 cm high. Mice were placed at the end of a pseudorandomly chosen arm as a starting point and allowed to explore the maze for 5 min. Each arm visit was referred to a mouse moving all four paws into the arm, and each alternation was defined as a consecutive entry into three different arms. The sequence and the number of entries to each arm were video recorded. The percentage alternation was calculated based on the formula: total number of alternations/(total number of arm entries-2)*100, as previously described. 23 The spontaneous alternation Y-maze test measures hippocampus-based working memory 24,25 and was used to evaluate the effect of drug treatment on TBI-induced working memory deficits.
Morris water maze
The Morris water maze test was performed at 24–29 days post-TBI to examine spatial learning and memory, as described previously, with some modifications. 26 Briefly, a circular water tank with a diameter of 1.5 m was used to perform the Morris water maze. For extramaze visual cues, the four walls around the tank were permanently hanged with different colors and shapes for orientation. A round transparent platform (with a diameter of 11.5 cm) was hidden 1 cm beneath the surface of the water at the center of a given quadrant of the water tank. Mouse received training in the Morris water maze for 5 days, and each session was composed of four trials, plus a probe trial 24 h after the last hidden platform test. For each trial, the mouse was released facing the wall of the tank and allowed to search, find, and stand on the platform for 15 sec within the 60-sec trial period. The time it took to find the platform was calculated as latency. In each training session, the starting quadrant point and sequence of the four quadrants from where the mouse was released into the tank were randomly chosen so that it was varied among the separate sessions for each animal and was different for individual animals. In the latter case, the mouse who could not find the platform was guided to the platform and remained there for 15 sec before being returned to the home cage. The probe trial was conducted by removing the platform and releasing the mouse facing the wall exactly from the opposite or parallel side where the platform was hidden previously. The task performance was recorded for 60 sec. Tracking of animal movement was achieved with the ANY-maze video-tracking system (Stoelting Co.).
Quantitative real-time polymerase chain reaction
Mice were sacrificed on day 4 after three impacts to extract total RNA from the ipsilateral cortex using TRIzol (Sigma-Aldrich), following the manufacturer's instructions. RNA yield and purity were evaluated using a NanoDrop spectrophotometer. RNA (1 μg) was reverse transcribed to complementary DNA (cDNA). First-strand cDNA was synthesized with the Maxima First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA). Amplified cDNAs were diluted at 1:10 in ultrapure water and subjected to SYBER green master mix (Applied Biosystems, Grand Island, NY) based real-time polymerase chain reaction (PCR) on a light cycler 480 II Roche System (Roche, Indianapolis, IN) with primers interleukin (IL)-1β, IL-6, tumor necrosis factor alpha (TNF-α), glial fibrillary acidic protein (GFAP), ionized calcium-binding adaptor molecule 1 (Iba1), CB1R, CB2R, activating transcription factor 3 (ATF3), cyclooxegenase (COX)1, COX2, microsomal prostaglandin E synthases 1 and 2 (mPGES1/mPGES2), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The primer sequences are as follows: IL-1β (BC011437): forward, 5′-gcaactgttcctgaactcaact-3′ and reverse, 5′-atcttttggggtccgtcaact-3′; IL-6 (NM031168.2): forward, 5′-tagtccttcctaccccaatttcc-3′ and reverse, 5′-ttggtccttagccactccttc-3′; TNF-α (NM013693): forward, 5′- ccctcacactagatcatcttct-3′and reverse, 5′-gctacgacgtgggctacag-3′; GFAP (NM001131020.1): forward, 5′-ctggctgcgtatagacagga-3′ and reverse, 5′-gaactggatctcctcctcca-3′; Iba1 (NM001361501.1): forward, 5′-cagactgccagcctaagaca-3′ and reverse, 5′-aggaattgcttgttgatccc-3′; CB1R (NM001355020.2): forward, 5′-aagtcgatcttagacggcctt-3′ and reverse, 5′-tcctaatttggatgccatgtctc-3′; CB2R (NM001305278): forward, 5′-tgctgtcatatgctggtc-3′ and reverse, 5′-atcctggctcctaggtggtt-3′; ATF3 (NM007498.3): forward, 5′-agctgagattcgccatccagaa-3′ and reverse, 5′-ctcgccgcctccttttcct-3′; COX1 (YP001686700.1): forward, 5′-atgagtcgaaggagtctctcg-3′ and reverse, 5′-gcacggatagtaacaacaggga-3′; COX-2 (BC052900): forward, 5′-gtggaaaaacctcgtccaga-3′ and reverse, 5′-gctcggcttccagtattgag-3′; mPGES1 (NM022415.3): forward, 5′-tgtccaaatcctgtcttcca-3′ and reverse, 5′-ggttctggagcacaccctat-3′; mPGES2 (NM133783): forward, 5′-acttccactccctgccctat-3′ and reverse, 5′-gttgcaagctgtctccttcc-3′; and GAPDH (GU214026): forward, 5′-aggtcggtgtgaacggatttg-3′ and reverse, 5′-tgtagaccatgtagttgaggtca-3′. PCR reactions were conducted as follows: 95°C for 15 sec, 75°C for 1 min, and 60°C for 30 sec for 40 cycles, followed by a melting point determination or dissociation curves. All samples were run in triplicate. Relative levels of gene expression were determined by the 2−ΔCt method normalized to the expression of GAPDH.
Prostaglandin E2 assay
Prostaglandin E2 (PGE2) assay was performed in the ipsilateral cortex tissue isolated from day 4 post-TBI and subjected to homogenization with 40 μL of 0.02% trifluoroacetic acid (TFA) and 100 μL of acetonitrile on ice. To extract maximal lipid, 140 μL of tissue homogenate was dispersed in 1 mL of acetonitrile by vortex and left at 4°C overnight. On the next day, the homogenate-acetonitrile mixture was centrifuged at 2000g for 5 min to remove the debris and the supernatant was transferred to a silanized glass tube. The supernatant was evaporated under the nitrogen gas streaming in a water bath (approximately 35°C) and then reconstituted with acetonitrile. The levels of PGE2 in the lipid extract were measured using a PGE2 enzyme immunoassay (EIA) kit, following the manufacturer's protocol (Cayman Chemical, Ann Arbor, MI).
