
Abstract
Traumatic brain injury (TBI) and spinal cord injury (SCI) present a significant contribution to the global disease burden. White matter tracts are susceptible to both the physical forces of trauma and cascades of pathological secondary degeneration. Oligodendrocytes, the myelinating cells of the central nervous system (CNS), and their precursors are particularly vulnerable cell populations and their disruption results in a loss of white matter, dysmyelination, and poor myelin repair. White matter aberrations in TBI and SCI can be visualized in vivo using a number of magnetic resonance imaging (MRI)-based modalities. Recent advances in diffusion MRI allow researchers to investigate subtle abnormalities in white matter microstructure and connectivity, resting state networks, and metabolic perturbations associated with injury. Damage to oligodendroglia underlies white matter aberrations and occurs as a result of glutamate excitotoxicity, intracellular calcium ion (Ca2+) overload, and oxidative damage to lipids, proteins, and DNA. Structural changes to myelin include myelin decompaction, loosening of myelin lamellae, and disruption to the node of Ranvier complex. Neuronal and functional loss accompany dysmyelination together with an increase in astro- and microgliosis. Remyelination is often partial, and more work is needed to understand deficits in remyelination post-injury to develop strategies to both protect and repair myelin and thereby preserve function. This review covers disruptions to oligodendrocyte function and white matter tract structure in the context of TBI and SCI, with an emphasis on Australian contributions in recognition of the International Neurotrauma Symposium held in Melbourne, Australia in 2020.
Introduction
Injury to the central nervous system (CNS) is a global health concern, with 27 million individuals suffering from traumatic brain injury (TBI) and approximately one million new cases of spinal cord injury (SCI) per year. 1,2 The main causes of TBI and SCI are falls, motor vehicle accidents, assault, and conflict-related blast injuries. 1 –3 Unsurprisingly, conflict-affected countries have the highest incidence rates of TBI and SCI. 2 The economic impact of CNS injury is significant in developed nations, with the lifetime burden of TBI costing up to $4.8 million per person in severe cases and exceeding $9 million for patients with tetraplegia following SCI. 4,5 TBI has also been found to have an epidemiological association with Alzheimer's disease and Parkinson's disease, and thus may carry an even greater socioeconomic burden. 6,7 Individuals at most risk for CNS injury are young adult males and the elderly. 1,8
TBI can be classified along a continuum of mild to severe, with a substantially worse prognosis in severe cases (30–40% mortality). However, the vast majority of TBI cases (70–90%) are mild (mTBI). 9 Clinically, mTBI has been defined as ≤30 min loss of consciousness; ≤24 h of transient amnesia, confusion, or disorientation; and a Glasgow Coma Scale (GCS) score between 13 and 15. 9,10 The acute symptoms of mTBI include vestibular and oculomotor dysregulation, sleep disturbances, and emotional dysregulation and tend to resolve within weeks of injury. 11,12
However, patients suffering from mTBI can have deficits in memory and executive function as well as cognitive impairment persisting for months to years post-injury. 11,13,14 Patients exposed to repeated mTBI have a worse prognosis, with successive hits exacerbating inflammation and yielding significantly greater long-term cognitive deficits. 15 Sports persons and military personnel are at particular risk for repeated injury, along with subsequent neurodegenerative conditions that may include chronic traumatic encephalopathy (CTE), sleep disorders, and neuroendocrine disease. 3,16 Prognosis is substantially worse in severe TBI, with 38% mortality in younger patients, 17 and 74% for patients older than 55 years. 18 Persisting somatic symptoms years after injury include headaches, impaired balance, and motor disability. However somatic symptoms are less frequent than cognitive complaints, with 60–70% of patients presenting with memory and concentration deficits at 8-year follow-up. 19 In pediatric severe TBI, long-term social development is perturbed compared with mTBI and healthy peers, leading to family dysfunction and poorer caregiver mental health. 20
Neurotrauma comprises two components: the primary or mechanical damage, and the biochemical cascade known as secondary degeneration. 21 Secondary degeneration is a self-propagating series of metabolic and structural changes, associated with increased intracellular calcium ions (Ca2+), that results in oxidative stress and apoptotic cell death of initially undamaged tissue. 22 Because the initial insult is often unavoidable, treatments for functional recovery after TBI focus heavily on limiting secondary injury. 23
Patients with TBI and SCI are treated on a symptom-based approach, with priority given to reducing intracranial pressure (ICP), hemorrhagic complications, and edema. 1 Cerebral edema following TBI remains the most significant predictor of adverse outcome, with altered cerebral perfusion pressures leading to secondary ischemic injury. 24 Utilization of ICP monitoring reduces mortality rates in patients with severe TBI. 25 Decompression surgeries to correct raised ICP have mixed success, with reduced disability observed in pediatric patients but worsened neurological outcomes reported in adult patients. 26 Despite evidence in animal models for the use of osmotic transport devices to reduce edema, 27 craniectomy is still required for implantation and the surgery procedure itself may exacerbate injury. 24 Barbiturates are administered to 20% of patients with severe TBI to reduce ICP in the absence of surgical intervention. However, despite reducing ICP in 69% of patients, European studies indicate that barbiturate administration has no significant effect on outcome at any stage following injury. 28 In patients suffering from mTBI, treatments have little efficacy and patient education on the likely resolution of symptoms and the value of adherence to follow-up is currently considered one of the most effective approaches in regards to ameliorating persistent symptoms. 29 Despite extensive neurotrauma research, there is no effective pharmacological treatment currently available for preservation of white matter following injury. 30
Diffuse axonal injury (DAI) is a form of CNS injury following TBI that typically manifests in around 10% of patients, but is far more common (44.9%) in cases of severe TBI. 31,32 DAI frequently affects the corpus callosum, brainstem, and white/gray matter tissue interface. The Adams DAI injury classification utilizes both clinical presentation and identification of lesions to diagnose three grades of severity, ranging from microscopic white matter changes (Grade 1) to severe focal lesions (Grade 3). 31 A subset of DAI and an injury often observed in animal models is traumatic axonal injury (TAI), wherein damaged axons are dispersed among normal appearing axons in white matter tracts. 33 White matter injury can also manifest as demyelination or dysmyelination, wherein myelin structure is perturbed. 34 White matter is particularly vulnerable to mechanical injury, with studies indicating a greater inflammatory response and secondary degeneration compared with gray and cortical tissues following TBI and SCI. 35,36 White matter vulnerability following trauma has been extensively characterized with various magnetic resonance imaging (MRI) techniques, which provide insight into structural, metabolic, and connectivity changes in the brain. 37 –39 The cellular mechanisms behind this selective vulnerability have been explored using animal models, with evidence suggesting that specific predilection of oligodendrocytes to oxidative damage underlies white matter injury. 40 –43 This review focuses on white matter injury by first providing background on oligodendroglia and myelin, assessing macroscopic structural changes to white matter following trauma and then describing what is known of the cellular changes in oligodendroglia following injury. The review describes white matter injury through the lens of oligodendrocyte damage, as these cells are particularly vulnerable and critical to myelin integrity. In celebration of the International Neurotrauma Symposium being held in Australia in 2020, the review features Australian research where appropriate.
