
Abstract
Recent studies indicate that circular ribonucleic acids (circRNAs) are involved in a variety of human diseases. The roles of circRNAs in traumatic spinal cord injury (SCI) remain unknown, however. We performed RNA-seq to analyze the circRNA expression profile in rat spinal cord after SCI and to investigate the relevant mechanisms. In all, 150 circRNAs were significantly differentially expressed in rat spinal cord after SCI by a fold-change ≥2 and p value ≤0.05. Among these, 99 circRNAs were upregulated, while 51 were downregulated. Gene ontology, Kyoto Encyclopedia of Genes and Genomes pathway analyses, and circRNA/miocroRNA (miRNA) interaction networks were conducted to predict the potential roles of circRNAs in the process of SCI. In addition, the expression levels of six selected circRNAs were verified successfully by quantitative real-time polymerase chain reaction. Further study identified circRNA_07079 and circRNA_01282 as being associated with SCI, and they may participate in the pathophysiology of SCI through circRNA-targeted miRNA-messenger RNA axis. In summary, the results of our study revealed the expression profiles and potential functions of differentially expressed circRNAs in traumatic SCI in rats; this may provide new clues for studying the mechanisms underlying SCI and also present novel molecular targets for clinical therapy of SCI.
Introduction
Traumatic spinal cord injury (SCI) is one of the most devastating injuries that are caused mainly by traffic accidents or high falls and often results in motor impairment, sensory deficit, or autonomic nervous system dysfunction. 1 Although considerable effort has been made to improve the outcome of patients with SCI, few effective therapies targeting neurologic regeneration and functional recovery are available because of the limited understanding of the pathogenic mechanism of SCI. 2,3
Recent studies have shown that dysregulation of non-coding RNAs (ncRNAs), including long non-coding RNAs (lncRNAs) and microRNAs (miRNAs) after SCI are involved in the critical processes of SCI physiopathology, 4,5 and they could become potential targets for SCI therapy. 6,7 For the other classes of ncRNAs, however, their modes of action and physiological functions in SCI still remain to be elucidated.
Circular RNAs (circRNAs) are a class of endogenous non-coding RNAs that are characterized by covalently closed loop structures with neither 5′ to 3′ polarity nor a polyadenylated tail. 8 CircRNAs are derived mainly from exons of protein coding genes via “backsplicing” and exhibit cell-type or tissue-specific expression. 9 Recent studies have revealed that circRNAs are expressed widely in human cells and have important roles in the regulation of gene expression at the post-transcriptional level. 8,9
Presently, accumulating evidence indicates that circRNAs are associated closely with certain human diseases, such as diabetes mellitus, 10 acute myeloid leukemia, 11 osteoarthritis, 12,13 and several types of cancers, 14,15 and two independent articles report that circular RNA expression profiles also change significantly after traumatic brain injury. 16,17 The roles of circRNAs in traumatic SCI remain unknown, however.
In the present study, we used RNA sequencing (RNA-seq) to examine the expression profiles of circRNAs in rat spinal cords after SCI. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses based on the parental genes of differentially expressed circRNAs and circRNA/miRNA interaction networks were performed to predict the potential role of circRNAs in the process of SCI. The expression levels of several differentially expressed circRNAs were further validated by quantitative real-time polymerase chain reaction (qRT-PCR).
We also identified circRNA_07079 and circRNA_01282 as being associated with SCI, and they may participate in the pathophysiology of SCI through circRNA-targeted miRNA-messenger RNA (mRNA) axis. This study revealed the expression profiles and potential functions of differentially expressed circRNAs in traumatic SCI in rats and may provide new clues for studying the mechanisms underlying SCI and present novel molecular targets for clinical therapy of SCI in humans.
Methods
Animals
Adult male Sprague-Dawley rats weighing 200 to 220 g were used. Animals were maintained for at least seven days before the experiment in a temperature-regulated (23–25°C) and humidity-controlled (50% relative humidity) room on a 12 h light/dark cycle. Food was withheld, but the rats were allowed free access to water overnight before the surgical procedure. All experiments were performed in accordance with the guidelines of the Animal Ethics Committee of the Second Military Medical University (Shanghai, China).
Establishment of the SCI model
In this study, the SCI model was induced following the method described by Gruner. 18 Briefly, rats were anesthetized with an intraperitoneal injection of pentobarbital (1%, 35 mg/kg body mass). After anesthesia, a laminectomy was performed at the T9 level to expose the spinal cord without disrupting the dura. A MASCIS impactor was used, and the exposed dorsal surface of the cord was subjected to the impact of a dropped weight using a 10 g rod released from a height of 25 mm. The lesion severity was measured and verified by the impact velocity of the impact rod immediately after injury.
Animals with an impact velocity error of no more than 5% were selected for further study. The sham-operated animals received the same surgical procedures, but no impact was applied to the spinal cord. For rats that were housed more than 24 hours, bladders were expressed three times daily until spontaneous voiding occurred.
RNA isolation
To model the acute events, rats (n = 3 per group) were anesthetized and sacrificed for further RNA-seq analysis at 6 h after SCI and immediately after the sham operation. For RNA verification, rats (n = 6 per group) were anesthetized and sacrificed at a different time point after SCI. The spinal cords were harvested as described previously. 19 Briefly, a 1.5-cm segment of the spinal cord centered around the injury site of the SCI group and the same segment of the spinal cord of the sham group were harvested, rapidly frozen, and stored in liquid nitrogen. Total RNA was isolated from the different group samples using Trizol reagent as directed by the manufacturer. The RNA concentration was determined by OD260/OD280 using a NanoDrop ND-1000 spectrophotometer. The integrity of the RNA was assessed by electrophoresis on a denaturing agarose gel.