Western blot
To determine the levels of phosphorylated glycogen synthase kinase 3 beta (GSK3β), phosphorylated Tau (pTau), total Tau, amyloid precursor protein (APP), and p35/p25, mice were sacrificed on day 8 after injury. The ipsilateral cortical tissues adjacent to the site of injury were removed and homogenized in radioimmunoprecipitation assay buffer supplemented with 1 × protease and phosphatase inhibitor cocktail (catalog no.: 1861280; Thermo Scientific, Rockford, IL). Protein concentration was determined for each sample, and equal amount of proteins were run on 4–15% sodium dodecyl sulfate/polyacrylamide gel electrophoresis (Bio-Rad, Hercules, CA). Thereafter, proteins were transferred onto nitrocellulose membranes, blocked for 1 h with 5% bovine serum albumin at room temperature, and then incubated overnight at 4°C with the primary antibodies. These antibodies included anti-rabbit GSK3β (Y216; 1:1000; catalog no.: ab75745; Abcam, Cambridge, MA), antirabbit pTau (S404; 1:1000; catalog no.: D2746; Cell Signaling Technology, Danvers, MA), anti-mouse Tau46 (1:1000; catalog no.: 4019S; Cell Signaling Technology), antirabbit p35/p25 (1:1000; catalog no.: C64B10, Cell Signaling Technology), and antirabbit APP (1:1000; catalog no.: ab15272; Abcam). The expression of pre- and post-synaptic markers in hippocampus at 30 days post-TBI were examined using the antimouse Synaptophysin (1:1000; catalog no.: 7H12; Cell Signaling Technology) and antirabbit PSD95 (1:1000; catalog no.: D7403; Cell Signaling Technology), respectively. For assessing the expression of SNARE complex proteins, the antirabbit cysteine string protein alpha (α-CSP; 1:1000; catalog no.: AB1576; EMD Millipore, Burlington, MA) and antimouse synaptosome-associated protein of 25 kDa (SNAP-25; 1:1000; catalog no.: 850302; BioLegend, San Diego, CA) were applied. After probing with a goat antirabbit or antimouse immunoglobulin G (H + L)/horseradish peroxidase–conjugated secondary antibody (1:2500; Bio-Rad) for 1 h at 25°C, protein bands were detected using the Supersignal West Pico Chemiluminescence (Thermo Fisher Scientific). Membranes were subsequently probed for the expression of β-actin as a loading control. Images were acquired with a Chemidoc Touch image system (Bio-Rad) and analyzed using ImageJ software (NIH, Bethesda, MD).
Liquid chromatography with tandem mass spectrometry analysis
The cortical tissues obtained from day 4 TBI mice were subjected to liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) to measure the levels of 2-AG, AEA, and arachidonic acid (AA). The tissue was homogenized with 40 μL of of 0.02% TFA, 250 μL of acetonitrile, and 250 picomoles of 2-AG-d5 and AA-d8 and 0.5 picomoles of AEA-d4 (Cayman Chemical) using a Potter homogenizer at 4°C. The homogenate was dissolved completely in 2.5 mL of acetonitrile by vortex and kept at 4°C overnight. The homogenate was subjected to centrifugation at 2000g for 5 min to remove the debris, and then the supernatant was evaporated under nitrogen gas streaming in a water bath (approximately 35°C). The lipid was resuspended with 100 μL of acetonitrile and stored at −80°C until use.
A high-performance liquid chromatography (HPLC) system (1200 Series; Agilent Technologies, Santa Clara, CA) was used with a reverse-phase guard column (Wide Pore C18 [ODS], 4 × 2 mm ID; Phenomenex, Torrance, CA), and the column (Sephasil Peptide C18, 5 μ, ST, 100 × 4.6 mm ID; Pharmacia Biotech, Piscataway, NJ) was maintained at 40°C. The mobile phase was composed of solvent A (0.2% formic acid in water) and solvent B (0.2% formic acid in methanol), and the following gradient was used: 62% A/38% B isocratic for 30 sec, ramp to 90% B in 60 sec, isocratic at 90% B for 18.5 min, ramp back to 62% A/38% B in 60 sec, and re-equilibrate at 62% A/38% B for 8 min. The flow rate was 0.4 mL/min. The HPLC output was directed into the TurboV electrospray ionization source of a Q-Trap 4000 mass spectrometer (AB Sciex, Framingham, MA). The injection volume was 20 μL. LC-MS/MS analysis was performed in a positive mode with the ion source temperature of 600°C, a spray voltage of 5.5 kV, and a declustering potential of 45 V. Multiple reactions monitoring was performed on the transitions m/z 379 → 287 for 2-AG, 348 → 62 for AEA, and 304 → 121 for AA. The concentrations of 2-AG, AEA, and AA were determined by calculating the corresponding peak area ratio to the internal standard using a linear fit weighting to the calibration curve.
Immunohistochemistry
On day 8 post-TBI, mice were anesthetized using a combination of ketamine and xylazine (90 mg/kg of ketamine/10 mg/kg of xylazine in a volume of 10 μL/g of body weight, intraperitoneally) and then intracardially perfused with ice-cold phosphate-buffered saline (PBS) followed by 4% paraformaldehyde (PFA). The brains were quickly removed from the skulls and post-fixed in 4% PFA at 4°C overnight. On the following day, the whole brains were cryoprotected in 30% sucrose in PBS at 4°C until sinking, embedded in Tissue Tek OCT, and stored at −80°C until use. Coronal sections of the brain were cut at 30 μm using microtome (Leica SM2000R; Leica Microsystems, Wetzlar, Germany) and the series sections (10–12 slices) were collected per well in a 24-well plate and stored in cryoprotectant solution at −20°C until for immunostaining. Primary antibodies used for immunostaining included the antigoat Iba1 (1:300; catalog no,: ab48004; Abcam), antimouse GFAP (1:500; catalog no.: 3670; Cell Signaling Technology), and antirabbit p-Tau (S404; 1:1000; catalog no.: D2746; Cell Signaling Technology). Free-floating sections were immunostained using the specific antibodies listed above, followed by incubation with the corresponding fluorescent-labeled secondary antibodies. The sections were washed with 1 × PBS twice, mounted on slides, and then covered with fluoroshield mounting medium with 4’,6-diamidino-2-phenylindole (DAPI). Immunofluorescence images were obtained with a fluorescence microscope (Nikon Eclipse TE-2000U; Nikon Corporation, Tokyo, Japan). The cells with both DAPI and expected fluorescence were defined as positively stained cells. Negative controls were routinely performed in which the primary antibodies were omitted. All immunofluorescence data were obtained in a minimum of five to seven serial sections from the brain tissues of each animal. GFAP, Iba1, and p-Tau immunostaining images were taken, analyzed, and quantified using the NIH ImageJ software.
