
Abstract
Autophagy is the degradation process of dysfunctional intracellular components and has a crucial function in various human diseases. There are three different types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). CMA is a major route for the elimination of cellular aberrant proteins and can provide a cytoprotective effect. The present study investigated the expression of lysosome-associated membrane protein type 2A (LAMP2A), which is the hallmark of CMA activity, in damaged neural tissue after spinal cord injury (SCI) in mice.
The number of LAMP2A-expressing cells was significantly increased at the lesion following SCI. The increased number of LAMP2A-positive cells was observed from 24 h and peaked at 3 days after injury. A western blot analysis confirmed that the level of LAMP2A protein was significantly increased in the injured spinal cord compared with the uninjured cord. On double staining for LAMP2A and different neural cell type markers, the increased expression of LAMP2A was observed in neurons, astrocytes, oligodendrocytes, and microglia/macrophages following injury. An electron microscopic analysis showed that secondary lysosomes were increased in damaged neurons at the lesion site. Immunoelectron microscopy revealed that the gold particles with anti-LAMP2A antibody were frequently localized at the secondary lysosomes in the injured site. These findings indicated that CMA was clearly activated in damaged neural tissue after SCI. The activation of CMA may contribute to the elimination of intracellular aberrant proteins and exert a neuroprotective effect following SCI.
Introduction
Autophagy is the lysosome-mediated intracellular degradation system of the cytoplasmic proteins and organelles for material recycling to maintain cellular homeostasis. 1,2 In mammalian cells, there are three different types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). 3 Macroautophagy is characterized by the formation of double-membrane vesicles (autophagosomes) that fuse with lysosomes and degrade their contents. 4 In microautophagy, intracellular proteins are trapped in the vesicles directly formed via invagination and/or septation of the lysosomal membrane. 5 Both macroautophagy and microautophagy can be selective or non-selective degradation. 6
In contrast, CMA involves selective degradation wherein the targeted proteins are directly translocated into the lysosomal membrane. 7,8 During the CMA process, a KFERQ-like motif in the amino acid sequence of the substrate proteins is recognized by heat shock proteins such as heat shock cognate 70 kDa protein (HSC70). The substrate/chaperone complex then binds to the lysosomal membrane, assisted by lysosome-associated membrane protein 2A (LAMP2A) and the substrate is unfolded and translocated across the lysosomal membrane where the substrate is degraded rapidly. 3,9 The level of LAMP2A is the biochemical hallmark of CMA activity, so many studies have used LAMP2A to investigate CMA activity. 8,10
Selective protein degradation conducted by CMA helps maintain cellular homeostasis under various stress conditions, such as starvation, 11 hypoxia, 12 and exposure to toxins. 13 Under such stress conditions, CMA removes substrate proteins selectively, thereby contributing to the elimination of altered proteins and recycling amino acids via proteolysis. The timely degradation of specific proteins by CMA can regulate multiple cellular functions, such as glucose and lipid metabolism, DNA repair, and cellular reprograming. 8 Notably, previous studies have found that CMA malfunction is linked to the pathogenesis of various diseases in humans, such as neurodegeneration, 14,15 cancer, 16 kidney pathology, 17 or lysosomal disease. 18,19
Numerous studies have shown that an altered CMA activity is associated with different pathologies in various human diseases affecting the central nervous system (CNS). In Parkinson's disease (PD), for example, the aggregation of alpha-synuclein protein due to CMA dysfunction is involved in the pathological mechanism. 20 In other neurodegeneration diseases such as Alzheimer's disease and Huntington's disease, a decrease in CMA activity causes the accumulation of pathogenic proteins such as amyloid-β and Htt. 15,21 On the other hand, other studies have showed that CMA activity is upregulated in damaged neural tissue after traumatic brain injury and cerebral ischemia, 22,23 suggesting the neuroprotective function of CMA against CNS damage. In addition, CMA activation exerts a neuroprotective effect against inflammatory disease of the peripheral nervous system. 24,25 Therefore, CMA may play an important role in the neuroprotective mechanism following spinal cord injury (SCI).
However, no study has yet examined CMA activity after SCI. The present study investigated changes in LAMP2A expression, which is a hallmark of CMA activity, in damaged neural tissue following SCI using a spinal cord hemisection model in mice.
Methods
All experimental procedures were approved by the Institutional Animal Care and Use Committee of Tohoku University.
Animals
This study used adult female C57BL/6J mice (10–12 weeks of age; Japan SLC, Inc., Shizuoka, Japan). The animals were kept in specific-pathogen-free animal facilities, under a 12-h light-dark cycle. The animals were housed at four or five per cage in a room kept at 24°C with free access to water and food before and after SCI. All efforts were made to minimize the number of animals used and to decrease their suffering.
SCI model
For the surgery, the animals were anesthetized using 4% sevoflurane. A 15-mm midline skin incision was made, and then the laminae were exposed at the T9-10 levels. Laminectomy was performed at T10, exposing the dorsal surface of the spinal cord with the dura intact. The spinal cord was hemi-transected on the left side only using a sharp scalpel. 26,27 The sham control mice that received laminectomy without SCI were prepared in the same way. The skin and muscles were closed in layers. Following the surgery, mice were placed on a heating pad to maintain normal body temperature until they awoke. Bladders were manually expressed daily until spontaneous voiding began. Mice were attentively monitored for infection, autophagia, and decubitus.
Tissue preparation
At 4 or 24 h, or 3, 7, or 21 days following SCI and immediately after the sham operation, the animals were overdosed with an intraperitoneal injection of 100 mg/kg sodium pentobarbital. The animals were transcardially perfused with normal saline, followed by 4% paraformaldehyde in 0.1 M phosphate-buffered saline (PBS), pH 7.4. To perform the immunohistochemical analysis, the spinal cord segments containing the injured site were obtained, post-fixed in the same fixative overnight at 4°C, and cryoprotected in 30% sucrose in PBS for 48 h at 4°C. The spinal cord segments were then embedded in optimal-cutting temperature (OCT) compound (Thermo Scientific Ltd.). Serial 15-μm transverse sections around the injured site were mounted on slides. A total of 13 sequential sections at 250-μm intervals, spanning a 3000-μm length of the spinal cord centered at the epicenter were collected.