RNA-seq
Ribosomal RNA was removed by the RiboMinus Eukaryote Kit (Qiagen, Valencia, CA) before the construction of the RNA-seq libraries. The libraries were constructed using the VAHTS Total RNA-Seq (H/M/R) Library PrepKit for Illumina (Vazyme, Nangjing, China) according to the manufacturer's instructions.
Briefly, 3 μg samples of ribosome-depleted RNA were purified using RNA purification beads and fragmented. After fragmentation, random hexamer primers were used to synthesize first- and second-strand cDNA. The cDNA fragments were then adenylated at the 3′ ends and ligated to sequencing adapters. Last, the purified cDNA fragments were evaluated using the Bioanalyzer 2100 (Agilent, Santa Clara, CA). The libraries were then sequenced on an Illumina HiSeq X Ten system, and 150 bp paired-end reads were generated.
Identification of circRNAs
The sequencing results were processed further, and new circRNAs were predicted using CIRI software. 20 Briefly, the raw reads from the RNA-seq libraries were mapped to the rat reference genome (Rattus_norvegicus.Rnor_6.0), and for the unmapped reads, 20-nucleotide (nt) anchors were extracted from both ends of the reads. Anchors that aligned in the reverse orientation (head-to-tail) indicated a back-spliced junction. Relative circRNA abundance was measured by the total number of reads supporting a particular back-spliced junction. We further calculated the SRPBM (spliced reads per billion mapping) for each circRNA to quantitate its expression level. The fold-changes (FCs) of circRNA expression in each sample were also estimated based on the SRPBM.
The p value significance threshold in multiple tests was set by the false discovery rate. The differentially expressed circRNAs were selected using the following filter criteria: p value ≤0.05 and FC ≥2. Scatter plot generation and hierarchical clustering were performed to assess and distinguish the variation in circRNA expression between the SCI group and the sham control group.
Bioinformatics analysis
As reported previously, 8 circRNAs are transcribed alternatively from the exons or introns of their parental genes and could regulate the expression of these genes via a feedback mechanism. We therefore analyzed the potential functions of the parental genes of differentially expressed circRNAs using DAVID (Database for Annotation, Visualization and Integrated Discovery).
The parental gene function was then predicted by GO functional annotation. Gene function is divided into three domains: “biological process” (BP), “cellular component” (CC), and “molecular function” (MF). The related pathways of the parental genes of the differentially-expressed circRNAs were analyzed by KEGG. The results of the KEGG analysis are presented in a scatter plot.
CircRNA/microRNA interactions were predicted with the Arraystar homemade miRNA target prediction software based on TargetScan 21 and miRanda. 22 The circRNA-miRNA coexpression network was constructed based on the correlation analysis between the differentially expressed circRNAs and miRNAs. To establish a circRNA-miRNA network, the putative target genes of these miRNAs were identified by Targetscan. A circRNA-miRNA-gene network was generated to visualize the interactions using Cytoscape.
Quantitative qRT-PCR analysis
The qRT-PCR was performed to verify the accuracy of the circRNA-seq data. The target RNA and internal parameters of each sample were subjected respectively to RT-PCR analysis on an ABI PRISM® 7500 Sequence Detection System (Applied Biosystems, Forster, CA). Total RNA was reverse-transcribed to synthesize first-strand cDNA using a Prime Script RT Reagent Kit (Perfect Real Time; TaKaRa, Osaka, Japan). The data were analyzed by the 2−ΔΔCT method.
The qRT-PCR amplifications were performed as follows: 95°C for 10 min, followed by 40 PCR cycles of 95°C for 10 sec and 60°C for 60 sec (fluorescence emission collection). After the amplification reaction was finished, a denaturation procedure was performed as follows: 95°C for 10 sec, 60°C for 60 sec, and 95°C for 15 sec. The temperature was increased slowly from 60°C to 99°C (ramping rate of 0.05°C/sec) to establish the melting curve of the PCR products. The PCR primers used to amplify the six selected circRNAs, miRNAs, and the internal parameters of the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) control gene are shown in Supplementary Table 1.
Statistical analysis
All statistical analyses were performed in SPSS 21.0. Data were expressed as the mean ± standard deviation (means ± SD). Student t test was used to assess differences between the two groups. A p value <0.05 was considered to be a significant difference.
Results
Identification of differentially expressed circRNAs in the rat spinal cord after SCI
The RNA-seq was used to study circRNA expression profiles in the spinal cord after SCI. The scatter plot identified differentially expressed circRNAs at different p values and FC between two groups (Fig. 1A), and hierarchical clustering revealed that circRNA expression levels were distinguishable (Fig. 1B). Compared with the sham group, there were 150 significantly differentially expressed circRNAs (FC ≥2 and p value ≤0.05) in the spinal cord after SCI, of which 99 circRNAs were found to be up-regulated and 51 circRNAs were down-regulated (Supplementary Data 1). The top 10 up- and down-regulated circRNAs are listed in Supplementary Table 2 by log2 FC.