Fluoro-Jade C staining
Fluoro-Jade C (FJC) staining was applied to discern the degenerated neurons. Free-floating sections mounted on slides were incubated in the solution with FJC (0.0004% solution; EMD Millipore, Temecula, CA, USA) and DAPI (0.5 μg/mL) for 20 min, followed by 3 × 1 minute wash with distilled water. The slices were dried naturally at room temperature without light. The images were taken using a fluorescence microscope (Nikon Eclipse TE-2000U; Nikon Corporation), and the FJC-positive cells were counted and expressed as mean cell numbers per mm 2 .
Statistical analysis
Statistical analysis was performed using Prism software (version 7; GraphPad Software, Inc., San Diego, CA). For multiple comparison, analysis of variance followed by Tukey-Kramer's post-hoc tests were used, when appropriate. Differences in significance were defined at p < 0.05, and data are expressed as means ± standard error of the mean (SEM).
Results
Treatment with PF04457845 improved locomotor function and working memory in mice with repetitive closed head injury
To determine the role of a novel FAAH inhibitor, PF04457845, on locomotor function and working memory, different doses of PF04457845 (1, 2.5, and 5 mg/kg) were intraperitoneally administered at 30 min after each impact and then once-daily for 5 additional days. To evaluate the fine motor movement affected by TBI, the beam walk test was conducted and the number of foot faults over a total of 50 steps were counted. The missteps were greatly increased at 4, 6, and 10 days after TBI compared to sham control animals, and the missteps were completely prevented in the 5 mg/kg PF04457845 treatment group (Fig. 1B). Although there was a trend toward locomotor improvement in the 1 and 2.5 mg/kg treatment groups, the significant differences were not reached when compared to the TBI/vehicle group. Similarly, the working memory assessed by Y-maze test was significantly decreased in the TBI-vehicle group demonstrated by the reduced spontaneous alternation. The impaired spontaneous alternation was reversed by PF04457845 treatment at 5.0 mg/kg, but not at 1 and 2.5 mg/kg (Fig. 1C). These results clearly suggested that treatment with PF04457845 at 5 mg/kg was the optimal dose to reduce the symptoms associated with TBI, and therefore this dose was used for the rest of the experiments.
PF04457845 treatment selectively elevated the endogenous levels of anandamide in traumatic brain injury mouse brain
To determine the selectivity of systematically administered PF04457845 in mouse brain, the brain levels of AEA and 2-AG were measured by LC-MS/MS on day 4 post-TBI. Although there were no differences in the brain levels of AEA and 2-AG between the TBI and sham control animals, administration of PF04457845 selectively elevated the brain levels of AEA, but not 2-AG (Fig. 2A,B). Given that the hydrolysis of AEA by FAAH results in the production of AA, we also measured the brain levels of AA and PGE2, one of the major proinflammatory eicosanoids derived from AA oxygenation by COX-1/2 in PF04457845-treated animals. There were no differences in the amount of AA and PGE2 among the sham controls, TBI/vehicle, and PF04457845-treated groups (Fig. 2C,D). This result is consistent with the findings that blockade of 2-AG, but not AEA, hydrolysis contributes to the reduced brain levels of AA and PGE2. 27

Treatment with PF04457845 selectively elevated brain levels of AEA, but not 2-AG, in TBI mouse brain. Fresh ipsilateral mouse cortical tissues on day 4 post-TBI were subjected to LC-MS/MS analysis. (
Traumatic brain injury–induced neuroinflammation is reduced by PF04457845 treatment
Neuroinflammation is an important secondary event that contributes to neurodegeneration and neurological impairments in TBI. Neuroinflammation is characterized by astrocyte and microglial activation, leukocyte recruitment, and upregulation of proinflammatory cytokines. 28 On day 4 post-TBI, ipsilateral cortex adjacent to the impact site was isolated to assess the messenger RNA (mRNA) levels of proinflammatory cytokines and other mediators, including IL-6, IL-1β, TNF-α, COX-1, COX-2, mPGES-1 and mPGES-2, and ATF3, a marker of axonal injury. 29 The mRNA expression of IL-6, IL-1β, and TNF-α was significantly increased in the TBI/vehicle group and was almost completely blocked by PF04457845 treatment (Fig. 3A–C). The elevated expression of ATF3 in the TBI/vehicle group was also significantly blocked by drug treatment (Fig. 3D). Consistent with the reports that COX-1 is primarily expressed in reactive microglia and COX-2 can be constitutively expressed in neurons, 30 the expression of COX-1 and mPGES-1 was increased, whereas the expression of COX-2 and mPGES-2 did not change, even though a slight reduction for both enzymes was observed in the TBI and drug treatment groups (Fig. 3E–H). The expression of CB1 receptor predominantly expressed in neurons was not altered (Fig. 3J), but the expression of CB2 receptor, that is mainly located in microglia, was significantly increased (Fig. 3I). In agreement with these findings, the mRNA expression of GFAP, Iba1, and CD68, the respective markers of astrocytes and microglia, was dramatically increased in TBI animals and reversed in the PF04457845 treatment group (data not showed). Consistent with the results from mRNA expression, the accumulation of microglia and astrocytes examined by immunohistochemistry was also increased in the ipsilateral cortex and hippocampal dentate gyrus at 8 days post-TBI (Figs. 4 and 5). Of note, Tau phosphorylation, a marker of neurodegeneration, was accumulated in regions with highly expressed microglia (Fig. 4), suggesting that neurodegeneration is likely correlated with the increased inflammatory response.