Immunohistochemistry
Immunostainings of LAMP1, LAMP2A, and HSC70 were performed using the sections obtained at different time-points following SCI. The sections were washed in PBS for 15 min, followed by PBS containing 0.3% Tween for 10 min. The sections were then blocked with 3% milk and 5% fetal bovine serum (FBS) in 0.01 M PBS for 2 h. The sections were incubated with mouse anti-LAMP1 antibody (1:100; H4A3, Abcam), and either rabbit anti-LAMP2A antibody (1:250; 51-2200, Thermo Fisher) or rat anti-HSC70 antibody (1:200; ab19136, Abcam), and diluted in PBS overnight at 4°C. After rinsing with PBS, the sections were incubated with goat anti-mouse IgG Alexa Fluor 488 secondary antibody (1:500; A11029, Invitrogen), and either goat anti-rabbit IgG Alexa Fluor 594 secondary antibody (1:500; A11012, Invitrogen) or goat anti-rat IgG Alexa Fluor 594 secondary antibody (1:500; A11007, Invitrogen) for 1 h at room temperature. The stained sections were then covered with mounting medium containing DAPI to label the nuclei (Vector Laboratories). The three immunostainings (LAMP1, LAMP2A, and HSC70) from each time-point were performed at the same time.
Counting of LAMP2A- and HSC70-positive cells
Following the immunostaining of LAMP1, LAMP2A, and HSC70, the entire transverse spinal cord sections were scanned at 10 × objective lens magnification using a laser microscope (BX 51, Olympus). To quantify the LAMP2A and HSC70 expressions in the spinal cord, the number of LAMP2A- and HSC70-positive cells were counted using serial transverse sections at 250-μm intervals. The scanned image of the stained section was displayed on a monitor with a grid using the Photoshop CS4 (Adobe, CA) and then LAMP2A- and HSC70-positive cells in an entire section were counted using a manual counter. The positive cells were defined as cells double-labeled with each antibody and DAPI. The sections with the highest number of positive cells on the injured side and the 250-μm rostral and caudal sections in each animal were selected for the analysis. The sum of the numbers in the three sections was determined and then compared among the injured side, the contralateral side, and the sham group.
To evaluate the expressions of LAMP2A and HSC70 in the cells expressing LAMP1, the percentages of LAMP2A- and HSC70-positive cells among the LAMP1-positive cells were calculated in the injured side of the spinal cords at 3 days. The sections at the epicenter and at 250-μm rostral and caudal to the epicenter were selected. The percentages were calculated in the sum of three sections.
Western blotting of LAMP2A and HSC70
To assess the activity of CMA, the levels of LAMP2A and HSC70 in the spinal cord were evaluated by a western blot analysis. The mice were sacrificed at 4 or 24 h, or 3, 7, or 21 days after SCI, and spinal cords of 5 mm in length containing the injured site were removed. The enriched lysosomal fractions from the spinal cord were obtained by a density gradient centrifugation with a Lysosome Isolation Kit (ab234047l, Abcam). The spinal cords were homogenized on ice with Dounce homogenizer (30 strokes, 4°C) in 10 volumes of lysosome isolation buffer with 500 μL of lysosome enrichment buffer added. Homogenates were centrifuged at 500g at 4°C for 10 min to obtain supernatants. The supernatants were diluted with lysosome gradient by mixing 1 part of lysosome gradient with 3 parts of the supernatants. The diluted supernatants were added to the top of the density gradient, which was made up of five gradient solutions using lysosome gradient and lysosome enrichment buffer, and then centrifuged using an ultracentrifuge at 145,000g at 4°C for 2 h. The lysosome fractions were withdrawn from the top 1/10th of the gradient volume. To further purify the fractions, they were mixed with 2 volumes of PBS, and then centrifuged at 18,000g at 4°C for 30 min.
The pellets containing the purified lysosomes were resuspended in PBS. 28 The proteins in the samples were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) in 10% gels and then electrophoretically transferred to polyvinylidene difluoride membranes. The membranes were blocked for 1 h in TBST buffer (0.01 M Tris HCL, pH 7.5, 0.15 M NaCl, and 0.05% Tween 20) containing 3% milk and incubated overnight at 4°C with either rabbit anti-LAMP2A antibody (1:1000; 51-2200, Thermo Fisher) or rat anti-HSC70 antibody (1:200; ab19136, Abcam) diluted in TBST buffer. The membranes were then incubated with horseradish peroxidase conjugated secondary antibodies (DAKO) for 1 h at room temperature. The immunoreactive bands were developed using enhanced chemiluminescence reagent (ECL Prime, Amersham plc). The band density was quantified using a scanned densitometric analysis with the Image Lab software program, version 6.0 (Bio Rad Laboratories). The quantities of the band densities were normalized by β-tubulin.
Double staining for LAMP2A and neural cell type markers
We investigated the LAMP2A expression in a specific population of neural cells in the injured spinal cord. Double staining for LAMP2A and different neural cell type markers, such as NeuN for neurons, glial fibrillary acidic protein (GFAP) for astrocytes, Olig2 for oligodendrocytes, and CD68 for microglia/macrophages, was performed using the sections obtained at 3 days after SCI. The sections of the spinal cord were incubated with a mixture of rabbit anti-LAMP2A antibody (1:250; 51-2200, Thermo Fisher) and either rat anti-CD68 (1:100; BioLegend), goat anti-Olig2 (1:100; Santa Cruz Biotechnology), mouse anti-GFAP (1:50; DAKO), or mouse anti-NeuN antibodies (1:100; Chemicon) diluted in PBS overnight at 4°C. After being washed with PBS, the sections were stained with a mixture of goat anti-rabbit IgG Alexa Fluor 594 secondary antibody (1:500; A11012, Invitrogen) and either goat anti-rat IgG Alexa Fluor 488 (1:500; ab150157, Abcam), donkey anti-goat IgG Alexa Fluor 488 (1:500; Molecular Probes), or goat anti-mouse IgG Alexa Fluor 488 antibodies (1:500; Molecular Probes) for 1 h at room temperature. After the staining, the sections were covered with mounting medium containing DAPI (Vector Laboratories).