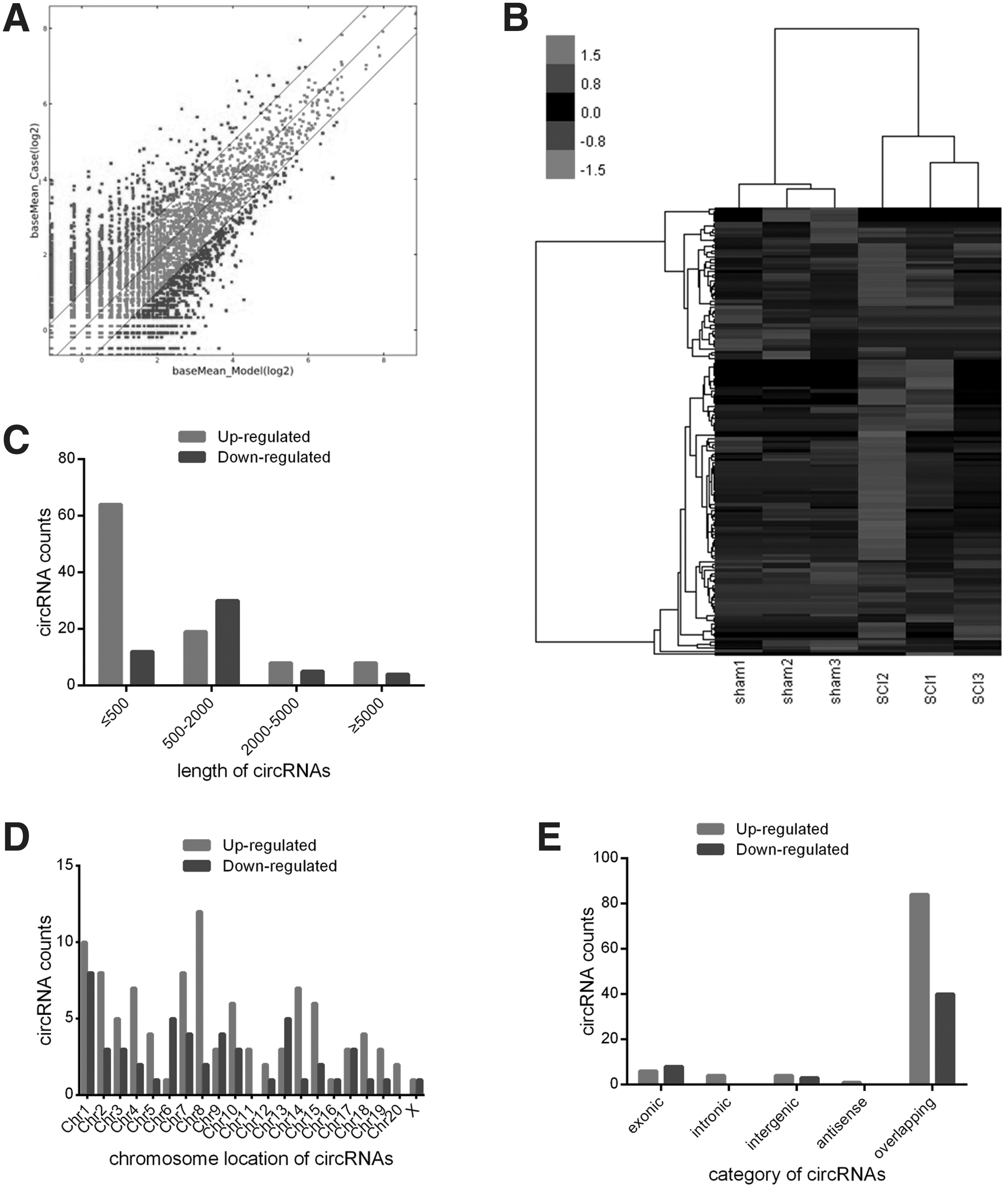
Differential expression profiles of circular ribonucleic acids (circRNAs) in the rat spinal cord after spinal cord injury (SCI). (
Approximately 83% of the 150 identified circRNAs are <2000 nts in length (Fig. 1C). The distributions of differentially expressed circRNAs on the rat chromosomes are shown in Fig. 1D. The up-regulated circRNAs were transcribed from all chromosomes, while the down-regulated circRNAs were transcribed from all chromosomes except chr11 and chr20.
Based on the sequence structure origin of the circRNAs, they can be divided into five categories, including exonic circRNAs, intronic circRNAs, intergenic circRNAs, antisense circRNAs, and sense overlapping circRNAs. Of the circRNAs that were differentially expressed after SCI, 84 circRNAs were transcribed from the sense overlapping regions, six from the exonic regions, four from the intronic regions, four from the intergenic regions, and one from the antisense region among the up-regulated circRNAs, while for the down-regulated circRNAs, 40 were sense-overlapping circRNAs, eight were exonic circRNAs, and three were intergenic circRNAs (Fig. 1E).
GO and KEGG pathways analysis of the parental genes of the differentially expressed circRNAs
To investigate the potential functions of the circRNAs after SCI, GO analysis and KEGG pathway analysis were conducted based on the parental genes of the differentially expressed circRNAs. The GO analysis was performed on the three major domains: BP, CC, and MF. The top 10 enriched GO entries for the up-regulated and down-regulated circRNAs were ranked and selected by enrichment score (-log10 (p value)).
For the up-regulated circRNAs, the top three enriched GO terms in each domain were “gene silencing by RNA,” “chromatin silencing by small RNA,” and “protein export from nucleus” in BP; “centromeric region,” “P-body,” and “nuclear envelope” in CC; “siRNA binding,” “Rab guanyl-nucleotide exchange factor activity,” and “iron ion binding” in MF (Fig. 2A).