Increased expression of neuroinflammatory molecules and the CB2 receptor in the TBI mouse brain was suppressed by PF04457845 treatment. The fresh cortex tissues from mice 4 days post-TBI were subjected to qRT-PCR analysis. The mRNA expression of proinflammatory mediators, including IL-6, IL-1β, TNF-α, ATF3, COX-1, PGES1, and CB2 receptor (
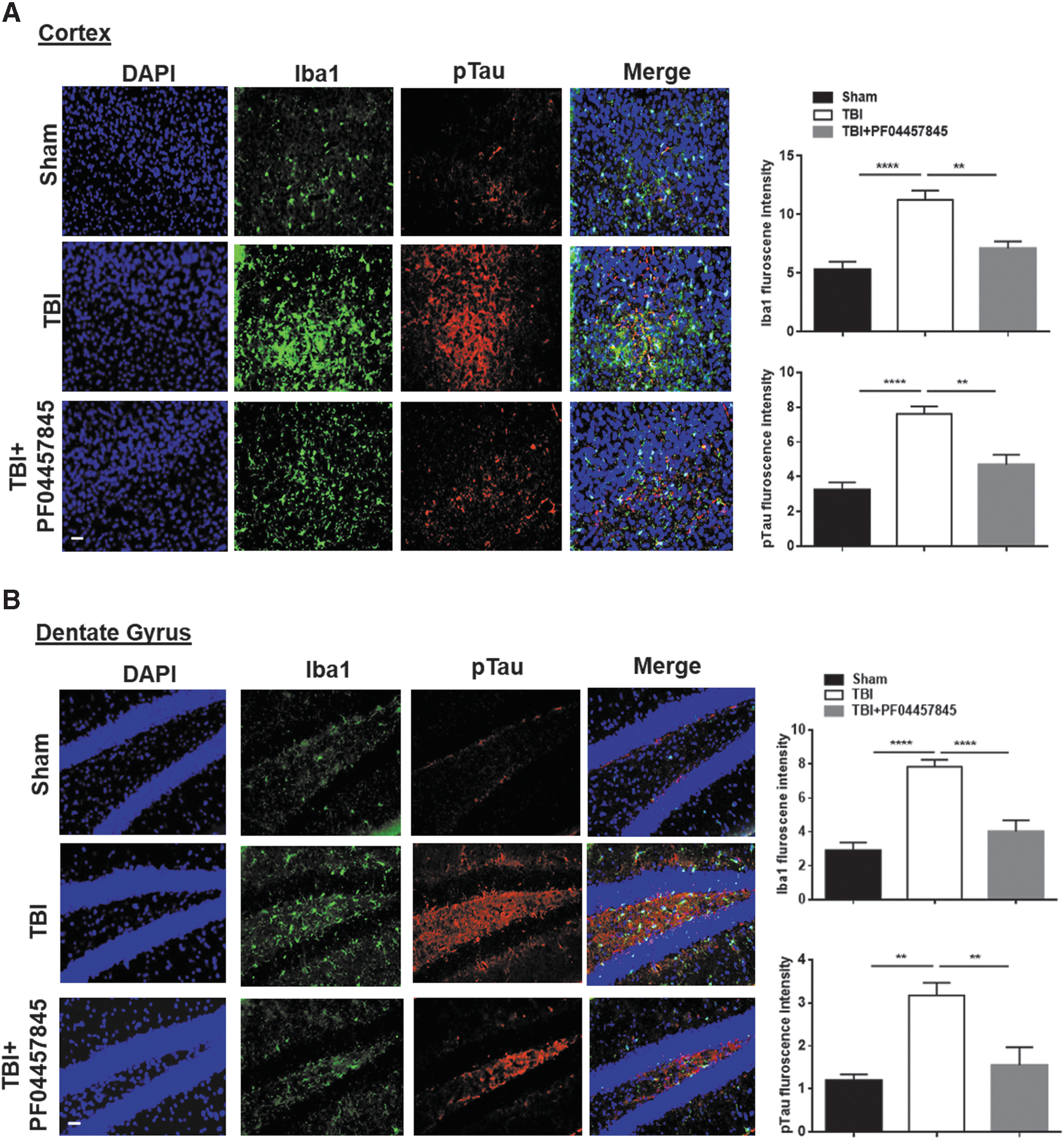
PF04457845 treatment reduced microglia activation and Tau phosphorylation in the ipsilateral cortex and dentate gyrus (DG) of TBI mouse brain. At 8 days post-injury, the Iba1-positive microglia and p-Tau were determined using immunostaining (n = 3 animals in each group; **p < 0.01 and ****p < 0.0001). Bars represent SEM. Scale bar = 50 μm. DAPI, 4’,6-diamidino-2-phenylindole; Iba1, ionized calcium-binding adaptor molecule 1; SEM, standard error of the mean; TBI, traumatic brain injury. Color image is available online.
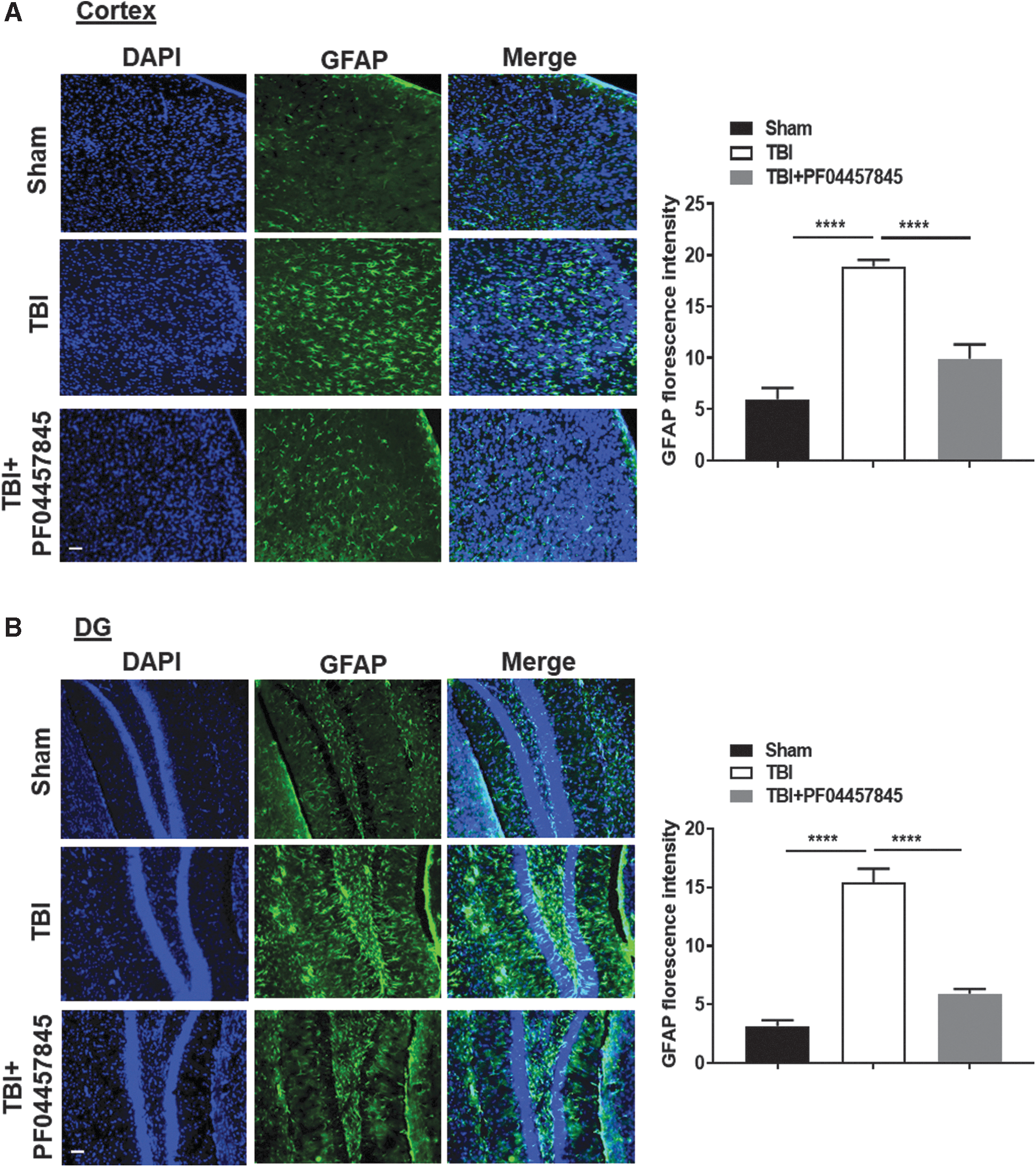
Inhibition of AEA metabolism reduced the accumulation of astrocytes in the ipsilateral cortex and dentate gyrus (DG) of TBI mouse brain. At 8 days post-injury, the GFAP-positive immunostainings were significantly increased in the ipsilateral cortex (
Fatty acid amide hydrolase Inhibition reduced neurodegeneration, tau phosphorylation, and amyloid precursor protein formation following repeated closed head injury
Several studies have shown that the expression of APP and phosphorylation of Tau are increased after TBI and serve as hallmarks of CTE and Alzheimer's disease. 24,25,31 –37 To examine the changes of these neuropathological biomarkers, western blotting analysis in the ipsilateral cortex at 8 days post-TBI was performed for APP, phosphorylated and total tau (p-Tau and Tau46), pGSK3β, and p35/p25 proteins. The expression of APP, phosphorylated, but not total Tau, pGSK3β, and p35/p25 subunits was significantly increased in TBI animals and suppressed by PF04457845 treatment (Fig. 6A). Neuronal cell death examined by FJC staining was increased in TBI mouse cortex and hippocampus and significantly reduced by PF04457845 treatment (Fig. 6B). These results suggested that FAAH inhibition suppresses neuroinflammation and signaling molecules associated with neurodegeneration.