Electron microscopy
To evaluate the histological feature of CMA in the spinal cord after injury, electron microscopic analysis was performed. At 3 days following hemisection and immediately after the sham operation, the animals were overdosed with an intraperitoneal injection of 100 mg/kg sodium pentobarbital. The animals were transcardially perfused with normal saline, followed by 25% glutaraldehyde. For electron microscopic analyses, the spinal cord segments containing the injured site were removed and post-fixed in the same fixative overnight at 4°C, and embedded in OCT compound. Serial 6-μm transverse sections around the injured site were mounted on slides, and viewed using the electron microscope (JEM-1400, JOEL, Tokyo, Japan).
Further, to confirm the expression of LAMP2A in lysosomes, an immunoelectron microscopic analysis was also performed. The animals were transcardially perfused with normal saline, followed by 4% paraformaldehyde in 0.1 M PBS, pH 7.4, and post-fixed in the same fixative overnight at 4°C, cryoprotected in 30% sucrose in PBS for 48 h at 4°C, and embedded in OCT compound. Serial 6-μm transverse sections around the injured site were mounted on slides. The sections were pre-incubated in a blocking solution of 10% bovine serum albumin (BSA) for 10 min at room temperature, and washed with 10% sucrose in PBS. They were then incubated in the primary antibody: rabbit anti-LAMP2A antibody (1:500; 51-2200, Thermo Fisher) as the primary antibody overnight at 4°C. After several washes with 10% sucrose in PBS, sections were incubated in the secondary antibody (5 nm nanogold) for 3 h at room temperature. The tissue was then washed and post-fixed in 0.5% glutaraldehyde for 1 min at room temperature. After that, the tissue was extensively washed and osmicated in 1.0% osmium tetroxide for 30 min. After being washed in 0.1 M PBS, sections were dehydrated in graded alcohol baths to propylene oxide and embedded in Epon resin. Ultrathin sections were prepared and evaluated using the electron microscope.
Statistical analysis
To minimize the number of animals used in this study, the sample size was calculated using a statistical power analysis software (G-Power, version 3.1.9.4; Franz Faul, University Duesseldorf, Germany). The power analysis suggested that a minimum of five animals per group were required at each time-point in each experiment (power = 0.80, α = 0.05, two-sided). All statistical analyses were conducted using the JMP software program, version 13 (SAS Institute Inc., Cary, NC, USA). The Mann-Whitney U test was used to compare the numbers of LAMP2A- and HSC70-positive cells between the injured and the contralateral sides at each time-point. A one-way analysis of variance followed by Tukey's post hoc test was used to compare the number of LAMP2A- and HSC70-positive cells, as well as the band densities obtained from the western blot analyses among the groups at different time-points. P-values of <0.05 were regarded as statistically significant. All data are presented as the mean ± standard deviation (SD).
Results
Immunohistochemical staining of LAMP2A
LAMP2A-expressing cells were rarely observed in the uninjured spinal cord in the sham-operated animals (Fig. 1). In contrast, the number of LAMP2A-expressing cells was higher at the injured side than at the contralateral side of the spinal cord obtained at 3 days after hemisection (Fig. 1). LAMP2A-expressing cells were observed in both the gray matter and the white matter on the injured side of the spinal cord. The increase in the number of cells expressing LAMP2A generally appeared within 500 μm of the areas rostral and caudal of the lesion epicenter (Fig. 1B). Double staining for LAMP2A and the lysosome marker LAMP1 confirmed that the LAMP2A-positive cells appeared to be LAMP1-positive as well (Fig. 2A,B). In addition, the cells expressing HSC70 were also LAMP1-positive (Fig. 2C,D). The percentages of LAMP2A- and HSC70-positive cells among LAMP1-positive cells in the injured side of the spinal cord obtained at 3 days were 87% and 35%, respectively (Fig. 2E).