Oligodendroglia
Collectively known as oligodendroglia, oligodendrocyte precursor cells (OPCs) and their progeny, oligodendrocytes, are one of the most abundant cell types in the CNS. In the adult rodent brain, OPCs and oligodendrocytes comprise approximately 5% and 20% of total brain cells, respectively. 44,45 OPCs are the main proliferative cell type of the CNS and are typically identified by their expression of the neuron glia antigen-2 (NG2), platelet-derived growth factor receptor alpha (PDGFRα) and/or oligodendrocyte transcription factor 2 (Olig2). 46 Lineage tracing of embryonic OPC development in mammals has shown that 80% of OPCs in the CNS arise from neuroepithelial progenitor cells located in the motor neuron progenitor (pMN), ventral midline region of the brain and spinal cord. 47 Dorsally derived OPCs, which originate from radial glia, consitute the remaining 20%. 48
Once generated, OPCs proliferate as they migrate laterally, dorsally, and ventrally from the midline, distributing evenly to various white and gray matter regions of the CNS. 49 Upon reaching their destination, OPCs can undergo two additonal stages of differentiation, initally becoming immature oligodendrocytes that express markers such as O4 and ectonucleotide pyrophosphatase/phosphodiesterase 6 (ENPP6). 50 Following this, oligodendrocytes mature progressively to express transcription factors and proteins including myelin regulatory factor (MyRF), NK6 homeobox 2 (NKX6.2), myelin basic protein (MBP), and the myelin proteolipid protein (PLP) that form myelin. 50 Myelin is a multi-layer lipid structure that ensheaths axons, thereby increasing the speed of electrical signal transmission across the CNS and providing structural support to neurons. 51,52 OPCs and oligodendrocytes have historially been considered homogenous cell populations. 53 However, accumulating evidence suggests that oligodendroglia are heterogenous, comprising various subpopulations with diverse characteristics, gene transcription profiles, and functions. 50
Oligodendroglia diversity
OPCs derived from different embroyonic regions produce oligodendrocytes that preferentially populate different regions across the adult CNS. 54 Indeed, oligodendrocytes originating from the ventral region of a developing mouse brain mainly populate the anterior commissure, pre-optic tract, and the lateral olfactory tract, whereas their dorsally derived counterparts predominatly localize to the cortex and corpus callosum. 48,54 Further, dorsally and ventrally derived oligodendrocytes preferentially myelinate dorsal and ventral regions of the adult CNS, respectively, 48 despite having a similar ability to generate mature oligodendrocytes. 55 Characteristic differences also exist between OPCs in white and gray matter, with the former having a fourfold faster rate of proliferation and a higher proclivity for maturing into myelinating oligodendrocytes. 44,52,56
With respect to transcriptional heterogeneity, single-cell RNA profiling of oligodendroglia in different regions of the mouse adult CNS identifed 12 transcriptionally distinct subpopulations, comprising OPCs, differentiation-committed OPCs, newly formed oligodendrocytes (NFOL), myelin-forming oligodendrocytes (MFOL), and mature oligodendrocytes (MOL). 57 In contrast to OPCs, differentiation-committed OPCs lack PDGFRα and NG2 but express genes such as sex-determining region y-box (SOX) 6, Neu4 and Bmp4 that keep oligodendrocytes undifferentiated. NFOL are distiguishable from MFOL by their expression of genes associated with early stages of differentiation (such as Tcf7l2 and contactin-associated-protein [Caspr]) and downregulation of genes involved in myelin formation, including myelin oligodendrocyte protein (MOG), proteolipid protein 1 (PLP1), and oplalin. MOL express genes associated with late oligodendrocyte differentiation and myelin formation simultaneously. Whereas OPCs and differentiation-committed OPCs were found to be transcriptionally homogenous, two populations of NFOL (NFOL1 and NFOL2) and MFOL (MFOL1 and MFOL2) and six populations of MOL (MOL1 to MOL6) with distinct gene expression profiles have been identified. 55
These subpopulations differed in spatial distribution within the regions of the CNS analyzed (i.e., amygdala, corpus callosum, cortex, zona incerta, dentate gyrus, striatum, dorsal horn, hippocampus, hypothalamus, and substantia nigra/ventral tegmental area; SN/VTA). NFOL1 and NFOL2 were found in every region investigated except the corpus callosum, where NFOL2 was absent. MOL subpopulations varied in region specificity, with some populations, such as MOL5, being present throughout the regions, whereas others were enriched in particular regions; for example, MOL2 were abundant in the spinal cord but almost absent in the brain. 55
Although the functional implications of the transcriptional heterogeneity and spatial preferences of oligodendroglia are still unclear, a recent study suggested that MOL subpopulations may fulfil distinct regenerative functions, as MOL2 were depleted at the site of SCI and, in contrast, MOL5/6 increased their contribution to lineage repopulation and thus remyelination. 55 The development of different oligodendroglia subpopulations and their progression from OPCs to mature myelinating oligodendrocytes is tightly regulated by complex interactions between several transcription, post-transcription, translation, and post-translation factors.