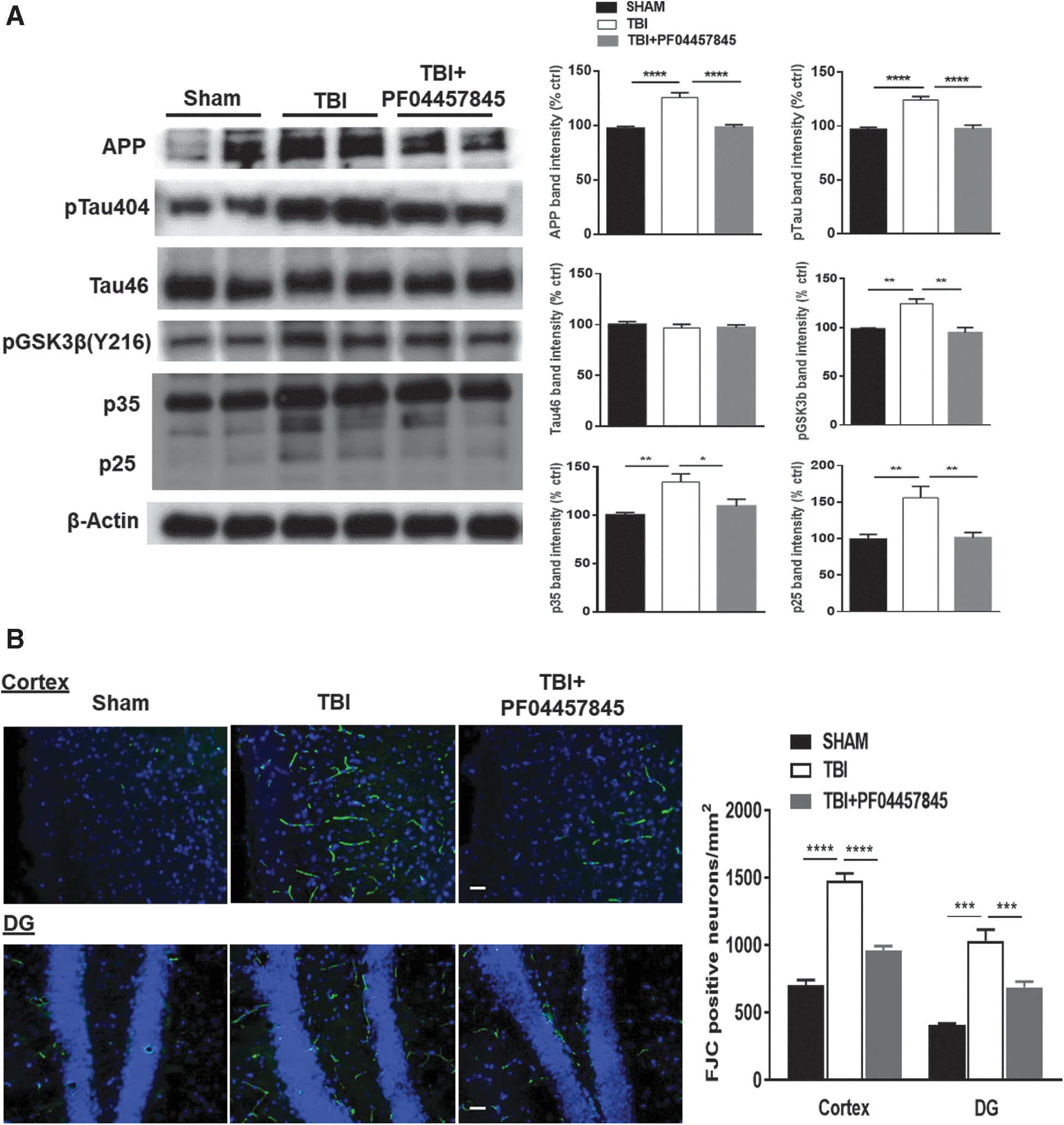
Treatment with PF04457845 attenuated neuronal death, APP formation, and Tau hyperphosphorylation in TBI mouse brain. (
The improvement of PF04457845 on motor function, learning and memory was partially mediated by activation of cannabinoid 1 and cannabinoid 2 receptors
To determine the contribution of the cannabinoid receptors in the therapeutic effect of PF04457845 in the current model system, the CB1 receptor antagonist, AM281, or the CB2 receptor antagonist, AM630, were coadministered with PF04457845. Similar to the results reported earlier (Fig. 1), TBI-induced motor disturbance was ameliorated by PF04457845 treatment, and the improved locomotor function on day 4, day 6, and day 10 was partially reversed by cotreatment with either AM281 or AM630 (Fig. 7A). Similarly, in the Y-maze test, the improved working memory by PF04457845 treatment was attenuated by coadministration of AM281 and AM630, despite that the significant difference was only observed in the AM281 and PF04457845-treated group when compared to the PF04457845-alone treatment group (Fig. 7B). These results suggested that activation of both CB1 and CB2 receptors was attributable to the therapeutic action of PF04457845 in mice with repetitive closed head injury.