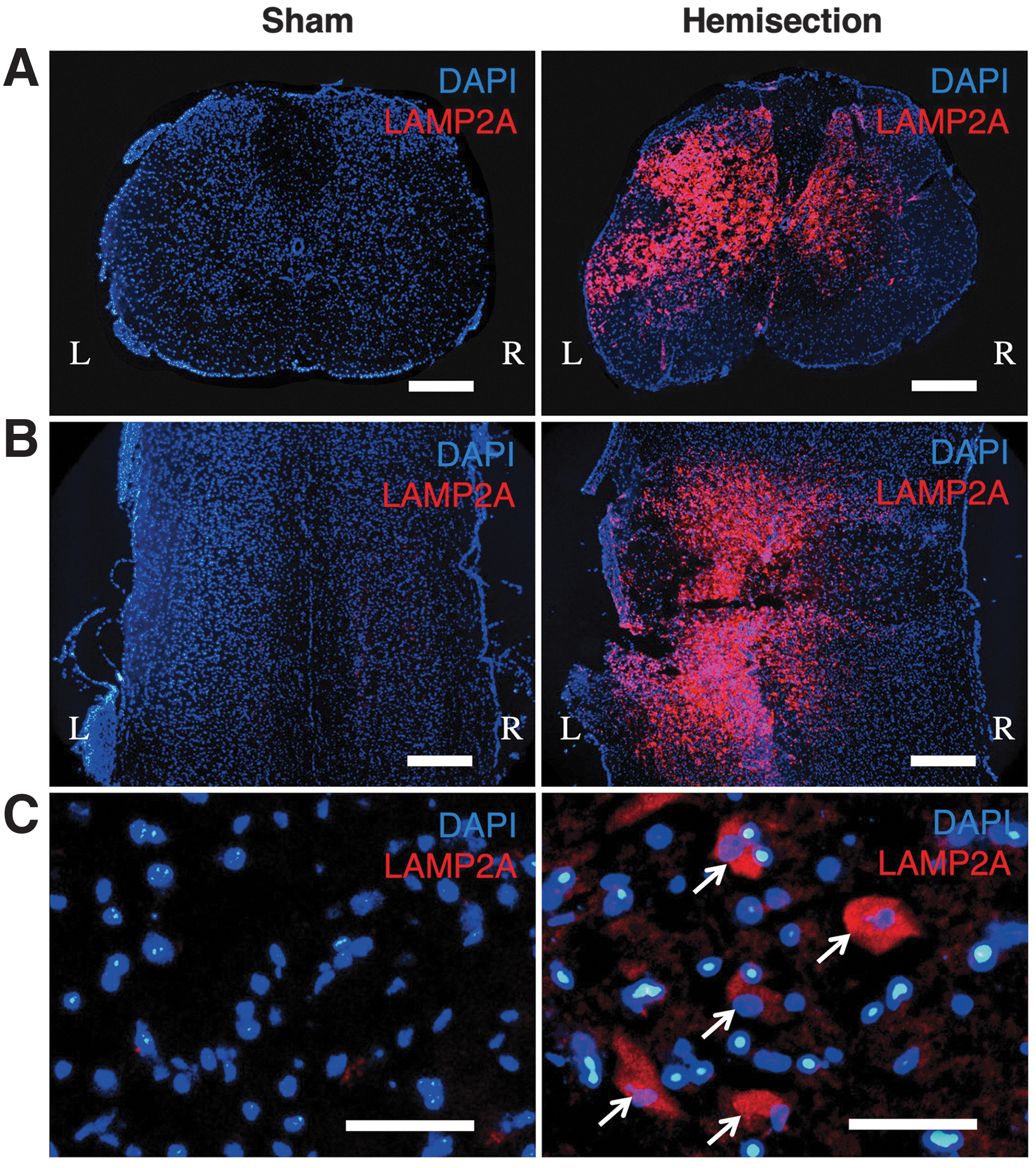
Immunohistochemistry of LAMP2A using the spinal cord sections at 3 days after hemisection and sham operation.
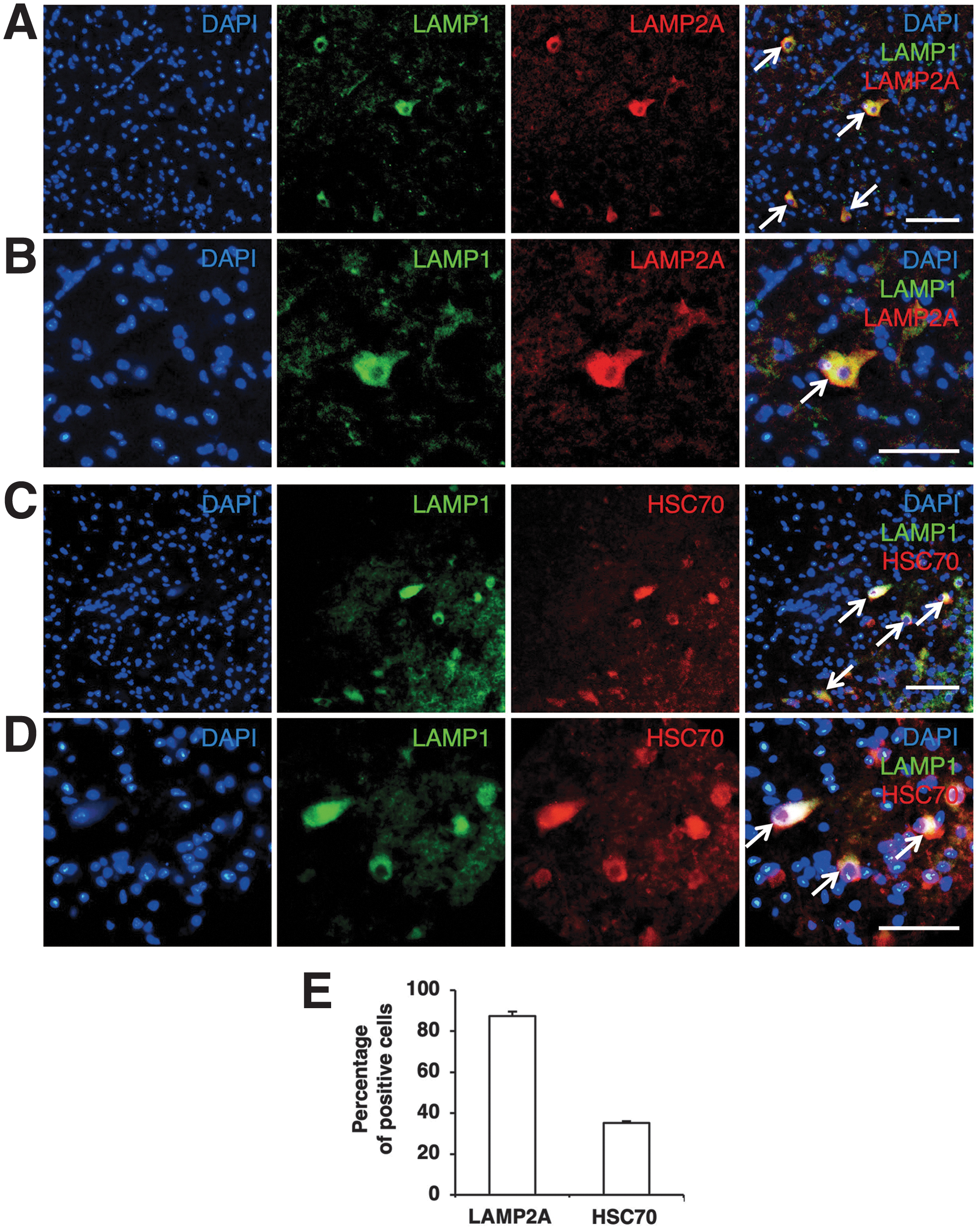
Double staining of LAMP1 with LAMP2A or HSC70.