Molecular control of oligodendroglia development
During embryonic development of the CNS, distinct molecular mechanisms control the production of dorsally and ventrally derived OPCs. Sonic hedgehog (SHH) signaling induces ventral OPC specification by driving NKX6.1, NKX6.2, NKX2.2, and Olig2 transcription in neural stem cells. 58 By contrast, dorsal OPC generation is regulated in a SHH-independent manner by Dbx1 and Ascl1 transcription factors and fibroblast growth factor and bone morphogenetic protein (BMP) signaling. 48 As OPCs migrate from their birthplace to populate the CNS, a complex network of transcriptional and translational changes coordinate to mediate OPC differentiation. Calcineurin, a calcuim-dependent phosphatase, was recently identifed as a key initiator of OPC differentiation. 59 Triggered by an increase in intracellular Ca2+ levels, calcineurin dephosphorylates the nuclear factor of activated T cells, cytoplasmic 2 (NFATC2) protein, resulting in its activation. In turn, activated NFATC2 interacts with SOX 10 transcription factor to allow Olig2 and NKX2.2 co-expression, which is a prerequisite for OPC differentiation. 60,61 Numerous transcription factors have been reported to play a role in oligodendrocyte differentiation over the years, including nuclear receptors such as retinoid X receptors, vitamin D receptors, and peroxisome proliferator-activated receptors that contribute to oligodendrocyte formation and myelination. 62 However, Olig2, SOX10, NKX2.2, zincfinger protein 24 (ZFP24), and MyRF are widely recognized as the main regulators of full oligodendrocyte differentiation and myelination. 62 SOX10 plays a central role in this process, binding and directly activating several genes involved in oligodendrocyte maturation, including MyRF, which mediates terminal differentiation of immature premyelinating oligodendrocytes. 60,61 In addition, SOX10 induces expression of NKX2.2 and Olig2, the latter of which activates ZFP24 to generate myelin transcripts. 60
Genes involved in lipid and cholesterol synthesis are also associated with myelination of mature oligodendrocytes. 63 The transcription factors sterol regulatory element binding proteins (SREBPs) have been reported to control the supply of fatty acids for myelin formation through the mammalian target of rapamycin (mTOR) signaling pathway. 63,64
Oligodendrocyte development can also be regulated epigenetically. At the OPC stage of lineage progression, the transcription factors and histone-modifying enzymes that respond to extracellular stimuli are mainly inhibitory, preventing differentiation. 65 Long noncoding RNAs (lncRNAs) are approximately 200 nucleotides long, and regulate gene expression but lack protein-coding potential. 66 Oligodendrocyte-specific lncRNAs such as lncOL1 and lnc158 can downregulate inhibitors of histone deacetylase activity and heterochromatin formation to promote expression of oligodendrocyte differentiation genes. 67,68 MicroRNAs (miRNAs) are another subgroup of ncRNA that have been shown to regulate myelination in oligodendrocytes. They are short nucleotides (20–25 nucleotides long) generated by enzymatic cleavage of long transcripts. 69 Numerous miRNAs with a role in oligodendrocyte development have been identified and reviewed in the literature. 62 Of note, a recent study showed that oligodendrocyte-specific miRNA-219 and miRNA-338 collaborate to downregulate negative regulators of oligodendrocyte myelination including the transcription factors SOX6 and ETS variant 5. 70 In addition, miRNA-219 represses expression of proteins such as PDGFRα that maintain OPCs in their progenitor state, thereby promoting oligodendrocyte differentiation. 70
After oligodendrocytes are generated, myelination is triggered by the downregulation of precursor proteins such as PDGFRα and transcription of MyRF and its target proteins, which are essential for myelin formation, including MBP, PLP1, myelin-associated glycoprotein (MAG), cyclic nucleotide phosphodiesterase (CNP), and MOG. 71 Myelinating oligodendrocytes use their expanded network of processes to make stable contact with neighboring axons, enwrapping segments of the neuron body with myelin sheaths. 72,73 The structure and function of myelin is central to white matter composition and physiology as white matter is mainly made up of myelinated axons.
Myelin structure and function
Myelin has a very high lipid content, comprising phospholipids, galactolipids, and plasmalogens 74 that have an affinity for, and bind, myelin-specific proteins. 72 The myelin sheath consists of compacted double bilayers of myelin, with myelin proteins, particularly MBP, acting as an adhesive between the bilayer membranes. 75 Once matured, myelin can consist of up to 160 layers of membranous, compacted lamellae that can be visualized as interperiodic lines using electron microscopy. This myelin sheath allows for saltatory conduction of action potentials and refined neural signaling, 76 with myelinated axons conducting signals at a speed 20 to 100 times that of unmyelinated axons of similar diameter. It has long been known that the thickness of the myelin sheath is proportional to the diameter of the axon 77 ; however, it was recently discovered that both axonal diameter and the corresponding myelin thickness can vary along the length of a single axon. 78
Myelin has a highly segmented structure to facilitate neuronal signaling. 79 The myelin sheath produced by mature oligodendrocytes along an axon comprises multiple myelin segments, called internodes, separated by unmyelinated regions called nodes. 80 Oligodendrocytes myelinate axons of all diameters above ∼0.4 μm, 72 and a single oligodendrocyte can provide up to 50 internodes of myelin on different axons. 81 The length of internodes and processes of an oligodendrocyte limits its ability to myelinate the same axon twice. 51
The spaces between internodes are referred to as nodes of Ranvier. 82 Localized at the node of Ranvier are specialized sodium channels that facilitate and propagate action potential signaling. 83 Thermosensitive and mechanosensitive K2P potassium channels (TREK-1 and TRAAK) are clustered at the nodes of Ranvier and are the principal drivers of rapid action potential repolarization. 84 Directly adjacent to the node is the paranode, which consists of loops of myelin lamellae that are anchored to axons via the paranodal junction, which comprises contactin, neurofascin 155, and Caspr. 85 The paranode has multi-faceted roles in maintaining saltatory conduction; including (1) segregating nodal sodium and juxtaparanodal potassium channels; (2) stabilizing and attaching the myelin sheath to the axon to prevent myelin damage from stretch stressors; and (3) allowing only specific nutrients to diffuse into and out of the internodal peri-axonal space. 86 The juxtaparanode, which lies between the paranode and the internode, contains a high concentration of voltage-gated potassium channels, 83 although the function of these specific to their juxtaparanode location is currently unclear. 82
Neuron-oligodendrocyte interaction is integral for axonal integrity. In addition to their well-established function of ensuring fast action potential propagation and axonal insulation, oligodendrocytes supply neurons with nutrients. 87 Oligodendrocytes transfer energy metabolites including pyruvate and lactate to neurons, through cytoplasmic channels and monocarboxylate transporters, allowing fast adenosine tri-phospate (ATP) synthesis. Further, oligodendrocytes contribute to the maintenance of axon integrity and survival of neurons by producing neuroprotective and trophic factors such as ciliary neurotrophic factor (CNTF), insulin-like growth factor-1 (IGF-1), and glial cell line-derived neurotrophic factor. 88 Indeed, partial ablation of oligodendrocytes causes axonal degeneration, even in the absence of widespread demyelination. 89 Loss of oligodendrocyte-specific proteins such as PLP and CNPase results in axonal swelling and degeneration, even if myelin structure remains normal. 90,91 These seminal studies confirm that oligodendrocytes play multiple roles in neuronal maintenance. A further means of interaction is the release of trophic factor exosomes from oligodendrocytes, which are taken up by neurons and improve their metabolic activity. 92 In addition, oligodendrocyte-neuron interactions are adaptive and plastic, with neural signaling promoting oligodendrogenesis and increased myelin thickness. 93 –96