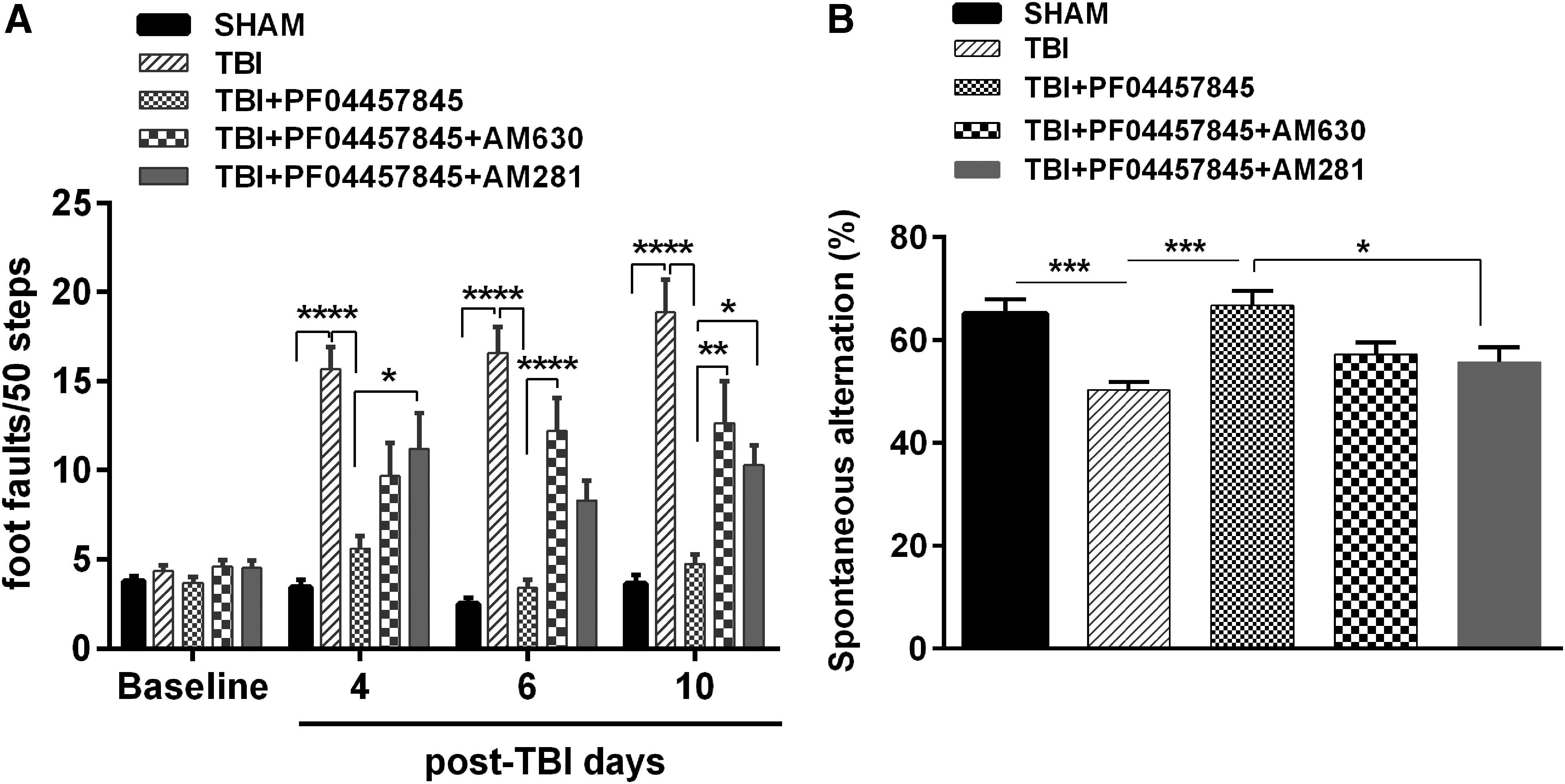
TBI-induced motor and working memory deficits were attenuated by PF04457845 through partial activation of CB1 and CB2 receptors. Mice were treated with PF04457845 (5.0 mg/kg) alone or in combination with AM281 or AM630, and the beam walk and Y-maze tests were conducted to examine the effect of PF04457845 on TBI-induced deficits on motor and working memory. (
Next, we examined whether the spatial learning and memory was affected by TBI and drug treatment. Using the Morris water maze test, the spatial learning and memory were measured based on the performance during a 5-day training trail at days 24–28 post-TBI and a probe test on the following day (day 29 post-TBI) to discern the submerged platform in a water tank. Our results showed that during the 5-day training period, TBI animals traveled a longer distance (Fig. 8A) and had an extended latency (Fig. 8B) to find the submerged platform compared to the sham control animals, but there was no difference in the swimming speed among sham, vehicle-, and drug-treated groups (data not showed). TBI-induced differences in the swimming distance and latency were reversed in the PF04457845-treated animals. The improved learning ability by PF04457845 treatment was reversed by the CB1 receptor antagonist, AM281, but not the CB2 receptor antagonist, AM630 (Fig. 8A,B). In the probe trial, the number of times a TBI mouse crossing the hidden platform region were counted. Animals in the TBI/vehicle group crossed the hidden platform less frequently than the sham and PF04457845-treated animals. Cotreatment with AM281, but not AM630, showed a tendency to hinder the effect of PF04457845 on the frequency of platform crossing in the probe trial (Fig. 8C). These findings suggested that treatment with PF04457845 improved the long-term learning and memory, and the therapeutic effect is likely mediated by activation of CB1 receptors.