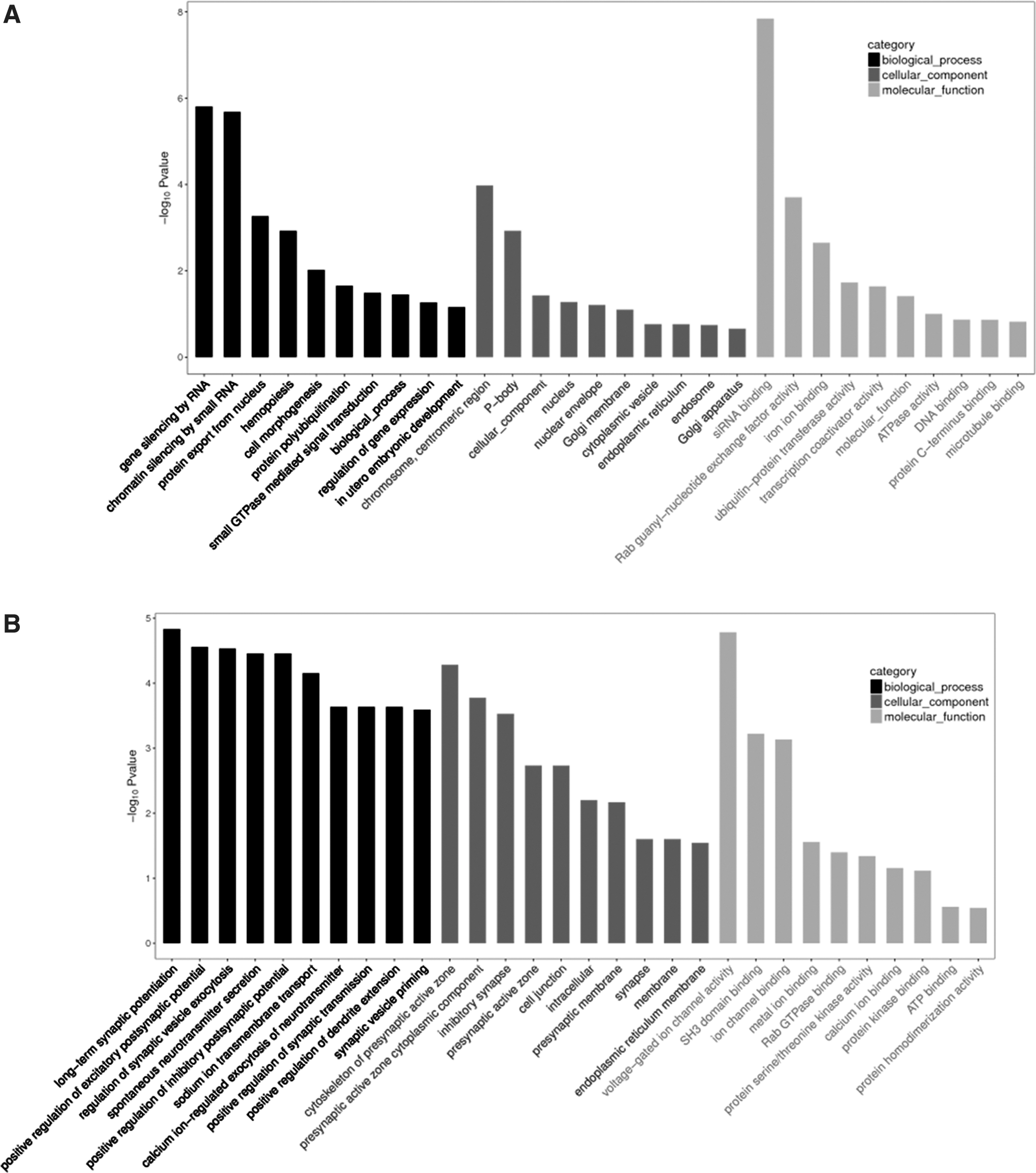
Gene ontology
For the down-regulated circRNAs, the major enriched and meaningful GO terms in BP were “regulation of synaptic vesicle exocytosis,” “spontaneous neurotransmitter secretion,” and “positive regulation of inhibitory postsynaptic potential.” In the CC domain, the majority of the parental genes of the differentially expressed circRNAs were found to be in the “synapse,” “membrane,” and “endoplasmic reticulum membrane” terms. As for MF, the most enriched terms were “protein kinase binding,” “ATP binding,” and “protein homodimerization activity” (Fig. 2B).
The KEGG pathway analysis results showed significantly enriched pathways that might be involved in the physiological and pathological processes of SCI. One hundred sixty-one pathways related to the functions of 150 differentially expressed circRNAs were identified by KEGG analysis (Supplementary Data 2). The top 20 pathways associated with these circRNAs are shown in Supplementary Table 3. Among these pathways, the most enriched and meaningful pathways were related to the peroxisome proliferator-activated receptor (PPAR) signaling pathway and extracellular matrix (ECM)-receptor interaction (Fig. 3A,B).
