
Abstract
An enduring deficit in neurogenesis largely contributes to the development of severe post-traumatic psychiatric disorders such as anxiety, depression, and memory impairment following traumatic brain injury (TBI); however, the mechanism remains obscure. Here we have shown that an imbalance in the generation of γ-aminobutyric acid (GABA)ergic and glutamatergic neurons due to aberrant induction of vesicular glutamate transporter 1 (vGlut1)-positive glutamatergic cells is responsible for impaired neuronal differentiation in the hippocampus following TBI. To elucidate the molecular mechanism, we found that TBI activates a transcription factor, Pax3, by increasing its acetylation status, and subsequently induces Ngn2 transcription. This event, in turn, augments the vGlut1-expressing glutamatergic neurons and accumulation of excess glutamate in the hippocampus that can affect neuronal differentiation.
In our study the acetylation of Pax3 was increased due to loss of its interaction with a deacetylase, histone deacetylase 4 (HDAC4), which was downregulated after TBI. TBI-induced activation of GSK3β was responsible for the degradation of HDAC4. We also showed that overexpression of HDAC4 before TBI reduces Pax3 acetylation by restoring an interaction between HDAC4 and Pax3 in the hippocampus. This event prevents the aberrant induction of vGlut1-positive glutamatergic neurons by decreasing the Ngn2 level and subsequently reinforces the balance between GABAergic and glutamatergic neurons following TBI. Further, we found that overexpression of HDAC4 in the hippocampus improves anxiety, depressive-like behavior, and memory functions following TBI.
Introduction
Traumatic brain injury (TBI) is one of the primary causes of mortality, and it affects more than 2.5 million people each year. 1 –4 Along with physical disabilities, a substantial number of TBI survivors experience TBI-related chronic neuropsychological conditions, including depression, anxiety, and memory loss, which can impede quality of life and cause long-term economic burden. 5 –9 Approximately 30% of patients suffering from mild or moderate TBI develop symptoms of depression within 1 year after injury, and this percentage rises to approximately 60% within a few years after injury. 10 –12
A compelling number of studies have suggested that neurogenesis, a dynamic process in the adult hippocampus, is essential for memory formation 13,14 and its proper homeostasis is sufficient to reduce anxiety and depression-like behaviors. 15 –17 Most research on adult neurogenesis has focused on the dentate gyrus (DG) area of the hippocampus, where neural stem cells (NSC) generate neural progenitor cells that differentiate into a neural lineage, which is critical to producing new granular neurons. 18 –20 An aberration in neuronal function in the DG affects the overall process of neurogenesis and results in impairment of memory function, contextual fear conditioning, and anxiety and depressive-like behavior. 13,21
At the molecular level, neurogenesis is regulated by the activation of numerous transcription factors, including the basic helix-loop-helix (bHLH) family of transcription factors, which determine the fate of newly born cells during neurogenesis. 22 –25 Among these factors, a bHLH transcription factor, Ngn2, differentially regulates the balance between progenitor maintenance and differentiation 26 –29 and specifies both the glutamatergic fate 26,28,30 and the γ-aminobutyric acid (GABA)ergic fate of newly generated neurons. 27,31 Glutamate is the primary excitatory neurotransmitter in the brain, whereas GABA is the principal inhibitory neurotransmitter. The balance of glutamatergic and GABAergic tone is crucial to normal neurological function. In some disease models associated with excitotoxicity, there is an increased expression of the vesicular glutamate transporter (vGlut) including vGlut1 32 –34 that results in increased glutamate release and toxicity. 35 –38 Ngn2 can transcribe vGlut, which is responsible for packaging glutamate into synaptic vesicles. Further, expression of Ngn2 can be regulated by a transcription factor, Pax3, and the functions of Pax3 have mostly been deciphered in developing brain and regenerative myogenesis. The activation of Pax3 mostly depends on its status of acetylation 39,40 ; however, the role of Pax3 in adult neurogenesis has not been elucidated yet.
Acetylation of histone and non-histone proteins can be regulated by several deacetylases including a class II HDAC, histone deacetylase 4 (HDAC4), which controls synaptic plasticity, as well as learning and memory. 41,42 Particularly, HDAC4 is implicated in controlling gene expression necessary for diverse cellular functions by mediating the transcriptional repression or interaction with proteins involved in the regulation of gene transcription, cell growth, survival, and proliferation. 43 HDAC4 serves as a nuclear co-repressor through the formation of multi-subunit complexes with other proteins 44,45 that regulate expression of genes in the brain. 45,46 A selective loss of Hdac4 in the brain results in impairments in hippocampal-dependent learning and memory and long-term synaptic plasticity 44 ; however, the molecular mechanism remains to be clarified.
In the present study, we found that aberrant activation of the Pax3-Ngn2 signaling pathway due to loss of HDAC4 led to overproduction of vGlut1-positive glutamatergic neurons and disrupted the balance of glutamatergic and GABAergic neurons upon TBI. The excess glutamate resulting from this cascade affects neuronal differentiation. Overexpression of HDAC4 can restore the balance and rescue the neurobehavioral abnormalities in the hippocampus if administered before TBI.
Methods
TBI procedure
The Committee on Animal Use for Research and Education at the University of Pittsburgh approved all animal studies, in compliance with National Institutes of Health guidelines. The procedures were conducted based on our previously published protocol. 47 –50 Briefly, 8–12-week-old adult male or female C57BL/6 mice (Jackson Laboratory) were anesthetized and subjected to sham or controlled cortical impact (CCI) injury. Mice were placed in a stereotaxic frame (Leica Impact One Controlled Impact Device #39463920 for CCI, Leica Microsystems) and mice were impacted at 4.5 m/sec with a 20-msec dwell time and 1.2-mm depression using a 3 mm in diameter convex tip, mimicking a moderate TBI. Sham-operated mice underwent identical surgical procedures but were not impacted. In another set of experiments, mice were treated with lithium (5mg/kg; intraperitoneal injection) immediately after TBI and treatment continued up to 3 days once per day. Then peri-contusional hippocampus was isolated after 72 h to carry out biochemical studies as described below.
Western blot and co-immunoprecipitation
The peri-contusional hippocampus was isolated and hippocampal lysates were prepared as described before to perform western blot and co-immunoprecipitation (Co-IP). 47 –50 Blots were incubated overnight at 4°C in the primary antibody Doublecortin (Dcx) (Santa Cruz Biotechology, 1:500 dilution), Ngn2 (Santa Cruz Biotechnology, 1:500 dilution), Mash1 (Santa Cruz Biotechnology, 1:500 dilution), vGlut1 (Santa Cruz Biotechnology, 1:500 dilution), HDAC4 (Santa Cruz Biotechnology, 1:500 dilution), and Actin (Sigma-Aldrich, 1:5000 dilution) followed by incubation with a LicorIRDye secondary antibody at room temperature. Blots were visualized using a Li-Cor Odyssey system and ImageJ software determined the intensity of each band, and the changes in the band intensity were represented as the fold change as described previously. 47 –50 For Co-IP, hippocampus collected from all sets of experiments was homogenized in lysis buffer (50 mm Tris, pH 7.4, 150 mM NaCl, 0.5% [v/v] Tween-20, 50 mM Tris [pH 7.5], 1 mM ethylenediaminetetraacetic acid (EDTA) with protease and phosphatase inhibitor) by passage through a 26-gauge needle, and 400 μg of the total protein for each sample was incubated overnight with either Pax3 (1:100) or NRF1 (1:100) antibody. The immune precipitates were resolved and analyzed by western blotting with either the HDAC4 antibody or acetyl-lysine antibody.