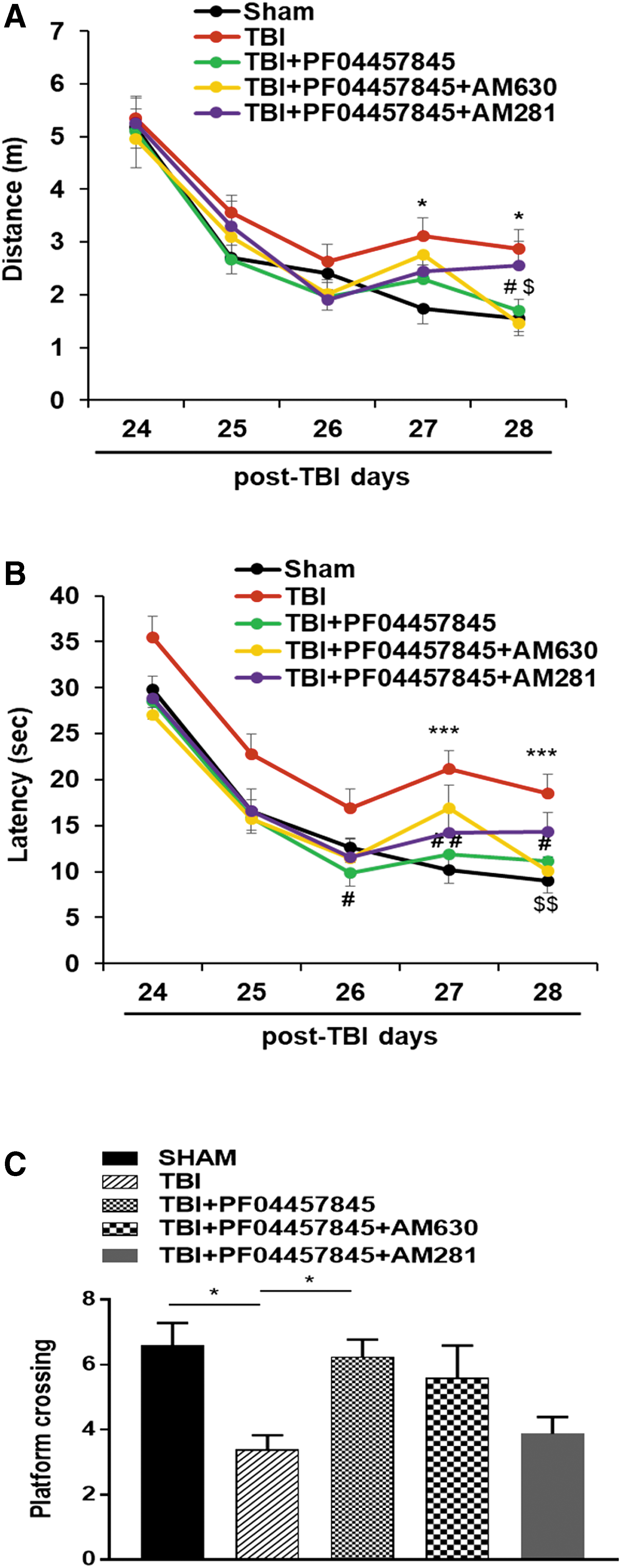
Treatment with PF04457845 improved spatial learning and memory in TBI mice. (
Treatment with PF04457845 attenuated the traumatic brain injury–induced reduction of the synaptic proteins
Studies have indicated that temporary or permanent memory loss is observed in many head injury patients, 38 and it may be caused by the impairment of synaptic plasticity in hippocampus. 39 Synaptic components have been shown to be highly vulnerable to damage, and the alteration varies from mild-to-severe TBI in different animal model systems and clinical settings. 40,41 We examined the expression of several pre- and post-synaptic proteins in hippocampus on day 30 post-TBI. The pre-synaptic proteins, such as synaptophysin, SNAP25, and α-CSP, showed a significant reduction in the TBI vehicle compared to the sham control, and these changes were normalized in the PF04457845-treated animals. The increased expression of synaptophysin by PF04457845 was significantly reversed by coadministration of the CB1 receptor antagonist, AM281, although the effect of PF04457845 on the expression of SNAP25 and α-CSP was cannabinoid receptor independent. Notably, the expression of the post-synaptic protein, PSD95, was not changed in control, TBI, and drug treatment groups (Fig. 9).

Expression of pre-synaptic proteins was reduced in TBI and reversed by PF04457845 treatment. The expression of synaptophysin, SNAP-25, α-CSP, and PSD95 in hippocampal tissue lysate at 30 days post-injury was performed by western blot analysis. Quantification of these images showed that the expression of synaptophysin, SNAP-25, and α-CSP was dramatically reduced in TBI animals compared to sham and reversed in the PF04457845 treatment group. The improvement of PF04457845 on the expression of synaptophysin was reversed by coadministration of the CB1 receptor antagonist, AM281, but not the CB2 receptor antagonist, AM630. The drug effect on the expression of SNAP25 and α-CSP was cannabinoid receptor independent (*p < 0.05 and **p < 0.01; n = 5 animals in each group). Bars represent SEM. CB, cannabinoid; α-CSP, cysteine string protein alpha; PSD95, post-synaptic density protein 95; SEM, standard error of the mean; SNAP-25, synaptosome-associated protein of 25 kDa; TBI, traumatic brain injury.
Discussion
In the present study, we investigated the role of FAAH inhibition in the repetitive closed head injury mouse model. Our data showed that the FAAH inhibitor, PF04457845, selectively elevated the endogenous levels of AEA and reversed TBI-induced deficits in locomotion, learning, and memory. Treatment with PF04457845 suppressed accumulation of microglia and astrocytes, the expression of proinflammatory cytokines, and other inflammatory mediators. The increased production of APP, phosphorylated tau, and the reduced expression of the synaptic proteins in TBI mouse brain were also significantly reversed. The therapeutic effects of PF04457845 seem to be mediated, at least in part, by activation of CB1 and CB2 receptors.