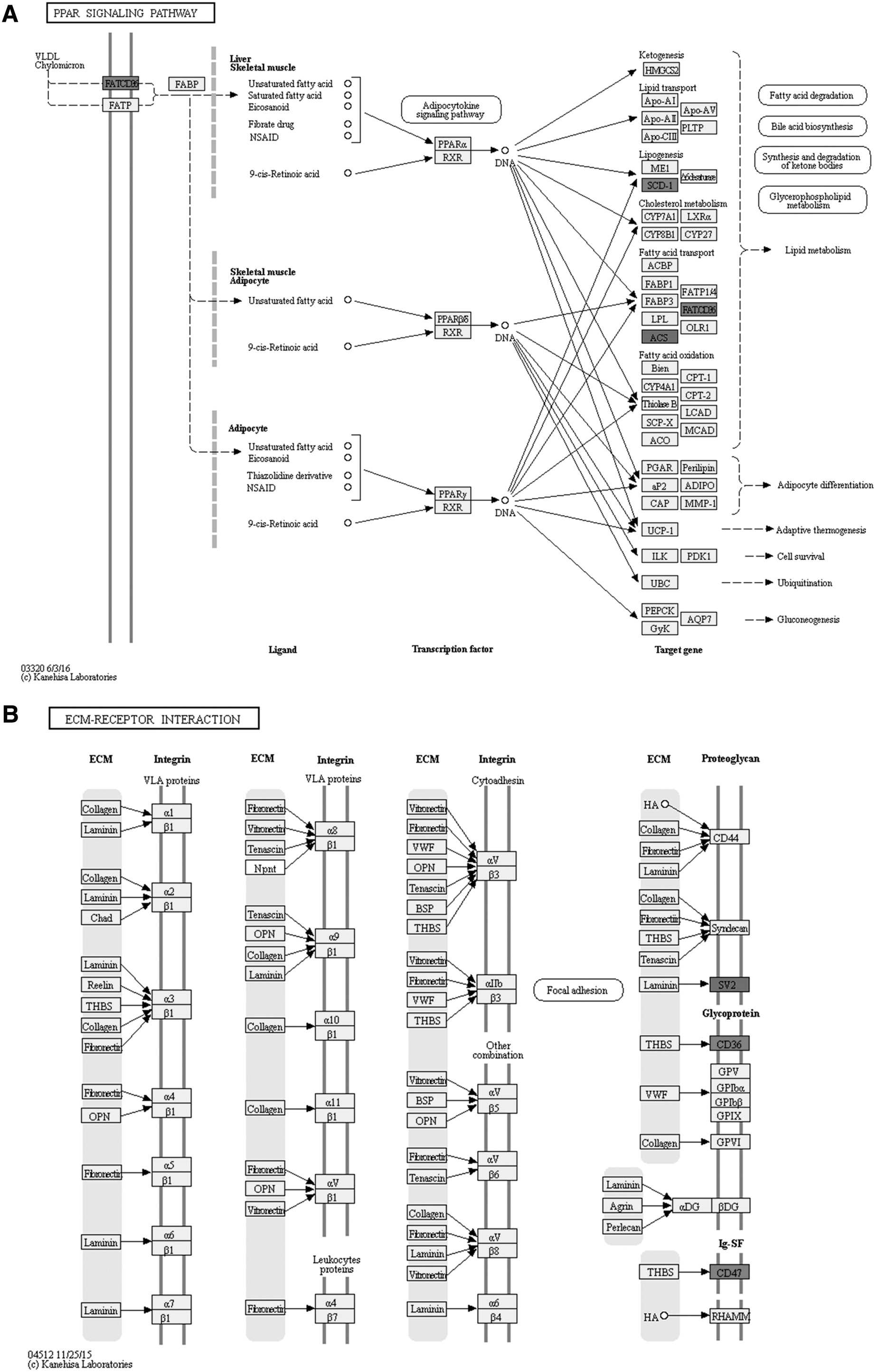
The peroxisome proliferator-activated receptor (PPAR) signaling pathway and extracellular matrix (ECM)-receptor interaction regulated by differentially expressed circular ribonucleic acids (circRNAs). (
Construction of the circRNA/miRNA interaction networks
There is mounting evidence to show that circRNAs can act as miRNA sponges that subsequently regulate the functions of their target mRNAs. To further evaluate the potential functions of circRNAs, the circRNA-miRNA networks were predicted based on the TargetScan and miRanda databases and visualized with Cytoscape. In all, 32 of 150 significantly differentially expressed circRNAs were predicted to have the complementary binding sites for 91 miRNAs (Supplementary Data 3), and a circRNA-miRNA network was constructed to illustrate interactions between circRNAs and their target miRNAs (Fig. 4).
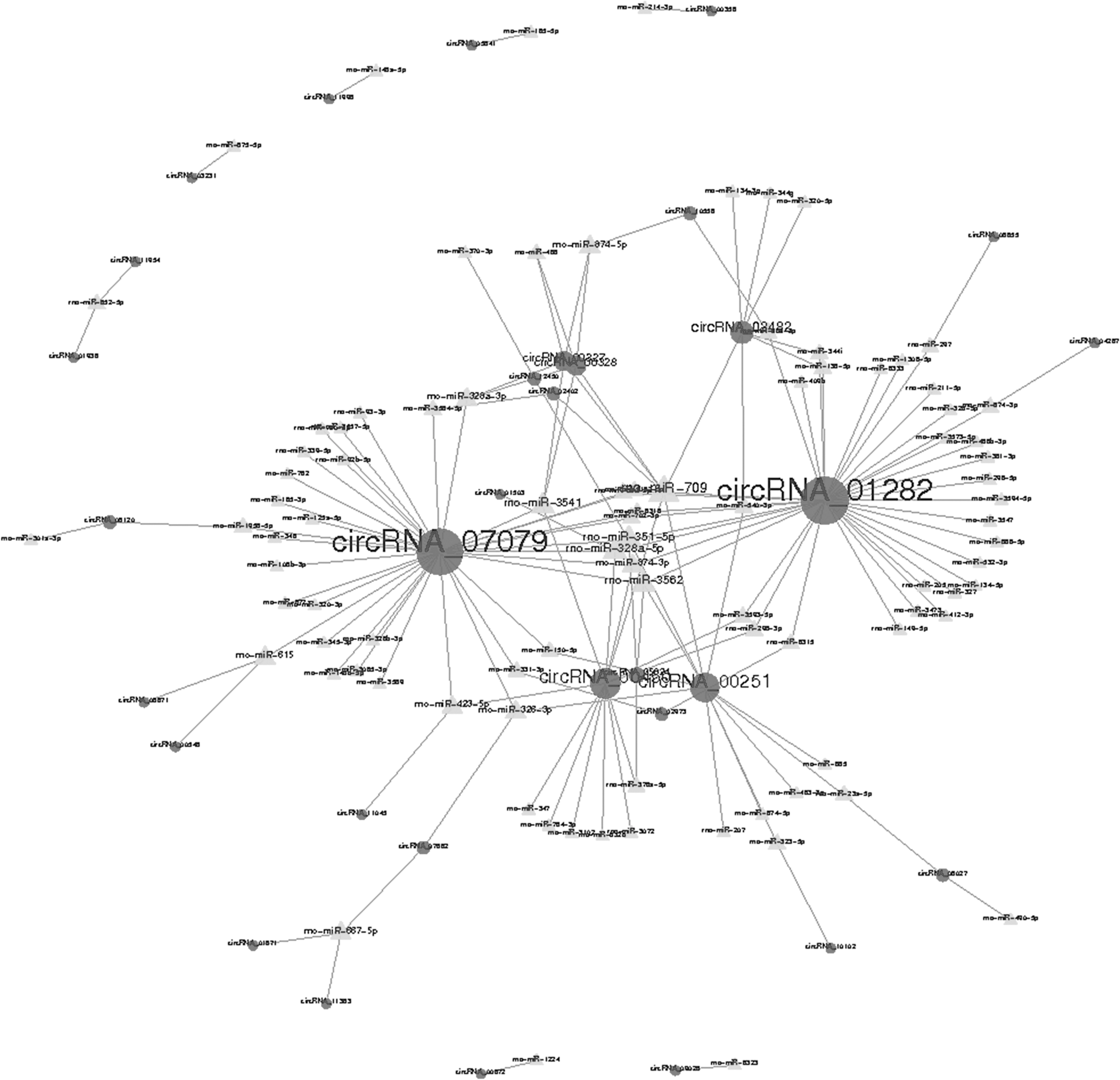
The circular ribonucleic acid/micro ribonucleic acid (circRNA/miRNA) interaction network for differentially expressed circRNAs. The interaction network of the differentially expressed circRNAs (circular nodes) and their complementary binding miRNAs (triangular nodes) was visualized using Cytoscape software.