Immunohistochemistry
The immunohistochemistry (IHC) was performed per our method as described previously. 47 –50 Seventy-two hour TBI brain samples were subjected to cryosectioning (20-μm thickness) and for immunofluorescence analysis sections were incubated 10 min at room temperature with 0.1% Triton X-100 in phosphate buffered saline (PBS), followed by overnight incubation at 4°C with the primary antibody against Dcx (1:100; Cell Signaling Technology), Mash1 (1:100; Santa Cruz Biotechnology), vGlut1 (1:100; Santa Cruz Biotechology), Flag (1:300, Abcam), or Ngn2 (1:100; Santa Cruz Biotechology). Sections were then washed three times with PBS and incubated with the appropriate Alexa Fluor-tagged secondary antibody for 2 h at room temperature in the dark. Sections were then washed three times with PBS and mounted with Prolong Glodantifade mounting media with DAPI (Invitrogen). Imaging was performed with the help of an Olympus fluorescent microscope (IX83).
Glutamate assay
Amounts of glutamate released during TBI were measured with an Amplex Red Glutamic Acid/Glutamate Oxidase Assay Kit (Molecular Probes, Eugene, OR) as described before 51 with modifications. Briefly, 50 μL of brain tissue lysate were mixed with 50 μL of a reaction buffer containing Amplex red, glutamate oxidase, glutamate pyruvate transaminase, alanine, and horseradish peroxidase. After incubation at 37°C for 30 min, the fluorescence of the reaction mixture emitted at 590 nm was measured every 10 min with a fluorescence microplate reader (Synergy H1 Hybrid Multi-Mode Microplate Reader) at an excitation wavelength of 555 nm.
Chromatin immunoprecipitation assay
The chromatin immunoprecipitation
In vivo transfection of HDAC4 in the dentate gyrus
Polyethyleneimine (PEI) complexes including Flag-HDAC4 (Flag-HDAC4 insert was in the pcDNA3.1 mammalian vector and under the CMV constitutive promoter from Addgene) were injected into the hippocampus as described previously. 52 –55 For in vivo transfection the plasmid was mixed within in vivo-jetPEI transfection reagent at an N/P ratio of 7, as suggested by the manufacturer (PolyPlus Transfection) to final doses of an oligonucleotide of 2.5 μg/mouse. C57BL/6 mice were injected in the DG at bregma: −0.2 AP, −0.13 ML, and 0.2 DV with 5 μL of DNA transfection reagent mix.
Quantitative real-time PCR
Quantitative real-time PCR (qRT-PCR) was performed per our method. 55 Total RNA was isolated using the kit (QIAGEN), and the complementary DNA (cDNA) was subjected to qRT-PCR analysis with fast-standard SYBR green dye (Applied Biosystems) using an AB7500 RT-PCR instrument (Applied Biosystems). Results were normalized using total input DNA and expressed as bound/Input (fold). Ipsilateral hippocampal tissues were taken 72-h post-TBI induction on the mouse brain. The primers used to carry out PCR analysis are as follows: vGlut1 (Forward 5'-TGC CAA GCT AAT CTG TAG GG3', Reverse 5'-GAA TGG GCG AAT CCT AAA AT-3'), Mash1 (Forward 5'-TCAACCTGGGTTTTG CCACC-3', Reverse 5'-TCGTTGGAGTAGTTGGGGGA-3'), Ngn2 (Forward-5'CTGTGGGAATTTCACCTGTC-3', Reverse 5'-CTAAATTTCCACGCTTGCAT-3'), ND1 (Forward 5'CGATTAGAGGCACGTCAGTT-3') Reverse 5'-TTCTTCCAAAGGCAGTAACG-3').
Preparation of primary neurosphere and glutamate treatment
Primary neurosphere culture was prepared from E14.5 C57BL/6 mice as described previously with some modifications. 47 –50 Briefly, the pregnant mouse was anesthetized, and the brain hemispheres were dissected out. Trypsinized tissue was washed and re-suspended in neurosphere complete medium. For differentiation of neurospheres, each neurosphere was re-suspended into neurosphere differential medium, and differentiated cells were treated with 100 μM glutamate. After 24 h, cells were subjected to IHC using MAP2 antibody (1:2000; Novus Biologicals). The quantitation of differentiated processes was measured by Sholl analysis plugin in ImageJ and data have been represented as the distance from soma versus several intersections.
Open field test
Anxiety-like behavior was assessed 2 weeks after TBI using an open field apparatus divided into a 5-cm2 grid by black lines (81 squares in total) as described previously with modifications. 56 –58 Briefly, mice were placed in the center of the open field, and activity was measured and recorded for 5 min. The total movement time and time spent in the center of the open field were analyzed by an investigator who was blinded to experimental conditions.
Spontaneous alternation behavior (Y-maze)
The Y-maze spontaneous alternation behavior (SAB) test, to assess hippocampal-dependent working (short-term) memory, was performed 2 weeks after TBI following previously published protocols with minor modifications. 59 Each mouse was placed at the end of a randomly chosen arm and allowed to explore the maze for 5 min freely. Visits to arms were scored, and an alternation was counted when the mouse visited three different arms consecutively; and the percent correct alternation was calculated as: (total number of alterations/total arm entries – 2) × 100.
Tail suspension test
Mice were individually suspended by the tail on a horizontal bar using adhesive tape placed approximately 4 cm from the tip of the tail as described previously with minor modifications. 60 The duration of immobility was monitored and recorded for 5 min.
Barnes maze
The maze consisted of a circular platform with 20 holes around the periphery (5 cm diameter) with an escape box attached to the bottom of one of the holes, following previous publication with minor modifications. 61,62 Briefly, during a spatial training period of 4 days, mice explored the maze freely for 3 min once per day. The escape latency and errors to find the holes were recorded. During the probe trial conducted 1 day after the last training trial, the escape tunnel was blocked, and mice explored the maze for 90 sec. The time spent around holes along with target was analyzed.
Statistical analysis
The biochemical studies, confocal analysis, Sholl analysis, and all behavioral tests except the Barns maze test were statistically analyzed using one-way analysis of variance (ANOVA) and multiple comparisons were performed using the Tukey-Kramer post hoc test (p < 0.05) unless noted otherwise. Two-way ANOVA with Tukey-Kramer post hoc test was used to assess the Barns maze test. Mean values were calculated for each biochemical experiment (n = 7) and each behavioral experiment (n = 10), and all the datawere depicted as the mean ± standard error of the mean (SEM).
Results
TBI increases vGlut1-positive neurons but decreases Mash-positive neurons, which results in an impairment of neuronal differentiation
To understand whether TBI has any influence on neurogenesis, we monitored the differentiation of newly born neurons by staining with Dcx (a marker of neuronal precursor cells and immature neurons) antibody, 72 h post-TBI, and Dcx-positive cells were counted by ImageJ software. Several studies have convincingly demonstrated that maximum decrease in neurogenesis (both in terms of Dcx+ cells and dendritic integrity) has been identified within 72 h post-TBI. 63 –67 Consistent with previous studies, we found that TBI leads to a decrease in the level of Dcx-positive cells in the hippocampal DG area (Fig. 1A, Supplementary Fig. S1A) indicating that TBI affects differentiation of newly born neurons in the subgranular zone (SGZ) of DG.