Neuroinflammation is an important mechanism of TBI pathogenesis 18,42 that can lead to locomotive and cognitive deficits. 43 PF04457845 treatment significantly reduced the expression of proinflammatory cytokines/mediators, including IL-6, IL-1β, and TNF-α, in the TBI mouse cortex. The expression of COX-1 and mPGES1 was increased, consistent with the notion that COX-1 is inducible in the activated microglia and macrophages. Several studies have shown that COX-1 is primarily expressed in microglia and actively involved in brain injury induced by proinflammatory stimuli, including amyloid-beta (Aβ), lipopolysaccharide, and interleukins. 44 –50 It has been reported that the clusters of COX-1–expressing microglia are detected around Aβ plaques in the Alzheimer's disease (AD) patient's brain. 51 A recent study in 3 × Tg-AD mice has also demonstrated that selective COX-1 inhibition can reduce neuroinflammation, neuropathology, and improve cognitive function. 51 Several studies have reported that the expression of COX-2 is elevated at the early stage after TBI injury and persists for an extended period of time. Such increased COX-2 levels have been associated with TBI pathogenesis, 52,53 and its inhibition with carprofen was shown to suppress neuroinflammation, improve neurological function, and induce gliogenesis in TBI animals. 54 Despite our current data showing that the mRNA expression of COX-2 and mPGES2 in the ipsilateral cortex was decreased at 4 days post-TBI, it is possible that the expression and function of COX-2 can be spatially and temporarily elevated, and therefore inhibition of arachidonic acid metabolism by both COX-1 and COX-2 may elicit protective effects. The close association of Tau phosphorylation in the ipsilateral cortex and hippocampus with the accumulated microglia and astrocytes suggests that the exaggerated inflammatory response might contribute to the phosphorylation of Tau and neuronal toxicity. Studies have shown that the expression of ATF3 is upregulated in response to injury, and it is closely linked to the survival and regeneration of axons following axonal damage. 29,55 The mRNA expression of ATF3, COX-1, and pTau proteins was increased in the TBI mouse brain and reduced by PF04457845 treatment, suggesting that PF04457845 treatment can mitigate inflammatory and neurodegenerative response after brain injury. These results are consistent with our previous findings showing that the FAAH inhibitor, PF-3845, attenuates inflammation and promotes neuronal survival in TBI mice induced by controlled cortical impact. 12 A very recent study also showed that treatment with the FAAH inhibitor, PF3845, alleviated the nitrergic and proinflammatory response in the rat hippocampus after acute stress. 56
Studies conducted in the post-mortem brain tissues of TBI patients and in the experimental animal models of TBI revealed the interplay between activated astrocytes, microglia/macrophages, and neuronal cell death. 57,58 Many studies have indicated that microglia are chronically activated after the initial brain trauma and contribute to the development of neurodegeneration and related neurological deficits. 59 Depending on their activation stages, the activated astrocytes and microglia/macrophages can be deleterious or beneficial in the context of amyloid deposition at the cerebral regions. 60 It has been reported that AD patients screened with amyloid deposition also show an increased level of phosphorylated tau, suggesting that amyloid deposition is functionally linked to tau phosphorylation. 61 In agreement to the findings using immunohistochemistry, the Tau phosphorylation examined by western blot was also significantly increased in the cerebral cortex at 8 days after TBI, and, concomitantly, the expression of APP was also greatly enhanced. Aβ peptide derived from APP is known to play a pivotal role in the pathogenesis of neurodegeneration. 62 We found a significant number of neuronal cell death in the cortex and dentate gyrus at 8 days post-TBI. The hyperphosphorylation of tau can disrupt microtubule assembly of neurons and lead to tau protein aggregation and neurofibrillary tangles formation. The formation of these abnormal protein aggregates causes neurodegeneration and results in cognitive impairments. 63,64 Our data showed an increased tau phosphorylation at serine 404 (S404), GSK3β phosphorylation at tyrosine 216 (Y216), and an elevated production of p35/p25 in the TBI mouse cortex. It has been demonstrated that GSK3β phosphorylation at Tyr216 can trigger tau phosphorylation at ser404 (active tau), result in the buildup of abnormal APP, and the disturbance of normal signaling cascades. 65 Studies also showed that CDK5 regulator subunit p25 was found to be elevated in the AD brain. 66 The abnormal function of these signaling molecules and the increased APP expression can also impede the hippocampal neurogenesis and further deteriorate the cognitive performance. 34 Notably, inhibition of AEA metabolism by the FAAH inhibitor, PF04457845, significantly mitigated the harmful factors involved in the TBI pathogenesis.
Patients with repetitive TBI appear to have cognitive deficits and are more likely to develop CTE and AD late in life. 67 Accumulating evidence indicates that repeated mild head trauma can lead to cumulative damage to hippocampus. 68 Our mice model also displayed behavioral deficits in learning and memory, suggesting that the function of hippocampus is compromised. It has been reported that cognitive defects are associated with loss of synaptic transmission and plasticity in hippocampus. 18 Consistently, we found a reduced expression of several SNARE complex proteins, such as synaptophysin, SNAP25, and α-CSP, in hippocampus at 30 days post-TBI. It has been reported that mice with a targeted gene deletion of synaptophysin exhibit behavioral and learning deficits, 69 further strengthening the common belief that the impairment in the pre-synaptic proteins is associated with TBI induced impairment on cognition. Our findings are also consistent with a recent report showing the loss of SNARE protein complex and the impairment of cognitive function in the fluid percussion–induced TBI mouse model. 40 Augmentation of the endogenous levels of AEA is able to normalize the expression of synaptic proteins in the TBI mouse hippocampus and restore the locomotor and cognitive function after brain injury.
Although blockade of the endocannabinoid hydrolysis is shown to be protective in several neurodegenerative disorders, the involvement of cannabinoid receptors is uncertain and is likely dependent upon the disease or model systems and cell types involved. 70 –72 By coadministration of CB1 and CB2 receptor antagonists, we found that the improvement of PF04457845 treatment on locomotor function and working memory is mediated, at least in part, by activation of both CB1 and CB2 receptors. However, the improvement of PF04457845 on spatial learning and memory seems to be attributable to the activation of CB1, but not CB2, receptors. Similarly, we found that the reduced expression of synaptophysin at 30 days post-TBI was also blocked by PF04457845 in a CB1-receptor–dependent manner, but the improvement on the expression of SNAP25 and α-CSP by PF04457845 treatment was not affected by the presence of either CB1 or CB2 receptor antagonists. Similar to our finding, it has been reported that treatment with ultralow dose of tetrahydrocannabinol in a lipopolysaccharide-induced sub brain damage mouse model improves cognitive function in a CB1 receptor- and peroxisome proliferator-activated receptor (PPAR)-γ–dependent manner. 73 These results suggest that the cannabinoid receptor–independent mechanisms, such as activation of transient receptor potential channels, PPARs, and other G-protein–coupled receptors, 74 –77 might be also involved in executing the therapeutic effects of PF04457845 in this model system.
Conclusion
Our findings demonstrated that boosting the endogenous levels of AEA is protective in the repetitive closed head injury mouse model. Selective inhibition of FAAH by PF04457845 elevated the AEA levels in the TBI mouse brain and attenuated neuroinflammation, neuronal death, and other signaling molecules that facilitate the chronic neurodegenerative process. Furthermore, our study also indicated that both CB1 and CB2 receptors are involved in the action of PF04457845 on improving the motor and cognitive function after TBI. These results suggest that selective inhibition of FAAH may have the therapeutic potential for patients with repetitive TBI.
Footnotes
Acknowledgments
This work was funded by the Department of Defense in the Center for Neuroscience and Regenerative Medicine (CNRM) (308049-14.01-60855). The authors thank Ms. Laura Tucker and Dr. Amanda Fu from the Center for Neuroscience and Regenerative Medicine for their help during behavioral tests and repetitive closed head injury surgery. We are also indebted to Dr. Sean Moran from the Biomedical Instrumentation Center for performing LC-MS/MS analysis.
Author Disclosure Statement
No competing financial interests exist.