Time course of the LAMP2A and HSC70 expressions
The immunostaining for LAMP2A at different time-points showed that the increase. in the number of LAMP2A-positive cells in the injured side started at 24 h after injury, peaked at 3 days, and lasted at least 7 days after hemisection (Fig. 3A). In counting the LAMP2A-positive cells (Fig. 3B), the number of LAMP2A-positive cells on the injured side at 24 h (84 ± 25 cells), 3 days (280 ± 54 cells), and 7 days (108 ± 28 cells) was significantly higher than that in sham control animals (36 ± 1 cells; p < 0.05). In addition, the number of LAMP2A-positive cells on the injured side was significantly higher than that on the contralateral side (123 ± 19 cells) at 3 days after injury (p < 0.05). In contrast, a similar population of HSC70-positive cells was observed at every time-point with no marked changes noted after SCI (Fig. 4A). Counting HSC70-positive cells showed no significant difference in the numbers of HSC70-positive cells at the different time-points (Fig. 4B). In addition, the number of HSC70-positive cells was not significantly different between the injured side and contralateral side at any time-point (Fig. 4B).
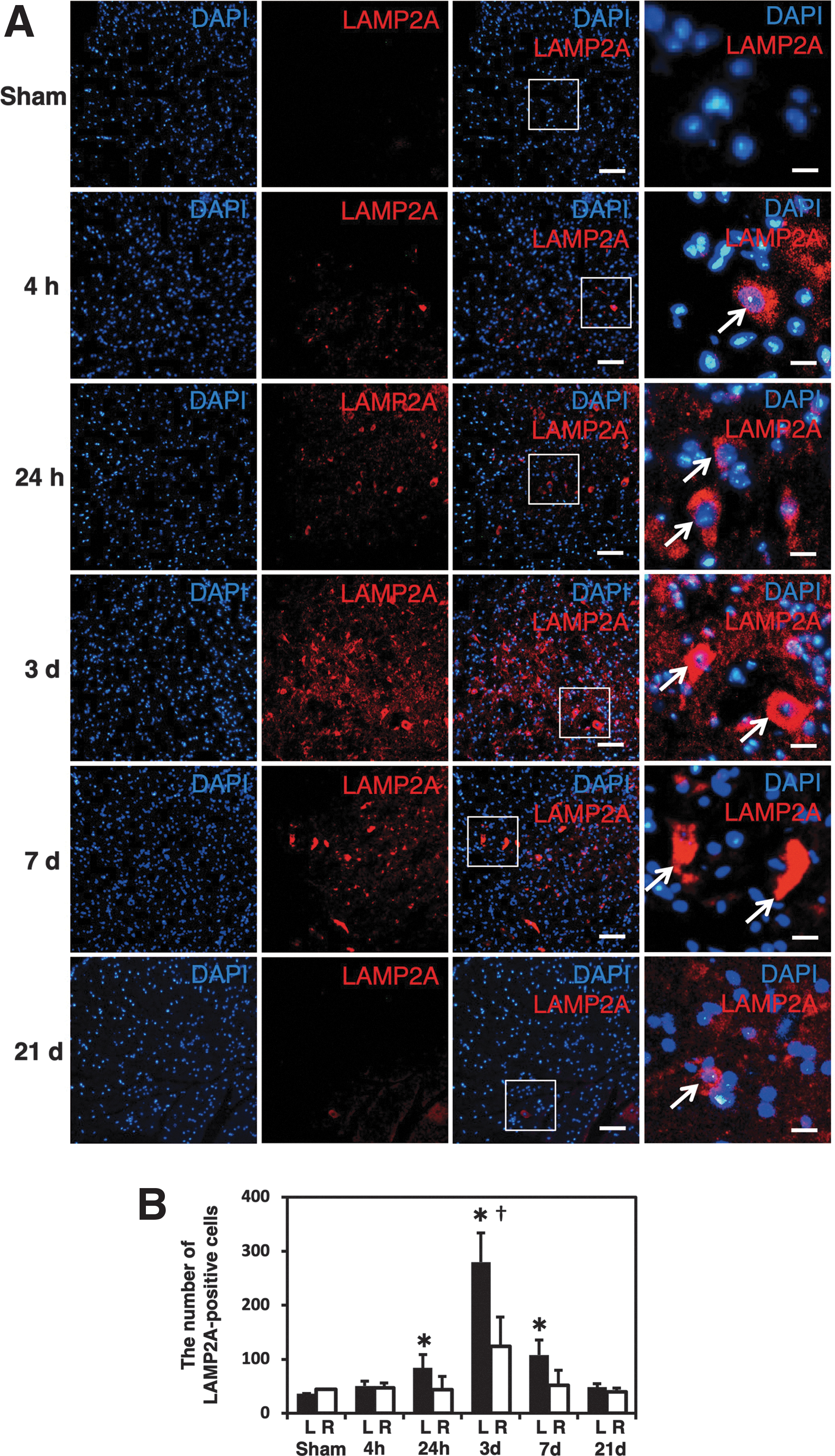
Counting of LAMP2A-positive cells at different time-points.