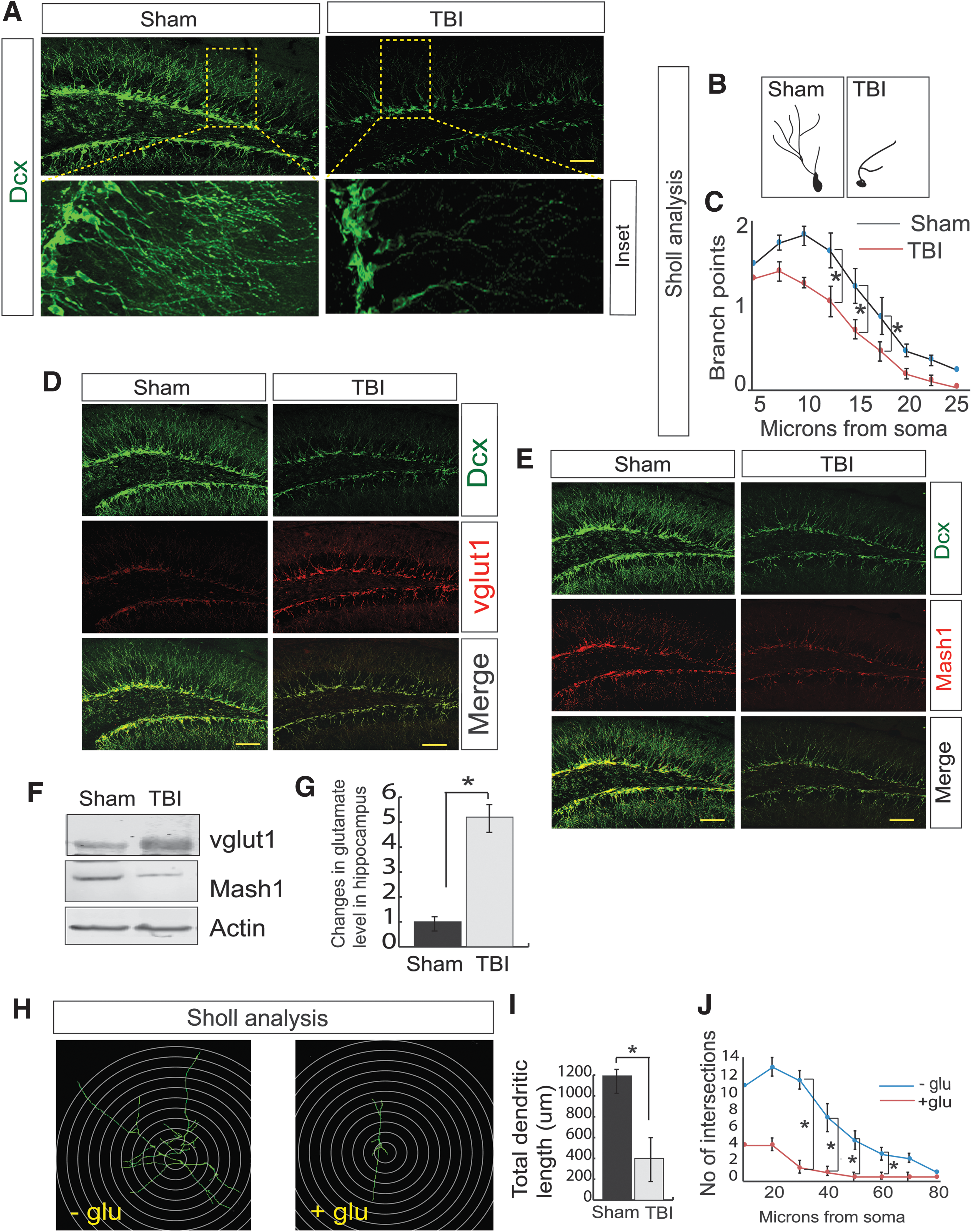
TBI leads to a decrease in neurogenesis in association with an increase in vGlut1-positive cells but a decrease in Mash1-positive cells.
We also performed Sholl analysis to obtain the number of branches (Fig. 1B, Supplementary Fig. S1B), the dendritic length (Fig. 1B, Supplementary Fig. S1C), and the branch points (Fig. 1B,C) of the Dcx-positive cells in the DG of both sham and TBI mice and found that TBI causes a significant decrease in branch points, number, and total dendritic length compared with sham. However, there is no significant decrease in the Dcx-positive cells on the contralateral side of mice 72 h following TBI (Supplementary Fig. S1D). Considering that formation of mature neurons from immature neurons is critical for memory functions, we found that the percentage of NeuN-positive, mature neurons was decreased after TBI (Supplementary Fig. S1E,F) along with a decrease in both Dcx- and NeuN-positive cells (Supplementary Fig. S1E,G) consistent with a reduction in Dcx-positive cells after TBI (Supplementary Fig. S1E,H).
To characterize the type of neurons affected by TBI, we monitored the GABAergic and glutamatergic neuronal progenitor cells by staining with vGlut1 and Mash1 antibody. vGlut1 stains for glutamatergic neuronal progenitor cells, whereas Mash1 indicated the GABAergic cells. We found that TBI leads to a substantial increase in vGlut1-positive cells (Fig. 1D, Supplementary Fig. S1I) compared with Mash1-positive cells in the DG (Fig. 1E, Supplementary Fig. S1K). Similarly, we observed that the number of vGlut1-positive cells within the population of Dcx-positive cells increased (Fig. 1D, Supplementary Fig. S1J), but the number of Mash1-positive cells within the Dcx-positive cell population decreased (Fig. 1E, Supplementary Fig. S1L) after TBI. These data indicate that the fate of Dcx-positive cells was more similar to that of vGlut1-positive cells compared with Mash1-positive cells. To further confirm, the expression of vGlut1 and Mash1 was monitored by western blot assay using the hippocampal tissue extracts from both TBI and sham mice. Consistent with the confocal data, we found that at 72 h post-TBI there was a significant increase in vGlut1 level but a decrease in Mash1 level (Fig. 1F, Supplementary Fig. S1M). Taken together, our data suggest that TBI augments the population of glutamatergic neurons compared with GABAergic neurons.
Previously, it was shown that an increase in the levels of vGlut1 protein and a loss of GABA-producing cells disrupts the balance of excitation and inhibition 68 and results in increased glutamate release. 36 –38,69 To monitor whether an increase in vGlut1- and decrease in Mash1-positive cells can increase glutamate level in the hippocampus, we monitored glutamate level in the hippocampal lysates isolated from both TBI and sham mice. Our data showed that glutamate level was markedly increased in the TBI samples (Fig. 1G) compared with sham. To confirm whether excess glutamate affects differentiation of newly born neurons, we prepared NSC and treated them with 100 μM glutamate. We found that treatment with glutamate decreases total dendritic length (Fig. 1H,I) and the number of intersections (Fig. 1H,J), indicating that neuronal differentiation was affected. Therefore, it is crucial to elucidate the underlying mechanism responsible for an increase in vGlut1 and decrease in Mash1 in the newly born hippocampal neurons following TBI.