Among these miRNAs, rno-miR-709, rno-miR-351-5p, rno-miR-328a-5p, rno-miR-3541, and rno-miR-3562 were found to be regulated by a greater number of circRNAs than other miRNAs, and the network showed that circRNA_07079 and circRNA_01282 were predicted to interact possibly with 34 and 36 miRNAs, respectively.
qRT-PCR analysis of the differential expression levels of the circRNAs
Six circRNAs, including two up-regulated circRNAs and two down-regulated circRNAs randomly selected from the group of 150 differentially expressed circRNAs, as well as two circRNAs (circRNA_07079 and circRNA_01282) that are predicted to regulate a large number of miRNAs, were selected to verify the circRNA expression profiles. The expression levels of circRNAs were determined in samples from the sham group and SCI group using qRT-PCR analysis and normalized to the expression of GAPDH.
As shown in Figure 5, circRNA_11070 and circRNA_08027 were significantly up-regulated in the SCI group compared with the sham group, while circRNA_01373 and circRNA_06211 were significantly down-regulated, and the relative expression of circRNA_07079 and circRNA_01282 were significantly up-regulated. The expression patterns of these selected circRNAs agreed with the circRNA-seq results, indicating that the circRNA expression profiles are highly reliable.

Quantitative real-time polymerase chain reaction (qRT-PCR) validation results of selected circular ribonucleic acids (circRNAs). The expression levels of six selected circRNAs (circRNA_11070, circRNA_08027, circRNA_01373, circRNA_06211, circRNA_07079, and circRNA_01282) were compared between microarray and qRT-PCR analysis (n = 6). The Y-axis of the columns in the chart represents the mean of fold change (log2 transformed) of each circRNA.
Prediction and annotation of circRNA_07079/circRNA_01282 targeted miRNA-mRNA network
The expression levels of circRNA_07079 and circRNA_01282, which were verified by qRT-PCR, were significantly increased in the rat spinal cord after SCI. Moreover, predicted splice junction of the selected circRNAs was validated by Sanger sequencing (Fig. 6A), thus confirming the existence of circRNA_07079 and circRNA_01282 in the spinal cord tissues. To further investigate the roles of circRNAs in SCI, we detected dynamic expression profiles of circRNA_07079 and circRNA_01282 in the rat spinal cord after SCI. We found that the expression of circRNA_07079 and circRNA_01282 was gradually up-regulated after SCI and remained a high level after three days (Fig. 6B).
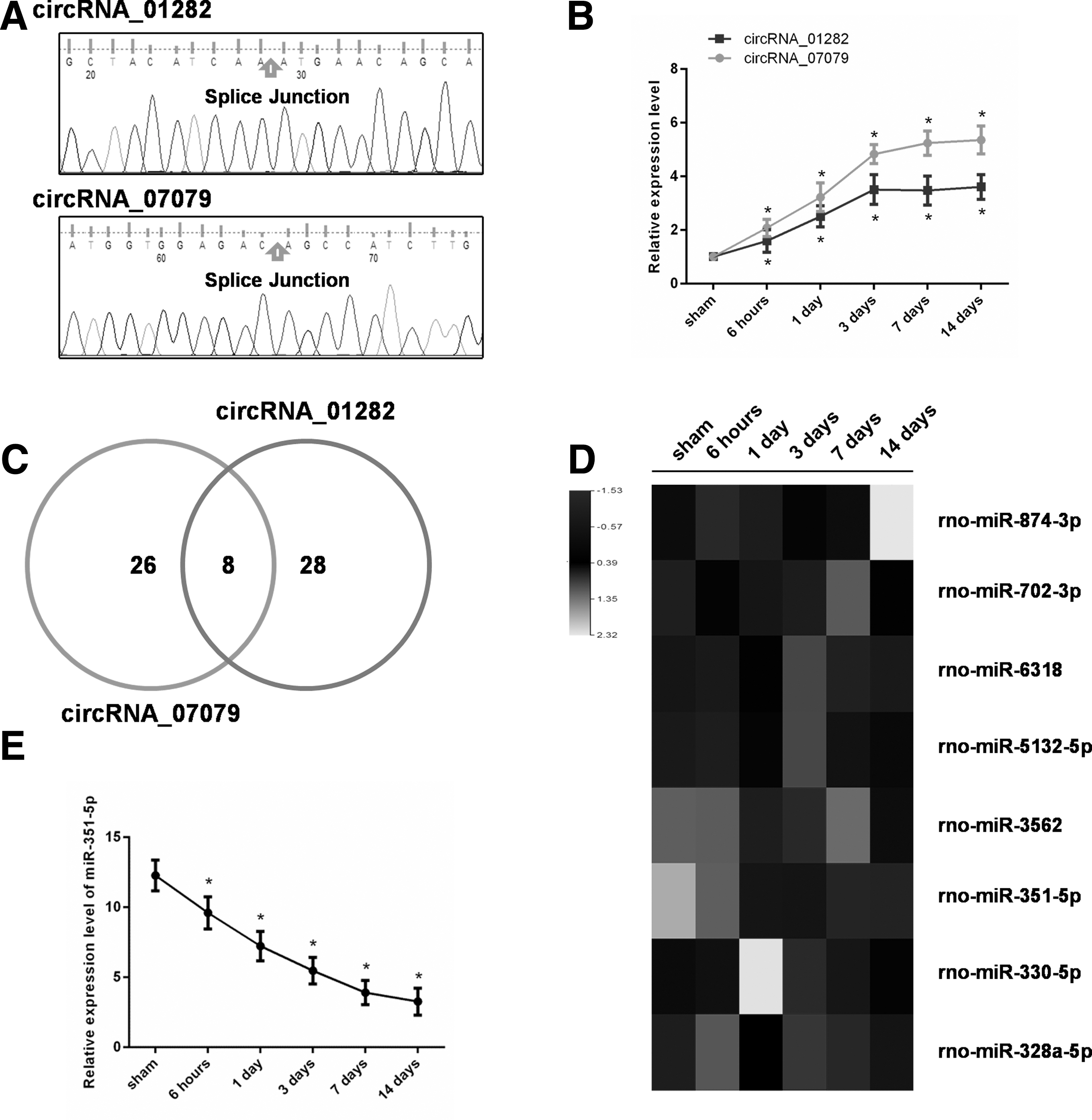
Expression patterns of circular ribonucleic acid (circRNA)_07079, circRNA_01282, and miR-351-5p in the rat spinal cord after spinal cord injury (SCI). (
Then, we focused on analyzing the target miRNAs of these two circRNAs and found that eight miRNAs could be targeted by both circRNA_07079 and circRNA_01282 (Fig. 6C). We detected the expression levels of these miRNAs by qRT-PCR and found that miR-351-5p expression was down-regulated gradually after SCI (Fig. 6D,E), which was negatively correlated with the expression of circRNA_07079 and circRNA_01282, suggesting that it might play a crucial role in the process of SCI.
Because both circRNA_07079 and circRNA_01282 have a complementary binding site for miR-351-5 (Fig. 7A), we assumed that circRNA_07079 and circRNA_01282 act as sponges of miR-351-5 to participate in the pathophysiology of SCI, and then the circRNA-miRNA-mRNA network were predicted by TargetScan and mapped by Cytoscape (Fig. 7B).