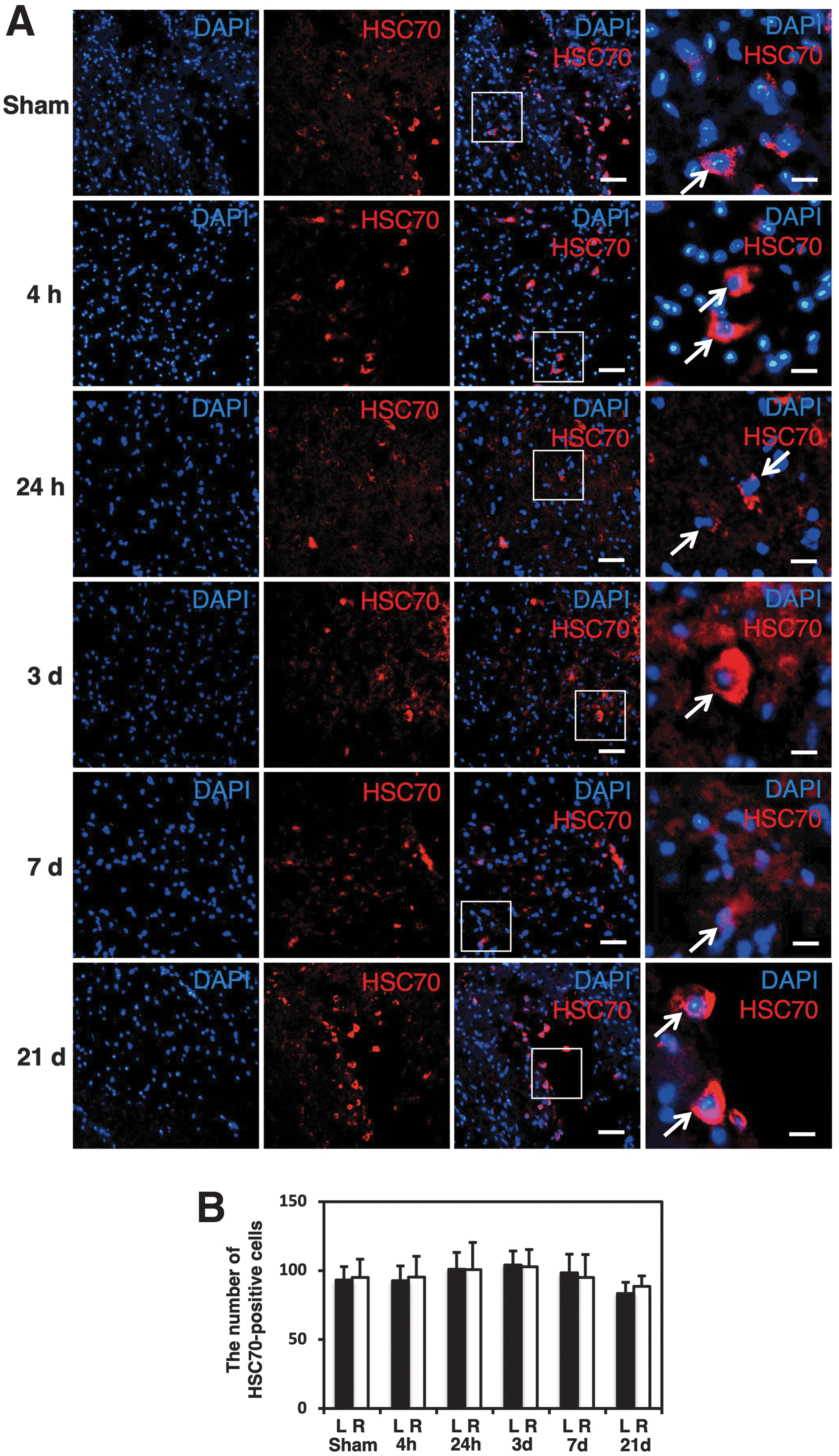
Counting of HSC70-positive cells at different time-points.
Western blot analysis results for LAMP2A and HSC70
In a western blot analysis, the LAMP2A protein expression gradually increased from 4 h after injury and peaked at 3 days, subsequently decreasing until 21 days (Fig. 5A). In the analysis of the band density, the level of LAMP2A expression at 3 days after injury was significantly higher in sham control animals (p < 0.05; Fig. 5A). Compared with the sham control, the levels of LAMP2A expression at 4 h, 24 h, 3 days, 7 days, and 21 days were 1.3-fold, 1.5-fold, 2.1-fold, 1.7-fold, and 1.2-fold higher, respectively. In contrast, the expression of HSC70 protein was almost unchanged following injury (Fig. 5B). The band density analysis showed that there was no significant difference in the level of HSC70 protein among the groups at different time-points (Fig. 5B). The levels of HSC70 expression at 4 h, 24 h, 3 days, 7 days, and 21 days were 1.0-fold, 1.0-fold, 0.8-fold, 0.9-fold, and 0.8-fold higher than in sham control, respectively.

Results of the western blot analysis of LAMP2A and HSC70 proteins.
Double staining for LAMP2A and various cell type markers
On double staining for LAMP2A and NeuN as a neuronal cell marker, LAMP2A-positive cells were observed among the NeuN-labeled cells on the injured side at 3 days after hemisection (Fig. 6A,B), indicating the LAMP2A expression in neurons at the lesion site. In addition, the expression of LAMP2A was also observed in GFAP-labeled cells (Fig. 6C,D), Olig2-labeled cells (Fig. 6E,F), and CD68-labeled cells (Fig. 6G,H), confirming the LAMP2A expression in astrocytes, oligodendrocytes, and microglia/macrophages, respectively, in the injured spinal cord.