TBI upregulates vGlut1 but downregulates Mash1 in Ngn2 dependent manner
To monitor whether the alteration in the expression level of vGlut1 and Mash1 results from the change in the messenger RNA (mRNA) level of vGlut1 and Mash1, we isolated total RNA from the ipsilateral peri-contusional hippocampus of 72-h post-TBI mice and corresponding sham brain samples and subjected it to quantitative RT-PCR analysis. Consistent with confocal and western blot data, we found that the mRNA level of vGlut1 was significantly increased (Fig. 2A), whereas the mRNA level of Mash1 was significantly decreased (Fig. 2B) in TBI compared with sham. These data suggest that TBI regulates vGlut1 and Mash1 in a transcriptional dependent manner.
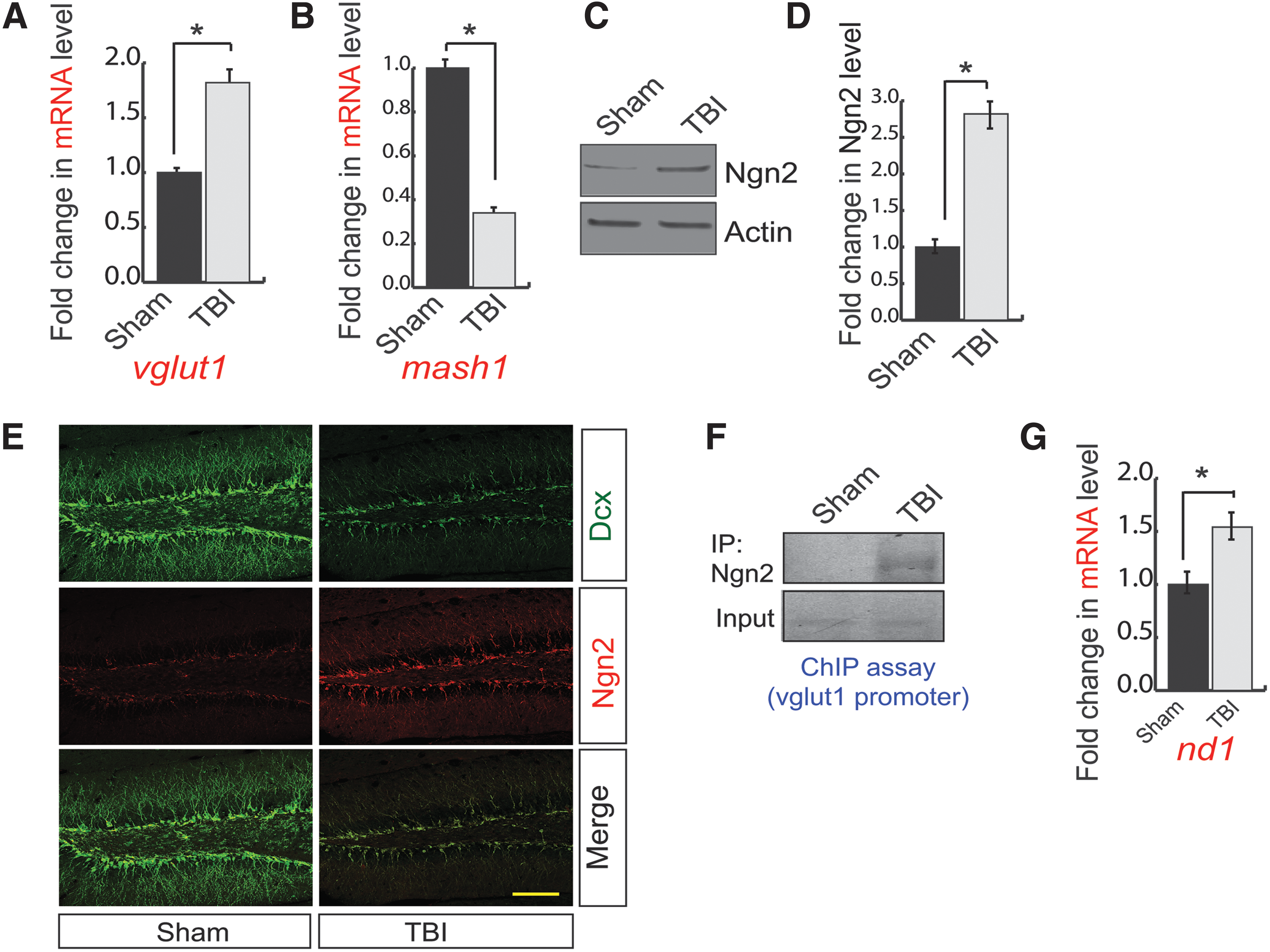
A transcription factor, Ngn2, regulates vGlut1 and Mash1 expression.
Previous literature suggests that a bHLH transcription factor, Ngn2, can transcriptionally regulate both vGlut1 and Mash1 during neurogenesis. 26 –28 We monitored the expression of Ngn2 by western blot analysis using hippocampal lysates isolated from 72-h post-TBI mice and sham mice. We found that the expression of Ngn2 was increased in post-TBI mice compared with sham mice (Fig. 2C,D). To further confirm the expression of Ngn2, we performed confocal microscopy analysis. Further, we counted the number of Ngn2-positive cells out of total Dcx cells, and we found that the expression of Ngn2 (Fig. 2E, Supplementary Fig. S2A) within the Dcx-positive cells (Fig. 2E, Supplementary Fig. S2B) was substantially increased in post-TBI compared with sham. ChIP assay suggests that binding of Ngn2 to the vGlut1 promoter was increased significantly in the hippocampus of 72-h post-TBI mice compared with sham mice (Fig. 2F, Supplementary Fig. S2C). Activation of Ngn2 also leads to an increase in ND1 (neurogenic differentiation factor 1, a neuronal differentiation factor), which in turn suppresses the transcriptional regulation of Mash1. 27,28 We monitored the mRNA level of ND1 using quantitative RT-PCR analysis using total RNA isolated from peri-contusional hippocampus of 72-h post-TBI mice. We found that the mRNA level of ND1 was significantly increased after TBI compared with sham (Fig. 2G). These data can explain how Mash1 level was decreased in the hippocampus following TBI. Taken together, our data suggest that an increase in Ngn2 is responsible for an increase in vGlut1-positive cells but decreases Mash1-positive cells during the neurogenesis. These data raised a question of how TBI upregulates Ngn2 level.
A reduction in HDAC4 triggers Ngn2 expression via acetylated Pax3 following TBI
Consistent with confocal and western blotting data, we also found that TBI leads to an increase in mRNA level of Ngn2 in the hippocampus compared with sham (Fig. 3A). Previously, it was shown that a transcription factor could regulate Ngn2 and Pax3, and activation of Pax3 mostly depends on its status of acetylation. 39,40 Therefore, to understand whether Pax3 transcriptionally upregulates Ngn2 following TBI, we performed the ChIP assay using peri-contusional hippocampal samples from 72-h post-TBI mice. We found that the binding of Pax3 to Ngn2 promoter was increased following TBI (Fig. 3B, Supplementary Fig. S3A). To see whether Pax3 was acetylated after TBI, we monitored acetylation of Pax3 using peri-contusional hippocampal lysates of both 72-h post-TBI and sham mice. We found that the acetylation level of Pax3 was markedly increased in the post-TBI compared with sham mice (Fig. 3C, Supplementary Fig. S3B). Thus, our data suggest that acetylated Pax3 transcriptionally upregulate Ngn2; however, it is not fully understood how the acetylation of Pax3 was increased following TBI.