Function annotations for target genes mediated by circular ribonucleic acid (circRNA)_07079/circRNA_01282-miR-351-5p-messenger (m)RNA axes. (
To gain further insights into the functions of circRNA_07079 and circRNA_01282, KEGG pathway analyses were utilized based on the predicted results. The top six enriched pathways were shown in Fig. 7C. Among these, the adenosine monophosphate-activated protein kinase (AMPK) signaling pathway and cyclic AMP (cAMP) signaling pathway had been reported to be involved in the pathogenesis of central nervous system injury.
Discussion
Taken together, our results demonstrate that a large number of circRNAs show significant differential expression in the rat spinal cord after SCI. The RNA-seq results showed that 150 circRNAs were differentially expressed in spinal cords from the SCI group compared with the sham-injured control group. Among these, 99 circRNAs were up-regulated and 51 were down-regulated. In addition, we performed GO and KEGG pathway analyses and constructed circRNA/miRNA interaction networks to predict the potential role of circRNAs in the process of SCI.
Moreover, the expression levels of six selected circRNAs were shown to be significantly differentially expressed by qRT-PCR analyses, and the results were consistent with the RNA-seq expression profiles, suggesting that the RNA-seq expression profiles are highly reliable. Further study identified circRNA_07079 and circRNA_01282 as being associated with SCI, and they may participate in the pathophysiology of SCI through the circRNA-targeted miRNA-mRNA axis.
Traumatic SCI is one of the most common and devastating injuries encountered in spine and neurosurgery departments. The SCI is caused usually by traffic accidents, high falls, sports injuries, and violence, and can cause permanent disabilities such as motor impairment, sensory deficit, or autonomic nervous system dysfunction. 1 A multitude of molecular and biochemical changes are involved in the cellular mechanisms of SCI, and many studies have advanced our understanding of the pathophysiology process after SCI.
At present, there is increasing evidence to suggest that ncRNAs, both lncRNAs and miRNAs, play important roles in the progression of SCI. Studies have demonstrated that the expression of lncRNAs and miRNAs changed significantly after SCI, and that dysfunction of certain lncRNAs and miRNAs is associated with the pathophysiological processes of SCI. 6,7 For example, it has been shown that down-regulation of lncRNA-XIST can inhibit neuronal apoptosis by up-regulating miR-494 in SCI rats. 23 LncRNA-Map2k4 can promote FGF1 expression and spinal cord neuron growth by sequestering miR-199a after SCI. 24
Further, microRNA-9 may be involved in the pathobiology of acute SCI by controlling apoptosis in neurons by targeting monocyte chemotactic protein-induced protein 125, and research has shown that lentiviral delivery of miR-133b can improve functional recovery after SCI in mice. 26 To date, however, little attention has been paid to the role of circRNAs in SCI. To the best of our knowledge, this is the first study to demonstrate changes in circRNA expression profiles in response to SCI in rats, and this could be an important first step in understanding the underlying molecular mechanisms involved in SCI.