Other than acting as a pool of progenitors for oligodendrocytes, OPCs play multi-faceted roles in the CNS. Ablation of OPCs results in excessive inflammation, suggesting OPCs may suppress neuroinflammation in a healthy system. 97 It has recently been discovered that peri-vascular OPCs wrap their processes around blood vessels in the brain and may be partly responsible for regulating and modulating the neurovascular unit. 98
Specifically, OPCs play a role in maintaining blood–brain barrier (BBB) integrity via communication with cerebral endothelial cells, 99 including via transforming growth factor (TGF)-β signaling. OPCs located around blood vessels secrete TGF-β1, which activates the mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinases (ERK) pathway in cerebral endothelial cells, increasing expression of tight-junction proteins and protecting BBB integrity. 100 In turn, cerebral endothelial cells have been found to release growth factors such as brain-derived neurotrophic factor and basic fibroblast growth factor, which promote the survival and proliferation of OPCs, both of which are significantly decreased following stroke. 101,102 This trophic coupling between the cerebral endothelium and neighboring OPCs has been dubbed the oligovascular niche. 102 In vitro studies have also shown that astrocytes can support OPCs, via MEK/ERK and PI3K/Akt signaling, 103 and OPCs and peri-cytes could potentially secrete factors to support each other. 104 Further, within the developing brain, OPCs secrete hypoxia-induced factor to promote angiogenesis of the CNS vasculature. 105
Etiology of White Matter Injury
The etiology of white matter injury is complex, heterogeneous, and its magnitude relates specifically to the nature of the primary injury. 15 White matter pathology manifests in a wide array of symptoms and presentations depending on the nature and timing of injury as well as stage of disease progression. 15 Generally, injury from the acute mechanical forces manifests as cortical focal lesions at the site of impact and the contrecoup region. More subtly, diffuse white matter injury can include myelin decompaction or unfurling of the myelin sheath, and TAI. Wallerian degeneration can take place following injury: an immune-mediated response where primary discrete axonal injury spreads distally leading to irreversible fragmentation distant from the injury site. 106,107 Oligodendrocytes are unable to clear myelin debris, leading to a prolonged and gradual reduction of myelin and long-term deposition of a glial scar. 108
In TBI, injury also results from both linear and rotational acceleration and impact deceleration forces on the brain. The brain's complex gyrencephalic structure and inhomogeneous tissue properties contribute to a non-linear dynamical behavior of deep white matter during trauma. Consequently, deep white matter tracts are particularly susceptible to injury, including: the corpus callosum, brainstem, thalamus, hippocampus, and the white/gray matter tissue interface. 109 Rotational direction-specific vulnerabilities exist due to the arrangement of fibers in these white matter tracts of the brain, 110 and as a result of the anisotropic effects of axonal arrangement, deep white matter tracts are particularly susceptible toTAI. 109
Resultant insults include mechanical damage from shearing, tearing, or stretching of axons, neurons, glia, and blood vessels. 15,107 Tearing of axonal tau-microtubule bonds by external mechanical forces damages the cytoskeleton, affecting axonal transport, ion flux, and initiating secondary axonal degeneration. 111 The initial injury may result in additional pathologies such as hemorrhage or cerebral edema; however, these complications only occur in approximately 10% of mTBI cases and could be considered “complicated” mTBI. 1 In SCI, raised intrathecal pressure due to edema or hemorrhage exacerbates the effect of secondary degeneration leading to deleterious functional outcomes. 112 Axons adjacent to the injury site are vulnerable to secondary damage due to the release of toxic factors by injured neurons and glia, associated with an inflammatory immune response. 113,114 The inflammatory immune response is greater in white matter compared with gray matter in both the brain and spinal cord, indicating a differential degree of secondary degeneration following injury. 35 Secondary degeneration is characterized by myriad reactive metabolic pathways, including astrocyte reactivity, microglial and macrophage activation and infiltration, glutamate and ATP induced excitotoxicity, intracellular Ca2+ overload, mitochondrial dysfunction, oxidative stress, and apoptotic cell death and dysmyelination. 22 Oligodendrocyte and OPCs are particularly vulnerable to these degenerative processes, and their death is associated with chronic dysmyelination after injury: 34 this phenomenon will be addressed in greater detail in sections below.
Models of CNS injury
For the mechanisms of white matter degeneration following CNS injury to be studied, it is important to utilize reliable animal models of injury. Animal models can be employed to replicate human TBI and SCI, and experimental models can be amended to study specific facets of heterogeneous pathologies, allowing mechanisms to be studied in a rigorous and controlled manner. 115,116 Rodent models of injury are typically employed by researchers as they are cost and time effective; however, many translational challenges exist due to inter-species differences in neurometabolism, lifespan, and anatomy. 116 Animal models of SCI include spinal contusion and compression, which best simulate the biomechanics and pathology of human injury. 115 Most SCI models use a thoracic injury site as opposed to cervical, as the thoracic SCI model tends to be more reproducible, has a lower risk of mortality, and permits the study of secondary white matter deficits. 115 Prominent models of TBI include: blast injury models, fluid-percussion injury (FPI), controlled cortical impact (CCI) models, and closed-head weight-drop models. 117 –119 Additionally, TAI can be induced in mice using closed-skull stereotaxic impactors delivering single or repeated mTBI to study microstructural changes with electron microscopy and observe long-term behavioral changes. 120 –122
Although all of the aforementioned models robustly elicit an inflammatory response and white matter injury, FPI and CCI models (which require the animal to be stationary) fail to mimic the human head kinematics following TBI. 15,116 The closed-head impact model of engineered rotational acceleration (CHIMERA) is a recent innovation that allows highly reproducible and accurate mTBI delivery. 123 The CHIMERA model induces DAI, behavioral changes, and energy dose-dependent increases in axonal damage. 124 Closed-head weight-drop models have also been employed in recent years, as they approximate the angular and rotational forces of human TBI and allow study of repeated TBI. 15,116,125 White matter reductions and demyelination are observed in the corpus callosum in the closed-head weight-drop model; therefore it remains an appropriate method to replicate human pathology. 15 A caveat of the rodent model is that there is proportionally less cerebrospinal fluid (CSF) as compared with humans; therefore rotational effects are not necessarily comparable. 126,127 Angular acceleration forces are reduced in rodent brains due to their small physical size; therefore white matter injury may be reduced and less clinically relevant. Large animals have a higher white-to-gray matter ratio due to their gyrencephalic structure, and therefore are better suited for translational validation of novel discoveries in rodent models. 127
In many models of neurotrauma, it is challenging to distinguish between primary and secondary damage, as there is often no clearly defined margin between tissue damaged by the initial insult and spared tissue. 128 Partial optic nerve transection is an established and useful model for investigating secondary degeneration of a white matter tract that features a primary injury of a penetrating nature that does not mechanically damage adjacent tissue. 129 In the model, the right dorsal optic nerve is partially transected, which allows for spatial separation between primary and secondary injuries. 129 The partial optic nerve transection model results in transient disruption of the BBB, which can be a useful feature for proof of principle investigations into secondary degeneration of white matter with a transiently open BBB. 130 The optic nerve stretch model allows for the study of wallerian degeneration often seen in CTE. 131 The stretch model and the transection model differ as the stretch model analyzes distal injured axons, whereas in the partial transection model uninjured axons ventral to the injury site are analyzed for secondary degeneration.