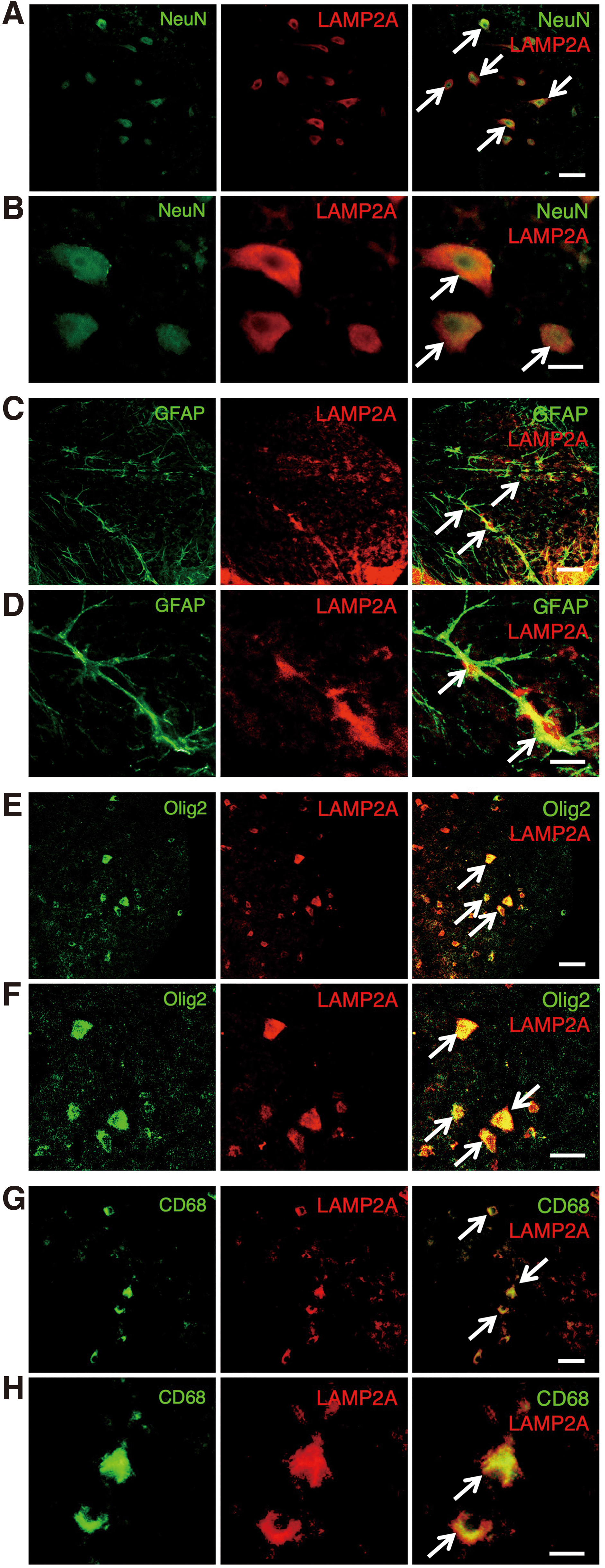
Double staining of LAMP2A and various cell type markers. In representative images on the injured side in transverse sections at 3 days after SCI, the expression of LAMP2A was increased in the NeuN- (arrows in
Electron microscopic analysis findings
In the electron microscopic analysis, neurons in the uninjured spinal cord of the sham control animals had bright normal nuclei and cytoplasm, suggesting they were undamaged (Fig. 7A). These cells possessed normal intracellular organelles, such as mitochondria and endoplasmic reticula (Fig. 7D,E). In contrast, the damaged neurons in the injured spinal cord showed condensation of the nucleus and cytoplasm (Fig. 7B,C). Further, the electron-dense secondary lysosomes were frequently observed in the damaged neurons at the lesion site (Fig. 7F–I). On immunoelectron microscopy, gold particles with anti-LAMP2A antibody were frequently localized in the lysosomes at the injured site (Fig. 7K,L,O–R). In contrast, gold particles were rarely seen in the uninjured spinal cord of the sham control mice (Fig. 7J,M,N).

Results of the electron microscopic analysis.
Discussion
In the results of the present study, the number of LAMP2A-expressing cells was significantly increased in the damaged neural tissue following SCI. This increase in LAMP2A-positive cells was observed from 24 h after injury, peaking at 3 days, and then gradually decreasing until 21 days. In contrast, the number of HSC70-positive cells did not change markedly after SCI and remained similar at time-points. A western blot analysis confirmed that the level of LAMP2A protein was significantly higher in the injured spinal cord than in the uninjured spinal cord. On double staining for LAMP2A and neural cell type markers, the LAMP2A expression was observed in neurons, astrocytes, and oligodendrocytes in the injured cord. The electron microscopic analysis additionally showed that secondary lysosomes were increased in damaged neurons at the lesion site. Further, on immunoelectron microscopy, gold particles with anti-LAMP2A antibody were frequently localized in the secondary lysosomes at the injured site. These results indicated both biochemically and histologically that CMA was activated in the damaged neural tissue after SCI.
Dysfunction of CMA is associated with the pathological mechanisms in various diseases such as neurodegeneration, 14,15 kidney pathology, 17 and lysosomal disease. 18,19 In neurodegenerative diseases, degenerative proteins, such as alpha-synuclein, a component of Lewy bodies in familial PD, fail to be degraded properly by CMA, and subsequently accumulate. 20 The reduced activity of CMA leads to the accumulation of other pathogenic proteins in diabetic-induced renal hypertrophy. 17 In contrast, several studies have shown that activity of CMA is upregulated at lesions in animal models of traumatic brain injury, 22 cerebral ischemia, 23 and Huntington's disease. 15 These studies suggested that the upregulated CMA reduces the level of aberrant proteins in the damaged neural cells and exerts a neuroprotective effect.