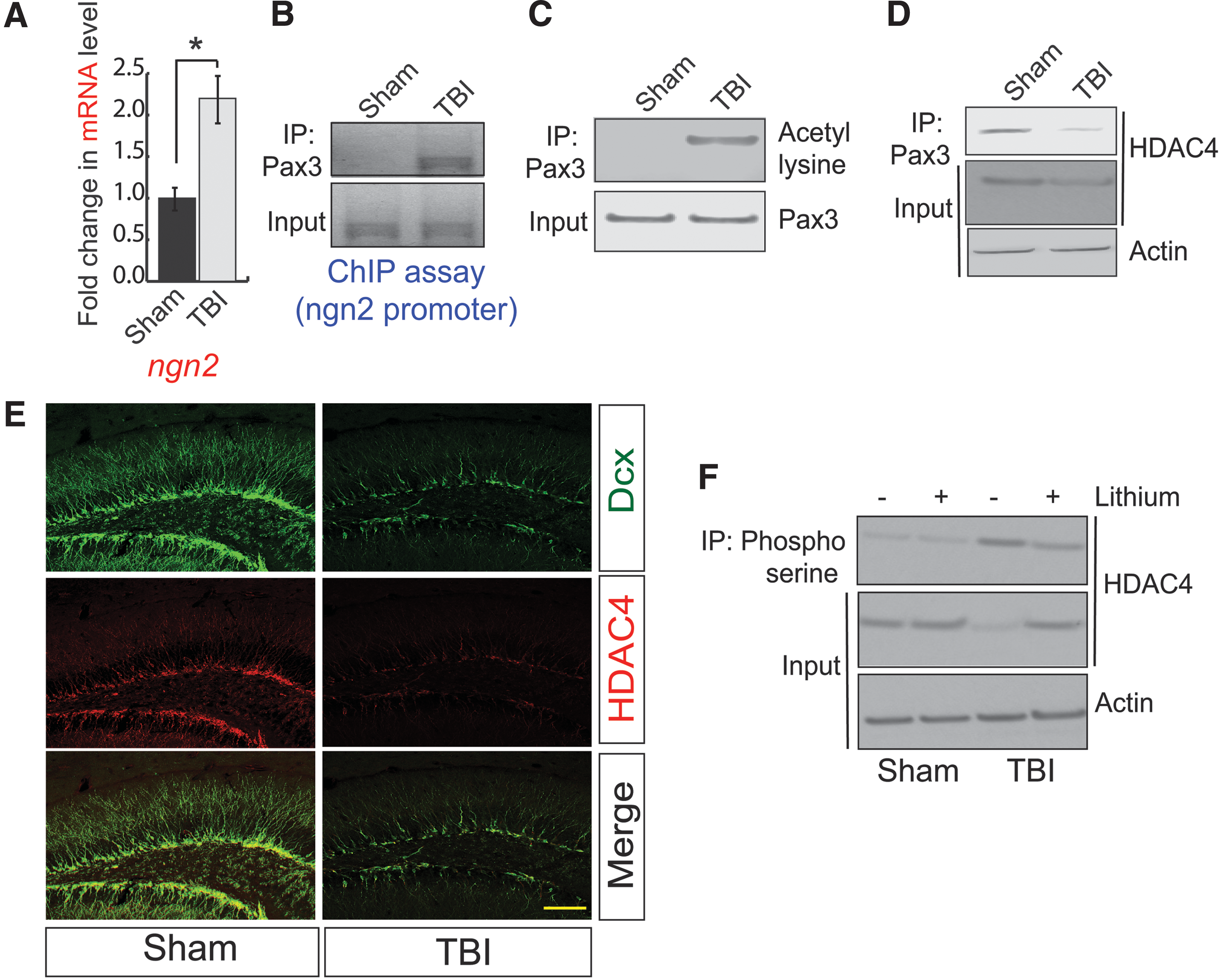
A decrease in HDAC4 facilitates Pax3 acetylation to transcribe Ngn2.
While screening for the Pax3 binding proteins, we found that the interaction between Pax3 with HDAC4 was markedly decreased 72-h post-TBI (Fig. 3D, Supplementary Fig. S3C). The decrease in the interaction between Pax3 and HDAC4 is associated with a decrease in HDAC4 level in the peri-contusional hippocampal lysates in TBI mice (Fig. 3D, Input; Supplementary Fig. S3D). To further confirm the loss of HDAC4 after TBI, we performed the confocal microscopic analysis using brain sections obtained from both sham and 72-h post-TBI mice. Consistent with our western blot data, we found that TBI leads to a decrease in HDAC4 in the DG of the hippocampus (Fig. 3E, Supplementary Fig. S3E). Moreover, the number of HDAC4-positive cells increases in Dcx-positive cells following TBI (Fig. 3E, Supplementary Fig. S3F). Collectively, our results show that a decrease in HDAC4 causes an increase in Pax3 acetylation, which is responsible for the induction of Ngn2 level following TBI.
The structural analysis of HDAC4 revealed that it contains a PEST domain, which acts as a signal to degrade the protein. 70 A previous study has shown that phosphorylation of serine residue (S298) of the PEST domain of HDAC4 by a kinase, GSK3β, leads to its degradation. 71 The previous study from our group has shown that TBI leads to activation of GSK3β 47,55 ; thus, we tested whether blocking GSK3β after TBI can prevent the downregulation of HDAC4. We found that treatment with an inhibitor of GSK3β, lithium (5 mg/kg), prevented the phosphorylation of HDAC4 (Fig. 3F, Supplementary Fig. S3G) and rescued the loss of HDAC4 following TBI (Fig. 3F, Supplementary Fig. S3H). It is important to note that we were unable to directly monitor the phosphorylation of HDAC4 at Ser298 residue due to the lack of a commercially available antibody. Thus, we used a pan-phosphoserine antibody to monitor the phosphorylation of HDAC4. Because HDAC4 is known to be phosphorylated, other serine residues that are independent of GSK3β treatment with lithium did not abolish the serine phosphorylation of HDAC4; however, it rescues the degradation of HDAC4—this further substantiates the importance of GSK3β in the stabilization of HDAC4 in the hippocampus following TBI.
Overexpression of HDAC4 rescues the deficit in neurogenesis following TBI
To understand whether overexpression of HDAC4 can reduce the Pax3 acetylation, we overexpressed HDAC4 in the DG area of the hippocampus and subjected it to TBI. We found that in mice overexpressing HDAC4, the acetylation level of Pax3 was decreased significantly following TBI (Fig. 4A, Supplementary Fig. S4A). Similarly, in mice overexpressing HDAC4, the interaction between Pax3 and HDAC4 was rescued after TBI (Fig. 4A, Supplementary Fig. S4B). The ChIP assay revealed that TBI-induced increases in the binding of Pax3 on the Ngn2 promoter and Ngn2 binding to the vGlut1 promoter were significantly decreased after overexpression of HDAC4 in the hippocampus before TBI (Fig. 4B,C, Supplementary Fig. S4C).