Imaging to Detect Changes to White Matter
Computerized tomography (CT) scans are the primary clinical diagnostic tool employed following TBI and SCI, 132,133 and total emergency room usage of CT has increased by 330% in recent years. 134 Although CT scans are critical and effective for detecting macroscopic lesions, edema, and hemorrhage, 90% of scans are negative for clinically relevant brain injury. 135 CT scans therefore lack the sensitivity to detect the more subtle, diffuse changes that occur following TBI such as TAI. CT scans have robust efficacy in detecting bone fractures, which are a correlate of worse outcomes following severe TBI. 136 Micro CT also provides a promising avenue to study microstructural changes in trabeculae of rodent skulls following TBI, allowing inferences to be made in regards to damage from repeated TBI. 137 Although CT scans following SCI have robust efficacy in detecting fractures, early collar removal following negative CT scans may lead to further injury, 138 and follow-up magnetic resonance imaging (MRI) scans are recommended in obtunded blunt trauma patients. 139 MRI has significantly greater sensitivity in detecting white matter lesions following mTBI compared with CT; therefore it remains the imaging modality of choice when researching structural correlates of clinical outcomes. 140,141
Detecting lesions and volume changes
MRI is a reliable tool that can provide high-resolution images allowing for the detection of white matter lesions, detected as changes in iron and fluids due to hemorrhages and swelling in the brain. In a blinded study in the acute phase of TBI or minor stroke (<48 h post-injury), clinicians were able to accurately discriminate TBI from acute minor stroke: MRI-facilitated detection of vascular abnormalities in 54% of patients with TBI, whereas only 14% of patients had positive vascular findings using CT. 142 Hyperintense white matter lesions can be detected following TBI using T2-weighted fluid-attenuated inversion recovery (FLAIR) sequences. FLAIR nullifies body fluids during acquisition, therefore suppressing CSF effects on image contrast and allowing peri-ventricular hyperintensities and meningeal focal enhancements common in TBI to be detected with greater accuracy. 142,143 Presentations of lesions following TBI are heterogeneous. Although the number and volume of lesions correlates with adverse outcomes, 144 the presence of white matter hyperintensities and microhemorrhagic lesions in mTBI are uncommon and do not necessarily predict persisting symptoms. 145,146
However, in cases of moderate and severe TBI, microhemorrhages are an independent predictor of disability (GCS-extended score ≤6; odds ratio = 2.5) in the absence of axonal injury. Therefore, caution should be taken when interpreting lesion data, as microhemorrhages have a significant role in TBI. 147 Additionally, T2-weighted MRI may be limited in its utility as lesions do not often appear in the acute phase following mTBI, but are more often detected at 6 month follow-up in cases of persisting symptoms. 148 However, in cases of severe TBI, lesions referred to by the authors as TAI in FLAIR sequences in the corpus callosum and the brainstem are predictors of reduced functional outcomes at 12 month follow-up. 149 Nevertheless, lesion presence alone does not provide sufficient prognostic clinical value.
Brain volume changes provide insight into gross structural changes to white matter in the brain. Reductions in cortical thickness provide an indication of widespread atrophy in patients with severe TBI and these changes are associated with deficits in declarative memory and auditory verbal learning. 150 Cortical thinning is exacerbated in patients with repeated mTBI and veterans exposed to blast injuries following mTBI. 151,152 Despite cortical thickness reductions often taking place in tandem with reductions in white matter integrity, 150 changes to white matter integrity can take place independent of cortical thinning. 153 Widespread atrophy is also inferred by total brain volume reductions, with patients with severe TBI having on average a 4% reduction in brain volume at 12 months post-injury, indicative of long-term TAI. 154 Additionally, atrophy is greatest in white matter compared with gray matter. 155,156 Regional brain volume reductions have been observed in the white matter of the anterior cingulate, 156 brainstem, thalamus, and corpus callosum. 154 In SCI, white matter volume loss is prevalent in the corticospinal tract, although gray matter atrophy appears to be more robustly evident compared with heathy controls. 157 Additionally, ventricular enlargement and CSF volume increases may serve as a surrogate marker of ascending neurodegeneration following SCI. 158 Taken together, it is clear that brain volume changes provide clinically relevant insight into longitudinal atrophy of both white and gray matter.
DTI connectivity changes
White matter fasciculi can be accurately mapped and characterized using diffusion tensor imaging (DTI) techniques. DTI utilizes the anisotropic nature of water displacement in white matter to derive values such as fractional anisotropy (FA), which characterizes diffusion anisotropy differences between intensity limits of zero and one. In highly organized white matter tracts, FA values are high (∼0.8) due to diffusion approaching unity along white matter tracts and diffusion in all directions perpendicular to tracts. In gray matter, FA values generally vary (∼0.10–0.25) depending on the structure of the region concerned. 159 In CSF, FA values approach zero as water diffusivity is equal in all directions. 160 Mean diffusivity (MD) is another common measurement and quantifies mean diffusion in each direction as opposed to FA, which quantifies the total magnitude of diffusion along axonal fibers. FA is calculated as a ratio of axial diffusivity (AD) and radial diffusivity (RD), which describe the magnitude of water diffusion parallel and perpendicular to the tract, respectively. 161 In cervical SCI, mean FA values are reduced at the site of injury and mean MD values are increased, compared with healthy controls. The decrease in FA value may reflect a decreased degree of myelination in the tracts or increased cell density, whereas higher MD may be indicative of white matter tract disorganization or fluid edema. 162
Diffusion basis spectrum imaging (DBSI) is a novel technique that shows promise in elucidating underlying pathology behind diffusion metric changes. 163 DBSI allows quantification of white matter tract density, as well as residual axon and myelin integrity and axonal volume while accounting for vasogenic edema. The DBSI technique has been validated in a traumatic SCI study in mice, wherein co-existing pathologies were identified non-invasively and then correlated with post-mortem histology markers. 163 Clinically, a decrease in FA values is also seen at the thoracic level below the injury site, 164 and persists in ascending tracts into cerebral white matter tracts, with reduced FA and increased MD in the corpus callosum, corona radiata, cingulate gyrus, and the thalamic radiation years post-injury. 165 White matter disruption is worse in patients with cervical compared with thoracic injury, and time since injury is negatively correlated with FA values in some brain regions. 165 These DTI changes are consistent with wallerian degeneration, with discrete white matter injury in the spinal cord causing demyelination and structural disorganization distal to the injury site. 106,164,165