The circRNAs formerly were considered to be the results of splicing errors that had no biological function. 27 With the development of new sequencing methodologies and bioinformatics tools, however, the biological roles of circRNAs are being revealed and investigated. Recent studies have shown that circRNAs are alternatively transcribed from exons, introns, or other regions of their parental genes, and may participate in feedback mechanisms to regulate their own expression levels. 9,28
To further determine the potential relevance of differentially expressed circRNAs in traumatic SCI, GO and KEGG pathway analyses were performed to predict the functions of the parental genes. The GO enrichment analysis of the down-regulated circRNAs revealed multiple biological functions, such as synaptic vesicle exocytosis, spontaneous neurotransmitter secretion, and positive regulation of inhibitory post-synaptic potential, all of which are related closely to the physiological functions of neurons. These data suggest that the down-regulated circRNAs might play important roles in SCI by mediating the above biological functions.
We also found that most of the up-regulated circRNAs were located in the nucleus, while the down-regulated circRNAs were located predominately in the cytoplasm. Current studies have shown that circRNAs in the cytoplasm may play important roles in post-transcriptional gene regulation by sponging miRNAs or RNA-binding proteins, 9,29 while the intranuclear circRNAs function mainly as cis-inducers of host-gene transcription. 30 Our data indicate that the up-regulated circRNAs and down-regulated circRNAs might function in the pathophysiological processes of SCI through different molecular mechanisms.
In addition, KEGG analysis revealed many pathways that are enriched after SCI, such as the PPAR signaling pathway and ECM-receptor interaction. The data presented here show that the differentially expressed circRNAs could very well act as regulators via these signaling pathways. Thus, they should be investigated further.
There is emerging evidence to confirm that circRNAs may function as competing endogenous RNAs, thereby modulating the function of miRNAs. 9 For instance, ciRS-7 was reported to be a miR-7 sponge that participates in gastric cancer and colorectal cancer. 31,32 The circ-HIPK3 enhances cell proliferation by interacting with multiple miRNAs, including miR-124. 33
In this study, the circRNA/miRNA interaction network analysis was conducted to explore further the potential functions of the differentially expressed circRNAs. Bioinformatic analyses showed that hundreds of circRNAs and miRNAs are predicted to have interacting relationships among the circRNAs that show changes in expression. An interaction network of the significantly differentially expressed circRNAs was predicted and visualized with Cytoscape.
A large amount of valuable information was provided by this network for the study of circRNAs and their targeted miRNAs. This network also revealed that 32 of 150 significantly differentially expressed circRNAs identified in our study may interact with more than one miRNA, and the majority was predicted to have multiple miRNA-binding sites.
In addition, among the confirmed circRNAs, circRNA_07079 and circRNA_01282 were predicted to interact possibly with 34 and 36 miRNAs, respectively. Given that a large number of binding sites for specific miRNAs and a large number of predicted miRNAs mean more potential function in sponging miRNAs for a certain circRNA, 34,35 all of the data suggests that these circRNAs might be involved in the pathogenesis of SCI by regulating miRNAs.
Previous studies demonstrated that the pathophysiology of SCI is separated commonly into primary and secondary mechanisms 2 : the primary mechanism encompasses the initial injury, whereas the secondary mechanism comprises a cascade of biochemical and cellular processes that are initiated by the primary process. 36 We found that the expression profiles of circRNA_07079 and circRNA_01282, as well as miR-351-5p, a miRNA that predicted to be cotargeted by two circRNAs, were dynamic changed after SCI, indicating they might also play important roles in the secondary mechanism of SCI.
Then a circRNA-targeted miRNA-mRNA network was constructed, and function annotations found that many pathways related to central nervous system injury were enriched—for example, the AMPK signaling pathway and the cAMP signaling pathway. Zhao and associates 37 reported that the SIRT1/AMPK signaling pathway is associated with the neuroprotective effects mediated by resveratrol, a plant polyphenol that activates neural autophagy and inhibits neural apoptosis after SCI. Song and colleagues 38 demonstrated that cAMP signaling pathway-related proteins may play important roles in the pathogenesis and recovery of traumatic brain injury, and Bales and coworkers 39 found that targeting the cAMP signaling pathway could be a promising therapeutic approach for the management of traumatic brain injury.
Therefore, we put forward the assumptions that the circRNA_07079 and circRNA_01282/-miR-351-5p-mRNA axis may be involved in the mechanism of SCI, both the primary and secondary stage. Further research is needed, however, to validate these mechanisms.
Conclusion
Our study has provided the first evidence of differentially expressed circRNAs after acute traumatic SCI of rats, and we were able to make preliminary predictions about the potential functions of these circRNAs by bioinformatics analysis and a circRNA/miRNA interaction network. Further study identified circRNA_07079 and circRNA_01282 as being associated with SCI, and they may participate in the pathophysiology of SCI through the circRNA-targeted miRNA-mRNA axis.
These findings may provide new clues for studying the mechanisms underlying SCI and might present novel molecular targets for clinical therapy of SCI. To further elucidate the role of circRNAs in SCI, however, additional studies will be required to investigate the function and regulatory mechanisms of these differentially expressed circRNAs.
Footnotes
Acknowledgments
This research was supported by the National Natural Science Foundation of China (grant numbers 81701215) and the Shanghai Sailing Program (grant number 17YF1425300). The funders had no role in the study design, data collection, data analysis, decision to publish, or manuscript preparation.
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Table 1
Supplementary Table 2
Supplementary Table 3
Supplementary Data 1
Supplementary Data 2
Supplementary Data 3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.