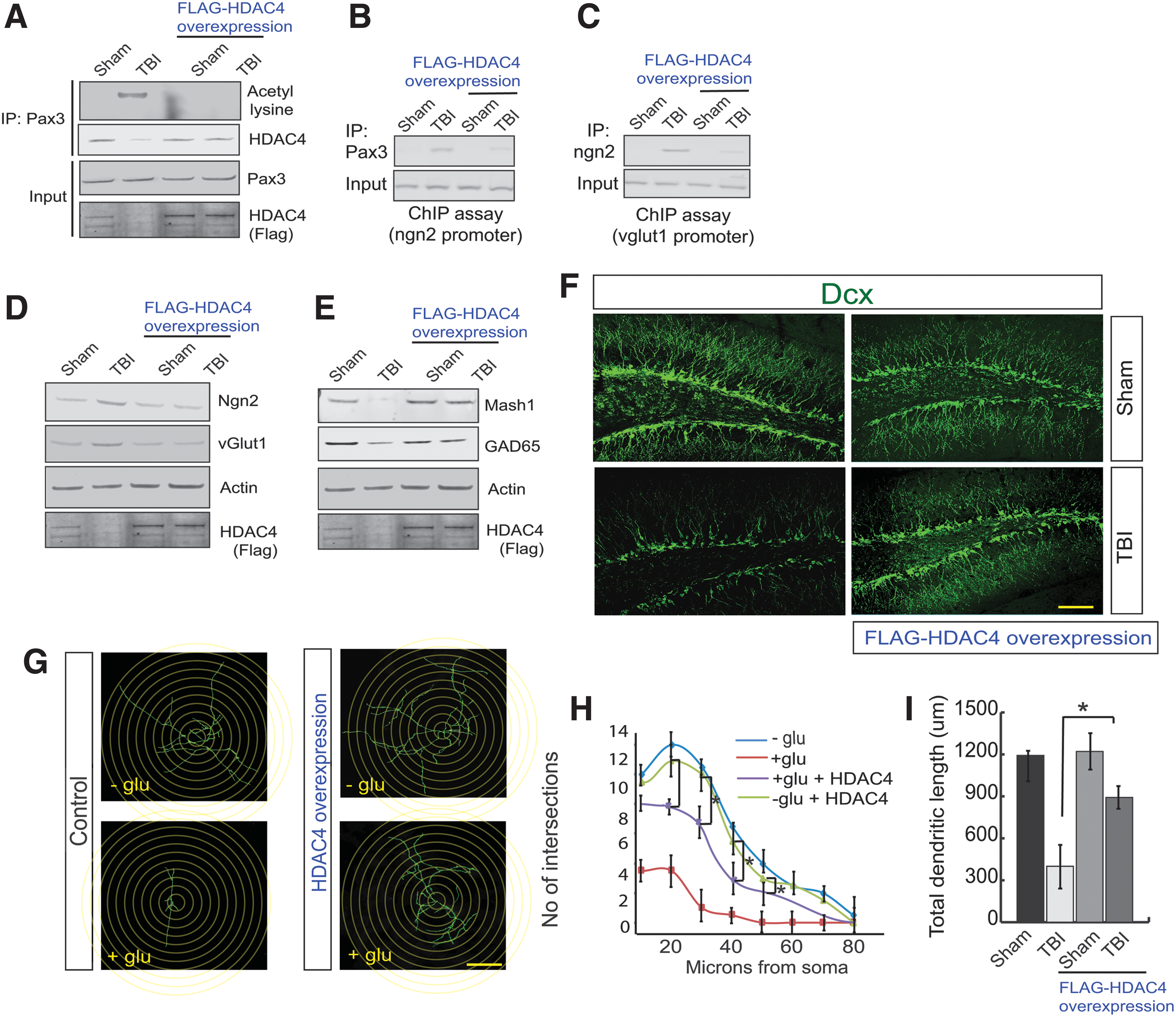
Overexpression of HDAC4 attenuates the transcriptional activity of Pax3 and Ngn2 and rescues neurogenesis following TBI.
TBI-induced increase in the expression level of Ngn2 (Fig. 4D, Supplementary Fig. S4D) and vGlut1 (Fig. 4D, Supplementary Fig. S4E) were significantly reduced in mice overexpressing HDAC4 following TBI along with an increase in the expression level of Mash1 (Fig. 4E, Supplementary Fig. S4F). Consistent with Mash1 data, we found that TBI leads to a decrease in the protein level of the GABA synthesizing enzyme, glutamic acid decarboxylase 65 (GAD65); however, its level was rescued in mice overexpressing HDAC4 72-h post-TBI (Fig. 4E, Supplementary Fig. S4G). We also monitored the overall neurogenesis in mice overexpressing HDAC4. We found that the loss of Dcx-positive cells was rescued in the DG of mice overexpressing HDAC4 following TBI (Fig. 4F, Supplementary Fig. S4H). Similarly, the number of branches (Fig. 4F, Supplementary Fig. S4I), and dendritic lengths of Dcx-positive cells (Fig. 4F, Supplementary Fig. S4J) were increased in mice overexpressed with HDAC4 in the hippocampus. Because HDAC4 was tagged with Flag, the overexpression of HDAC4 was confirmed by staining with Flag antibody in the DG by confocal microscopy (Fig. Supplementary Fig. S4K) and western blot analysis (Fig. 4A,D,E input). Consistent with this in vivo data, we found that overexpression of HDAC4 blocks glutamate-induced decrease in the number of intersections (Fig. 4G,H) and dendritic length of neuronal stem cells (Fig. 4G,I). These data suggest that HDAC4 is required to attenuate TBI-induced impairment of neurogenesis.
Overexpression of HDAC4 improves neurobehavioral outcomes following TBI
The open field test is used to assess locomotor, exploratory, and anxiety-like behavior in mice. We found that total movement time was similar between sham and 2-week post-TBI mice (Fig. 5A), indicating that there was no significant deficiency in locomotor activities. However, the TBI mice spent significantly less time in the center but more time on the periphery compared with sham mice (Fig. 5B,C), suggesting that TBI mice executed more anxiety-like behavior compared with sham mice. However, mice overexpressing HDAC4 in the DG spent more time in the central zone compared with the periphery (Fig. 5B,C), suggesting that overexpression of HDAC4 improves anxiety-like behavior following TBI compared with sham.