Unlike SCI, DTI findings from TBI studies lack clinical consensus and are far more heterogenous. 161 An extensive review by Asken and colleagues 166 postulates that variability in the changes in FA and MD values after injury are attributable to different characteristics of the study populations and comparative control groups. 166,167 In the acute phase of mTBI, FA values increase and MD values decrease in the days following injury. Specifically, FA values have been seen to increase in the corpus callosum, 168 fimbria, and the internal capsule. 169 These FA and MD changes are believed to be due to axonal cytotoxic edema in the initial phases of white matter degeneration following injury. 168 –170
Axonal swelling can occur due to interruption of axonal transport and subsequent accumulation of transported materials, or it can manifest in axonal bulbs following complete axonal disconnection. 171 In a rodent CCI study, acutely increased FA in the genu corpus callosum was accompanied by decreased RD and MD, with no changes in AD observed. The changes in FA observed using DTI were unable to be detected by anatomical MRI or hematoxylin and eosin staining. 168 FA decreases in the contralateral cortex, thalamus, and hippocampus at 7 days have also been observed in a recent Australian study following CCI in rodents, validated by cross-sectional histopathology. 172 Accompanying acute injury in rodents, increased FA values have been associated with increased expression of tumor necrosis factor α (TNF-α) and a reduction in MBP staining. This supports the hypothesis that increased FA values immediately following injury are a result of cytotoxic processes affecting myelin integrity and therefore diffusivity along white matter tracts. 169 Wright and associates recently observed similar changes in DTI measures associated with changes to telomere length. Repeated mTBI resulted in reduced telomere lengths and these changes were associated with reductions in AD and RD. 173
An inverse pattern is observed in mTBI as time passes, wherein FA values decrease and MD values increase. 174,175 Multiple injuries in animal models exacerbate changes in diffusivity metrics and are also associated with hypoconnectivity in the hippocampus, brainstem, and cerebellum. Interestingly, rats exposed to a single mTBI display hyperconnectivity between brain regions, suggesting an adaptive response to injury. 175 A similar finding was observed following single CCI, wherein the brain was connectively promiscuous post-injury in the thalamus and cortex, perhaps reflecting circuit reorganization. 176 Following repeated mTBI in mice, no changes in FA are observed acutely, but after 6 weeks FA is significantly reduced in the corpus callosum compared with sham animals. This corpus callosum FA reduction occurred in tandem with microglial activation detected by post-mortem immunohistochemistry, with activation peaking at 7 days post-injury. 174
Whereas DTI can be used to detect chronic white matter aberrations, the many changes in the tissue and cells within voxels may contribute to changes in DTI measures in ways not yet fully understood. The temporal inversion of FA values is reflected in the clinical findings of DTI. A longitudinal study of DTI metrics found FA values decreased from the acute phase following mTBI (<7 day) to the subacute phase (8 days to rehabilitation discharge). The variability of FA values in the acute phase means that DTI is more likely to be useful as a prognostic tool subacutely. 177 MD values are higher in athletes who have received multiple concussions compared with those who have had single or no concussions, in the absence of any major neuroanatomical differences. 178
In severe chronic TBI patients, increased FA values in the splenium of the corpus callosum have been associated with better performance in social inference tests. Despite a number of gray matter abnormalities, the changes in the corpus callosum were those most implicated in functional deficits in these TBI patients. 179 In combat veterans who suffered TBI while deployed, those with higher FA values and lower MD values have a greater likelihood of returning to work post-deployment. 180 In terms of long-term prognosis, higher FA values in the corpus callosum in the acute phase following injury are associated with higher disability rating scores. Whereas in the subacute period, higher FA values in the splenium correlate with lower disability rating scores. 177
Some brain regions, such as the internal capsule and the fronto-occipital fasciculus, show recovery from a transient reduction in FA values. Increased FA values at 1 month follow-up are positively associated with better performance on cognitive information processing speed at initial assessment. However, continued loss of integrity is observed in the corpus callosum, forceps major, and anterior corona radiata at 3 months, and these regions may serve as an objective biomarker of pathology that can predict persisting post-concussion symptoms (PPCS). 181 Hirad and co-workers 182 used accelerometers and DTI imaging on college football players and tracked head impacts across a competitive season in conjunction with an mTBI cohort of patients. FA in the midbrain was reduced post-season compared with pre-season, as was the case for the right midbrain in the mTBI patients. Reductions in midbrain white matter integrity in the football players were related to the amount of rotational acceleration sustained as opposed to linear acceleration, 182 and this is consistent with previous biomechanical models of TBI. 109,110 Serum-based tau, a marker of axonal injury and BBB integrity, was inversely correlated with midbrain FA values in the mTBI patients. 182
FA and MD metrics can also be used to add structural insight to neuropsychological outcomes in TBI patients. In a recent Australian study, 183 patients with severe TBI had widespread white matter pathology in the corpus callosum, longitudinal fasiculi, corticospinal, brainstem, and cerebellar tracts. Diffuse white matter integrity disruption in these patients, particularly changes in the thalamus and fornix, was associated with social information processing deficits. 183 Diffusion tensor techniques can also be applied to derive individual patient structural connectomes, in which cognitive associations can be explored. Mishra and associates 184 created a structural connectome using average FA values and brain atlas-defined nodes to assess differences between cognitively impaired and non-impaired professional fighters. White matter reorganization in the hippocampus, precuneus, and insula was detected due to repeated head injury in fighters exhibiting cognitive decline. 184 Recent work 185 in white matter plasticity used DTI techniques as well as myelin water fraction and longitudinal relaxation rate R1 to assess subtle changes in myelination and connectivity in healthy subjects following working memory training. The study highlighted the sensitivity of the R1 measure in detecting myelin remodeling following working memory training, which occurred in conjunction with changes in FA and RD parameters. 185 Given R1 values may be more specific in detecting changes in oligodendrocytes. 186 Future studies could employ this measure to assess white matter regeneration following cognitive rehabilitation in TBI patients.