In the brain ischemia model, activation of CMA contributes to the neuronal survival in response to hypoxic stimuli. 23 Therefore, CMA activation may serve as a protective mechanism for maintaining cellular homeostasis under conditions of neural tissue damage in the CNS. The present study using the spinal cord hemisection model in mice clearly demonstrated that the number of LAMP2A-positive cells was significantly increased at the lesion site. In addition, a western blot analysis confirmed that the level of LAMP2A was significantly higher in the injured spinal cord than in the normal spinal cord. Further, an immunoelectron microscopic analysis showed that the expression of LAMP2A in lysosomes was frequently observed in the injured spinal cord. These findings indicate that CMA is upregulated at the lesion site following SCI. Therefore, CMA activation may help maintain the cellular homeostasis and exert a neuroprotective effect following SCI, just as it dose for traumatic brain injury.
There are multiple pathophysiological processes occurring depending on the timing after the initial onset of SCI. 29 The spinal cord is initially damaged by the primary mechanical insult that causes hemorrhagic necrosis. The neural tissue damage then expands due to secondary injury. Previous studies have suggested that this secondary injury occurs between 24 h and 3 days after the initial insult. 30 –32 In our previous studies, the expression of Beclin 1 (promoter of macroautophagy) and light chain 3 protein (marker of macroautophagy) peaked at 3 days after SCI in mice. 33 –35 We also reported that macroautophagy contributes to the cytoprotection against secondary injury after SCI. 36,37 In a previous study investigating the activity of CMA in vitro, the LAMP2A expression in neuronal cells was significantly upregulated at 48 h under hypoxic stress conditions. 23 Another study found that the increased expression of LAMP2A peaked at 3 days after traumatic brain injury. 22 In the present study, the increase in the LAMP2A expression was observed from 4 h, peaking at 3 days, and then lasting for at least 7 days after injury. These results resembled the time course of the LAMP2A expression in traumatic brain injury. Taken together, these findings suggest that CMA might function in a protective manner by attenuating secondary neural tissue damage following SCI.
The upregulation of CMA was confirmed in neurons after traumatic brain injury, brain ischemia and early stages of Huntington's disease. 15,22,23 These previous studies suggested that CMA contributes to the removal of altered proteins and supports homeostasis to ameliorate neuronal damage in various pathological conditions affecting the CNS. In the present study, the increased expression of LAMP2A was observed in neurons in the injured spinal cord. Therefore, CMA activation might attenuate the damage of neurons at the lesion site after SCI.
In a previous study using a PD model, dysfunction in CMA led to the accumulation of pathogenic protein in astrocytes. 14 The activation of CMA can decrease the pathogenic protein level in astrocytes and improve the pathological condition of PD. It was also reported that the activation of macroautophagy promotes the survival and function of oligodendrocytes and prevents demyelination in the spinal cord. 38 The activity of CMA increases in microglia/macrophages at the lesion site after traumatic brain injury. 22 Microglia/macrophages can contribute to the cytoprotective function following CNS injury. 39 The present study showed that the expression of LAMP2A was increased in astrocytes, oligodendrocytes, and microglia/macrophages following SCI. Therefore, the upregulation of CMA in glial cells may exert a cytoprotective effect reducing the glial cell damage and ameliorating demyelination in the injured spinal cord.
HSC70 has an important function of recognizing the substrate proteins in the CMA process and has been frequently used to investigate the activity of CMA. 40,41 On the other hand, several studies have indicated that the function of HSC70 is not specific to CMA and is involved in the other molecular functions as well, including folding of nascent polypeptides, protein translocation across membranes, and prevention of protein aggregation under stress conditions. 42,43 Further, HSC70 is also involved in a multitude of housekeeping roles in non-stressed cells during normal conditions. 44 A previous study investigated the CMA activity in traumatic brain injury and demonstrated that the expression of LAMP2A but not HSC70 significantly increased in the damaged neural tissue. 22 In the present study, the LAMP2A expression was significantly increased in the injured spinal cord. However, the expression of HSC70 was not markedly changed at the lesion site following SCI. The multiple molecular functions of HSC70 might be one reason why the level of HSC70 was unchanged after SCI.
The present study has several limitations. First, this study used the spinal cord hemisection model but not the contusion injury model. The spinal cord contusion injury model is universally accepted to be more clinically relevant because the majority of human SCI is contusive in nature. The pathology of contusion injury should be different from that of hemisection or transection injury. Therefore, the results of the present study using the spinal cord hemisection model might not directly reflect the pathology of spinal cord contusion injury in humans. Second, the present study cannot clarify how much the CMA activity influences the pathological mechanism at the lesion site following SCI. It is difficult to understand precisely whether the proportion of LAMP2A-positive cells in the result has a significant impact on the pathology in the injured spinal cord. A study to modulate the activity of CMA at the lesion site may reveal the pathological significance of CMA in damaged neural tissue following SCI. Third, the present study did not perform the quantitative evaluation in the electron microscopic analysis. Further studies using electron microscopy to provide the quantitative data may support our results.
Previous studies have shown that overexpression of LAMP2A is associated with reduction in the levels of aberrant proteins in the CNS. 15,22,23 The administration of mycophenolic acid, a potent CMA activator, rescued hypoxia-mediated cell death in a brain ischemia model. 23 Another study reported that retinoid derivatives activate CMA and contribute to its cytoprotective function against oxidative stress and proteotoxicity. 45 In addition, the activation of CMA was shown to induce a neuroprotective effect in a model of inflammatory demyelinating disease of the peripheral nerve. 24,25 Our results suggest that the increased expression of LAMP2A activates CMA at the lesion site in SCI. Therefore, the upregulation of CMA may contribute to the mechanism of elimination of pathogenic proteins and tissue protection in secondary injury after SCI. Future studies will be needed to further elucidate the neuroprotective mechanisms of CMA following SCI.
Footnotes
Acknowledgments
We thank Ms. Yuzu Adachi, Ms. Aya Umeki, and Ms. Michiko Fukuyama for their technical assistance and the animal care team at the Institute for Animal Experimentation at Tohoku University for providing the animal care in this study.
Funding Information
The authors received no specific funding for this work.
Author Disclosure Statement
No competing financial interests exist.