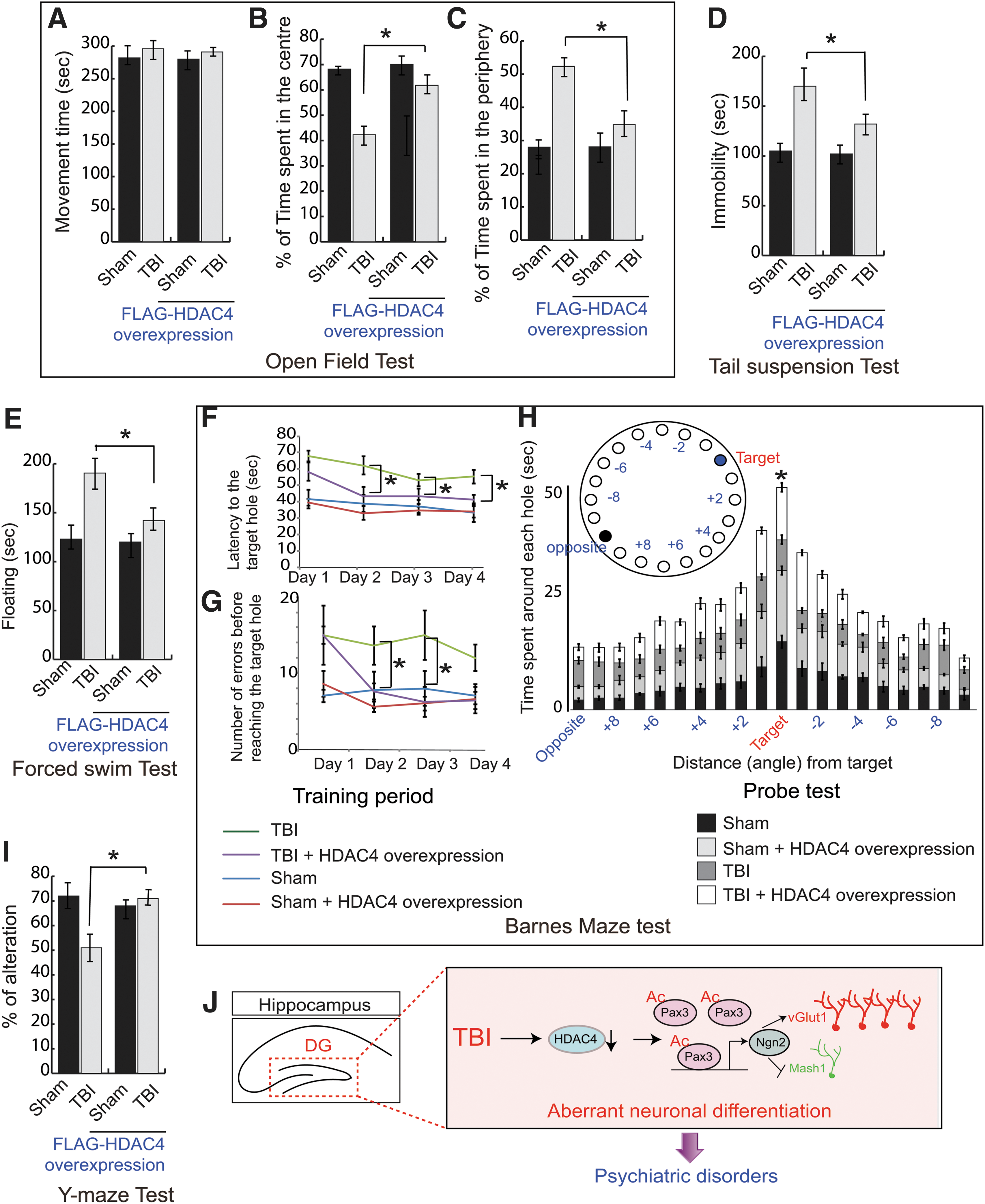
Overexpression of HDAC4 improves neurobehavioral outcomes following TBI.
The tail suspension test is specific for depressive-like behavior. We found that the period of immobility for 2-week post-TBI mice was significantly higher than sham mice (Fig. 5D); these data suggest that TBI mice execute depressive-like behavior. However, overexpression of HDAC4 in the hippocampus remitted in the reduction of the period of immobility significantly, implying that HDAC4 improves depressive-like behavior following TBI (Fig. 5D).
The forced swim test monitors the depression-like behavior in mice. Typically, “floating behavior” is used as a parameter to analyze depression-like behavior. We found that TBI mice spent more time floating compared with sham mice. However, in mice overexpressing HDAC4 before TBI, the floating time was decreased significantly compared with TBI mice (Fig. 5E), suggesting that HDAC4 is required to reduce depressive-like behavior following TBI.
The Barnes maze requires the use of hippocampal-dependent spatial memory to be able to locate the escape locations. This ability to remember the location of the target hole can be affected by the impairment of neurogenesis in the hippocampus. We tested the TBI mice where neurogenesis was significantly impaired compared with that of sham mice, and HDAC4 overexpressed sham mice and TBI mice. We found that the escape latency (Fig. 5F) and the number of errors before reaching the target hole (Fig. 5G) were increased significantly in TBI mice compared with sham mice during the training period of 4 days. The repeated two-way ANOVA shows the significance in Group effect (F[3,12] = 13.468, p = 0.001) as well as in Day effect (F[3,12] = 7.981, p = 0.003) for escape latency; and the significance in Group effect (F [3,12] = 17.917, p = 0.001) for number of errors, averaged over the 4 days. The significance was not found in Group × Day interaction (F < 0.464) for escape latency and Day effect (F < 2.541), and Group × Day interaction (F < 0.960F) for the number of errors.
Further, on the fifth day (Probe test) the time spent around the escape box and surrounding area was significantly decreased (p < 0.05) in TBI mice compared with sham (Fig. 5H). These data indicate that TBI affects spatial memory functions. However, in mice overexpressing HDAC4 in the hippocampus before TBI, the escape latency, as well as the number of errors before reaching the target, were minimized (p < 0.05, Fig. 5F,G) in comparison to TBI mice during the acquisition trial. During the probe test, mice overexpressing HDAC4 spent significantly more time (Fig. 5H) at the surrounding area of the escape box in comparison to that of TBI mice. These data confirm that downregulation of HDAC4 is responsible for impairment for spatial memory functions.
The Y-maze spontaneous alternation measures the willingness of rodents to explore new environments and hippocampal-dependent working (short-term) memory. Rodents typically prefer to explore a new arm of the maze rather than returning to one that was previously visited. We found that the percentage of alteration in the Y-maze was reduced significantly following TBI compared with sham (Fig. 5I); however, overexpression of HDAC4 augments the percentage of spontaneous alteration following TBI (Fig. 5I), suggesting that HDAC4 is necessary for short-term memory and willingness to explore new environments.
Discussion
A growing number of studies have highlighted the importance of HDAC in neurogenesis; however, its role in TBI remains controversial. Previously, it was shown that inhibitors of HDACs—such as sodium butyrate (SB), valproic acid (VPA), and trichostatin A (TSA)—induce neuronal differentiation in primary rat cortical cultures, presumably by inducing the neurogenic bHLH transcription factor NeuroD. 72 Moreover, chronic treatment of HDAC inhibitor has been shown to stimulate hippocampal neurogenesis in the ischemic brain. 73 In TBI, studies from independent groups have shown that neurogenesis can be increased, 74 –76 unchanged, 77 and decreased 63,67 after TBI. However, in our study, we provide evidence that disruption in balanced neurogenesis contributes to the TBI outcomes.
Briefly, we have shown that a decrease in the level of HDAC4 is responsible for triggering the newly born neurons to differentiate more into glutamatergic neurons via increased Pax3 acetylation following TBI. Thus, an imbalance in glutamatergic and GABAergic neurons causes glutamate toxicity, and this event leads to impairment of neurogenesis following TBI (Fig. 5J). Rescuing the deficiency in neurogenesis by overexpression of HDAC4 in the hippocampus before TBI improves neurobehavioral outcomes associated with anxiety and depressive-like behavior with an improvement in cognitive dysfunctions. Our data are consistent with another study where it was shown that treatment with the pan-HDAC inhibitor VPA blocks proliferation of adult hippocampal neural progenitor cells, 72 and that prenatal exposure impairs hippocampal function. 78 Also, the selective loss of HDAC4 is implicated in memory function and long-term synaptic plasticity. 44 Collectively, it can be interfered that implication of HDACs in neurogenesis is context-dependent. However, it is essential to mention that overexpression of HDAC4 does not selectively target the post-mitotic cells like newly born immature neurons; however, it can be considered that mitotic cells, which are relevant to the process of neurogenesis, do not express vGlut1; thus, the possibility of contribution of these cells to TBI-induced toxicity will not be directly relevant. Thus, we restrict our claim to the subpopulation of post-mitotic newly born Dcx-positive immature neurons.