Novel methods for increased sensitivity
The diffusion tensor model faces a number of limitations that restrict its sensitivity in detecting microstructural changes in white matter pathology. One of these limitations lies in the fact that DTI assumes a single fiber orientation and is confounded at regions of “crossing” fibers. This can be overcome by analyzing apparent fiber density (AFD), utilizing a DWI post-processing technique known as constrained spherical deconvolution. In a recent rodent FPI study conducted by Wright and colleagues, DTI and AFD measures were both able to detect white matter changes in the corpus callosum: a uniformly orientated tract. However, DTI was unable to detect any changes in the corticospinal tract, whereas there was an observed reduction in AFD in the same animals. 167 Tract weighted imaging (TWI) is a novel technique that utilizes constrained spherical deconvolution to better characterize the fiber orientation distribution in regions of interest, allowing characterization of tract density, curvature, and length as additional measures that may be more sensitive to white matter pathology. 187 Using TWI in an awake closed-head model of injury, Australian researchers 188 found that tract density is significantly increased in the fimbria and contralateral external capsule following injury. Additionally, TWI detected changes between sham and TBI group at day 1 post-injury, whereas DTI metrics only detected changes at 7 days. 188
Another inherent limitation of DTI is that it applies a Gaussian distribution model to tissue using a normative distribution. In reality, the complexity of brain tissue means that water diffusion deviates significantly from this pattern. 189 As an example, when oligodendrocyte apoptosis occurs in combination with vasogenic edema, the result is enhanced diffusivity across axons. This pathological state results in a discrepancy between RD and histological outcomes. 190
Diffusion kurtosis imaging (DKI) is a non-Gaussian distribution measure that accounts for tissue heterogeneity and provides improved characterization of white matter integrity. Common DKI parameters include: mean kurtosis (MK) and mean kurtosis tensor (MKT), which reflect the average diffusion in all directions; and axial kurtosis (AK) and radial kurtosis (RK), which quantify diffusion along the axial or radial direction of the diffusion ellipsoid, respectively. 189 In a rat CCI study utilizing both DTI and DKI techniques in vivo, animals were imaged acutely (>2 h) and subacutely (7 days) post-injury. 191 In the acute phase, both DTI and DKI parameters detected differences in the injury versus sham animals. FA increased in the hippocampus and the ipsilateral cortex and MD decreased across all regions. However, at subacute testing most DTI changes had returned to baseline with the exception being a reduction in FA in the ipsilateral cortex. DKI measures were able to distinguish changes in both white and gray matter microstructure at the subacute period, with an increase in MK observed. 191 In addition to being more sensitive than DTI metrics in this study, kurtosis abnormalities scaled inversely with distance from impact site, therefore providing some discriminatory power to injury severity. 191 However, at longer time-points post-injury (42 days), it was found that DKI metrics did not provide additional sensitivity for detection of abnormalities in the corpus callosum compared with DTI metrics. 174
Similar to DTI metrics, a time-dependent biphasic response has been observed in MK values following injury, whereby MK values initially decrease following injury and later reverse 7 days following injury. This biphasic change in MK values is believed to represent a shift from cytotoxic to vasogenic edema. 159 Another confounding factor pertains to astrocyte activity following injury, as increases in FA can be falsely interpreted as remyelinating processes when in reality they reflect glial scar formation. 192 Increases in MK have been associated with astrogliosis, 191 and occur in tandem with higher density of microglia and astrocytes. 193 A recent Australian study 194 used a longitudinal rodent weight-drop design and assessed both DTI and DKI metrics. The study commended the sensitivity of both diffusion metrics; however, it highlighted that kurtosis measures were highly sensitive to changes in astrocyte markers at 3 months. Additionally, axial measures were not changed to the same degree as RK and RD metrics in the injury group. Therefore it is postulated that axonal integrity is maintained and the radial diffusivity changes may reflect cellular aggregations from glia, although these changes may also be due to demyelination. 194 However, a recent clinical mTBI study found that AK values were positively correlated with working memory deficits, indicating axonal perturbations. 195 Previous rodent work has also found correlations between DKI parameters and neuron loss, microglia activation, and myelin disruption. In addition, changes in MK values in the ipsilateral cortex and hippocampus are associated with deficits in cognitive function. 196
Advances in diffusion imaging techniques allow neuronal projections, known as neurites, to be imaged in vivo. The model used is known as neurite orientation dispersion and density imaging (NODDI), and estimates the microstructural complexity including dendrites and axons. 197 When NODDI is employed in conjunction with traditional DTI metrics, greater insight into the pathophysiological underpinnings of injury can be achieved. Repeated mTBI in martial arts athletes results in decreased FA, an increase in orientation dispersion index (ODI), and increased intracellular volume fraction (VIC), therefore inferring a potential role of edema in traumatic injury. 198 In concussed athletes, increased FA and decreased MD was detected alongside increasedVIC and decreased ODI. The increase in neurite density and coherence of neurites in the white matter of sports-concussed athletes likely reflect an adaptive reparative response to injury, especially given the lack of functional impairment present. 199 In contrast, in a study conducted in athletes who suffered mTBI, reduced FA occurred concurrently with a reduction in VIC, in the absence of any significant changes in ODI at 7 days post-injury. These changes may be associated with glial-mediated edema and the initiation of a neuroinflammatory response to injury. In athletes scanned after being given medical clearance to return to play, ODI was increased in individuals with elevated symptoms at initial consultation. Therefore, the change in neurite geometry may reflect a longitudinal response to injury, and may be a sensitive measure of clinically asymptomatic neuroplastic changes in the brain. 200 A study comparing patients with mTBI with orthopedic trauma controls observed decreased FA and increased MD in anterior tracts at 2 weeks post-injury, with a concomitant elevated free water fraction (FISO) in white matter regions indicative of vasogenic edema. However, at 6-month follow-up, no significant differences were detected in DTI metrics, despite a longitudinal decrease in white matter neurite density on NODDI. Additionally, associations were found between ODI and FISO and rate and magnitude of functional improvement within the mTBI group. 201 In summary, NODDI augments the sensitivity of existing DTI metrics and further studies are warranted to explore its prognostic and research value.
Resting state and connectivity changes
Patients suffering trauma also have changes to functional connectivity within the brain that can be mapped and quantified using MRI-driven techniques analyzing the default mode network (DMN). The DMN spans the hippocampus, precuneus, cingulum, inferior parietal lobe, and the orbital frontal, medial prefrontal, lateral temporal, lateral parietal, cingulate, and parahippocampal cortices 202 –204 and is activated during rest or self-reflective periods but suppressed during attention or goal-oriented tasks. 202,205,206 The DMN is suspected to be involved in integrating multi-modal information and evaluating the cognitive and homeostatic environment, and is connected by white matter tracts across the brain. However, its exact function is still unknown. 203 The DMN is frequently used to characterize resting-state connectivity changes in neurological disorders, but has only recently been applied to examining functional outcomes of mTBI. 205,207 A reduction in resting state functional connectivity (rs-FC) within the DMN during the acute stage of mTBI injury has been demonstrated, 202 consistent with significant reductions in rs-FC in more severe TBI populations shortly after injury. 208,209