The activity of Class IIa HDACs including HDAC4 is known to be primarily regulated by their translocation to the nucleus and its deacetylase activity. 44,45,79 –81 Exclusion of HDAC4 from the nucleus causes an increase in histone acetylation and facilitates the transcriptional process. However, we found that the expression level of HDAC4 was reduced after TBI and as a part of the mechanism we have shown that degradation of HDAC4 is dependent on its phosphorylation by GSK3β. This was evidenced by the fact that the administration of a GSK3β inhibitor, lithium, prevents the loss of HDAC4 after TBI. Our study is consistent with a previous study, where it was shown that HDAC4 contains a PEST sequence that upon phosphorylation by GSK3β forces HDAC4 to degrade via ubiquitination. 71
Also, aberrant activation of GSK3β due to inactivation of a protein kinase Akt is a common underlying mechanism responsible for TBI pathology. 47,55 Given that an induction of oxidative stress has a strong correlation with TBI 82 –84 and reduced neurogenesis, 85,86 and preventing the induction of the oxidative stress in a TBI model can improve the neurogenesis, 87 there is a concern whether an induction of oxidative stress impairs neurogenesis, at least partially, through manipulating HDAC4; but it is not unlikely. A recent study showed that epigenetic regulation by HDAC4 plays a role upstream of oxidative damage markers, 88 and oxidative stress can modify HDAC4 by oxidizing cysteine residues. 89 In this study, we provided evidence that a deficiency of HDAC4 impairs neurogenesis, and it may be possible that oxidative stress is responsible for a decrease in HDAC4 after TBI. This notion was further strengthened by the fact that inhibiting GSK3β prevents downregulation of HDAC4. Further, studies have shown that ROS-induced activation of c-Jun N-terminal kinase (JNK), a stress kinase, reduces the phosphorylation of protein kinase B (Akt) 90 that may lead to an activation of GSK3β by reducing its phosphorylation as a downstream substrate of Akt.
Histone acetylation and deacetylation play a crucial role in regulating gene expression during the processes of cell proliferation, and differentiation and HDAC4 are instrumental in maintaining the balance. The pan-HDAC inhibitor has been shown to stimulate neurogenesis and differentiation of hippocampal progenitor cells with an increase in histone acetylation. 72 In contrast, our study has shown that change in the acetylation level of a non-histone protein such as Pax3 is critical for neurogenesis. In the nervous system, the influence of Pax3 is involved in neural crest development, and peripheral neuron differentiation 39,91,92 and acetylation of Pax3 is prerequisite for its transcriptional activity; however, it was not clear how the acetylation status of Pax3 could be regulated in the adult brain. Our study shows that a decrease in the interaction between Pax3 and HDAC4 is responsible for acetylation of Pax3. The identification of Pax3 as a substrate of HDAC4 is unique and provides another potential mechanism that can explain how HDAC4 influences neurogenesis by altering the acetylation status of non-histone proteins in the mature brain.
However, our study raised a concern about whether a change in the level of HDAC4 has any influence on histone acetylation. Histone acetylation can be regulated by an orchestrated activation of several acetyltransferases and de-acetyltransferases including HDAC4. Thus, we do not anticipate that a reduction in only one deacetylase, HDAC4, will significantly affect the histone acetylation and block the activity of critical transcriptional factors. Because alteration of the acetylation status of Pax3 is enough to affect its transcriptional activity, we did not explore whether a change in histone acetylation also contributes to the impairment of the transcriptional activity of Pax3. However, we can anticipate that a reduction in HDAC4 will cause an increase in histone acetylation, which will, in turn, further facilitate the aberrant binding of Pax3 to its promoters.
Another unique aspect of adult hippocampal neurogenesis is a balance between GABAergic and glutamatergic inputs to tightly regulate neuronal activity. 93 However, not many studies have been conducted to elucidate the mechanism of how the balance is maintained in the adult brain. Our study provides evidence that HDAC4 is essential to maintain the balance and a loss of HDAC4 is responsible for an imbalance in the generation of glutamatergic and GABAergic neurons that accounts for impaired neurogenesis, anxiety, and depressive behavior following TBI. Glutamate is the principal excitatory neurotransmitter in the brain and GABA, the principal inhibitory neurotransmitter in the cerebral cortex, maintains the inhibitory tone that counterbalances neuronal excitation. 94,95 However, excess glutamate can initiate seizure-like activity. Thus, our data can explain the previous finding of why an aberrant increase in neurogenesis can exhaust neurogenic potential and enhance epileptogenesis following TBI. 96
In general, neurogenesis can be classified into two significant steps: the proliferation of mitotic cells and differentiation of post-mitotic cells, which mostly consist of immature newly born neurons and mature neurons. The mature neurons need to be integrated into the existing neuronal network in the DG of the hippocampus as a downstream process of neurogenesis, and it generally takes weeks to execute their functional relevance in terms of memory functions. A compelling number of previous studies have shown that impairment of either proliferation, differentiation, or the integration of neurons significantly contributes to the memory impairment, which can be monitored by either Y-maze or Barnes maze test.
In our study, we provided evidence in support of the fact that overexpressing HDAC4 can attenuate glutamate-induced toxicity by restricting the vGlut1-positive newly born immature neurons, which is an upstream process of integration of newly synthesized neurons into the DG. We were also able to demonstrate that this event leads to an improvement of memory function, which was examined after 3 weeks post-TBI. Thus, it can be reasonably considered that the integration of newly born neurons did not interfere with facilitating the memory improvement executed by overexpression of HDAC4 in TBI mice. Therefore, we did not consider studying the integration of newly born neurons to prove the current paradigm as a first preference.
Recently, almost all Phase II/III clinical trials with HDACs in neuroprotection have failed to show any consistent improvement in outcome for TBI patients, mostly because of non-specificity of HDAC inhibitors. 97,98 Our study fulfills some of the critical gaps and suggests that induction of HDAC4 instead of downregulation of HDAC is responsible for impaired neurogenesis following TBI. Thus, our study provides a new hope for designing a novel therapeutic strategy based on HDAC4, which plays a critical role during the differentiation of neurogenesis and restores hippocampus-dependent anxiety and depressive-like behavior in mice. Also, elucidating the role of HDAC4 to regulate adult neurogenesis can be extended to other neurodegenerative conditions such as Alzheimer's disease or Parkinson's disease where impaired neurogenesis is a common mechanism of neurological deficits.
Acknowledgments
This work was partly supported by the National Institutes of Health (Grants RO1NS094516 and RO1EY025622 to N.S.) and funding from University of Pittsburgh and Copeland Foundation to R.G, T.S, and N.S.
Footnotes
Author Disclosure Statement
No conflicting financial interests exist.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.