Considerable variability exists in DMN rs-FC reductions in patients with mTBI during the subacute stage of injury across studies. These inconsistencies may be attributed to differing times of observation post-injury, heterogeneous inclusion criteria, and the innate differences in patients' rate of recovery. 210 Patients with mTBI have rs-FC within the DMN at the chronic stage of injury. 202,210,211 Interestingly, patients with mTBI at the chronic stage have increased rs-FC between the DMN and the task positive network (TPN). 202 Additionally, there is increased rs-FC between the TPN and the medial prefrontal cortex (MPFC) and dorsal posterior cingulate cortex (dPCC) nodes of the DMN, with concomitant increases in rs-FC between the DMN and the left dorsolateral prefrontal cortex (L-DLPFC). 202 An increase in rs-FC between the DMN and dorsal anterior cingulate cortex (ACC) and bilateral insula has been observed at both chronic 202 and subacute stages. 212 The bilateral insula is part of the salience network (SN), 213 and is sometimes co-activated with the TPN. 214 This increased connectivity between the DMN and regions of the SN led to suggestions that there is a demand for the SN to modulate the activity of the disrupted DMN following mTBI, 202 and that the increased modulation is a possible cause of increased cognitive fatigue seen in patients with mTBI. 215 The changes in functional connectivity were found to persist for up to 6 months post-injury and likely reflect DAI in white matter. 202
Cerebral blood flow (CBF) in the brain is also altered upon mTBI. 202 Imbalances between resting arterial spin labeling (ASL) perfusion within the DMN and TPN are found at the chronic stage, 202 leading to suggestions that recovery from mTBI is a highly dynamic mechanism that involves CBF changes to balance out the basal activity of the two networks. 202 The altered CBF allocation to the two networks may also contribute to the cognitive fatigue, 215 and attentional deficits seen in patients with mTBI. 216
Patients suffering from persistent post-concussive symptoms (PPCS) fail to maintain innate balance between perfusion to the DMN and TPN. 202 An increase in resting perfusion in the TPN nodes is seen in patients suffering from PPCS, 202 and patients with PPCS were unable to uphold network perfusion patterns comparably with non-PPCS mTBI and control participants. Therefore, it has been suggested that during the initial stages of injury, it may be possible to use network perfusion patterns to determine which patients will develop PPCS. 202 However, the relevant data involved two groups of patients with mTBI of varying ages and changes in perfusion may be due to the age difference, 202 further confounded by the fact that older patients are at greater risk for developing PPCS. 217
Following mTBI, common complaints include cognitive fatigue, headaches, and emotional and vestibular dysregulation. 11,12,218 Children may also experience mood changes, sleeping difficulties, and concentration and memory issues. 219 Despite this, neuropsychological tests rarely detect differences in objective performance on working memory tasks in children after mTBI. 218 Children and adolescents represent the population that is most at risk for incurring an mTBI, 220 hence the Australian Barlow group including the KidStim Laboratory, focus on mTBI in children. 218 Working memory is described by the Barlow group as the ability to hold recent information and transiently manage it for goal-directed behaviors. 218 Working memory-related cortical activation is decreased in children who have not recovered from mTBI at 1 month post-injury. 218 Additionally, similar regions of cortical hypoactivation have been found in children 41 days post-injury. 221 However, other studies have demonstrated hyperactivation in cortical areas. 222 –225 These inconsistent findings are possibly the result of heterogeneous memory tasks, inconsistent inclusion criteria, and differing age ranges between studies. 218 Although the Barlow group found there to be similar working memory-related cortical activation and neurocognition in patients with mTBI and healthy controls, children showing signs of PPCS demonstrated greater inhibition of the DMN and hypoactivation in the DLPFC during the working memory task, compared with children showing no sign of PPCS post-injury. 218 Despite these changes, no performance differences were detected. 218 Resting-state data therefore provide an insight into internodal activity and a useful platform to characterize changes post-injury; however, more research is needed to achieve accurate prognostic value.
Metabolic changes
In addition to structural and functional aberrations in white matter following injury, a number of metabolic changes reflective of gliosis or pathology can be detected using magnetic resonance spectroscopy techniques. Magnetic resonance spectroscopy has been used to detect reductions in ratios of N-acetylaspartic acid (NAA), choline (Cho), and creatine (Cr) in the genu of the corpus callosum following mTBI in athletes regardless of number of hits. 39 Athletes recovering from their first mTBI showed the greatest alteration in NAA/Cho and NAA/Cr ratios, perhaps indicative of a tolerance effect with further hits. 39 Decreases in NAA/Cr ratios have been observed in the thalamus in mTBI patients with impaired cognitive function, and an increase in thalamic Cho/Cr ratio was evident in mTBI patients with self-reported sensory symptoms. 226 However, the NAA/Cho and NAA/Cr ratios do not differ with the severity of symptoms. 227 A substantial literature explores the relationship between metabolite changes in the brain following TBI, including in white matter. These have been extensively reviewed, 228,229 and will not be covered further here.
Quantitative susceptibility mapping (QSM) is a novel imaging technique that allows in vivo measurement of magnetic biometals (iron) and high concentrations of Ca2+. 230,231 The technique has displayed sensitivity in detecting changes in neurodegenerative disease, such as Parkinson's disease 230 and Alzheimer's disease, 232 and has been used in TBI research. 233,234 Iron deposition in the cortex has been associated with cognitive decline, and is believed to induce oxidative stress and a pro-inflammatory response. 232 Abnormal accumulation of iron in white matter can be caused as a result of microhemorrhage following TBI, 233 and in patients with severe TBI, iron oxide targeting P-selectin microparticles can be used to identify endothelial activation in the cortex and hippocampus. 235 In a susceptibility-weighted imaging study in patients with chronic mTBI, abnormal iron depositions were detected in the hippocampus, thalamus, and the splenium of the corpus callosum at 6 months post-injury. Additionally, iron accumulations in the right substantia nigra are positively associated with cognitive impairments in patients with mTBI. 236
More recently, QSM has been utilized in a FPI rodent model to visualize thalamic Ca2+ influx in severe TBI, repeated mTBI, and single mTBI. No calcifications were detected 1 week post-injury; however, at 4 weeks calcifications were detected in all injury severities. The repeated mTBI group had the largest calcification volumes, a higher prevalence of calcifications compared with control and sham animals, and calcium increases in repeated hits were associated with neurological disability. 234 Another novel technique is flortaucipir positron emission tomography (PET), which allows the study of tau pathology in relation to TAI. Flortaucipir PET allows for in vivo detection of abnormal tau in neurofibrillary tangles, which traditionally has been used as a marker of pathology in post-mortem Alzheimer's disease tissue. In patients who received a single moderate TBI, flortaucipir binding was increased many years after injury compared with healthy controls. This increase in flortaucipir binding was associated with the presence of TAI, and with CSF markers of neurodegeneration T-tau and P-tau. 237 Future analysis of biometals and metabolites may provide insight into subtle changes in white matter in patients with TBI.
Detection of Cell-Specific White Matter Pathologies
Glutamate excitotoxicity and changes in calcium ions and reactive species
In addition to the primary mechanical insult, white matter injury induces a cascade of molecular changes within neuroglia that can be detected using biochemical and imaging based methods. Glutamate-induced excitotoxicity is a prolonged or excessive exposure to glutamate that ultimately results in neuronal and glial cell death. 238,239 In normal physiology, glutamate is rapidly removed from synapses by glutamic acid reuptake systems on glial cells and neurons. 240 Reuptake prevents a buildup of glutamate within synapses, sustaining glutamate levels at approximately 0.6 μmol/L. 241 However, after neurotrauma the concentration of extracellular glutamate can increase to up to 5 μmol/L. 241 Following TBI in humans, the levels of glutamate in the CSF are increased, and remain higher 1 week following injury, with these levels correlated with injury severity. 242 Further, by 2 days following a moderate midline FPI, tonic glutamate levels are increased by 178% in the striatum and 256% in the dentate gyrus. 243
Glutamate excitotoxicity has also been implicated in white matter damage following SCI. 244 Glutamate elevation following injury is caused by: 1) Ca2+-dependent excitotoxic release of glutamate from axons; 2) dysfunctional astrocytic and neuronal glutamate reuptake and recycling; 3) ATP-dependent P2X7receptor-mediated astrocytic glutamate release; 4) spillage from adjacent injured cells and; 5) glutamate release from activated microglia, associated with an inflammatory response. 241,245 –247 In addition, there is a positive feedback mechanism associated with glutamatergic excitotoxic insult. Glutamate interacts with N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors to increase the expression of inflammatory cytokines and chemokines. 248 These toxins stimulate nearby microglia and astrocytes to release more glutamate and inflammatory factors into the extracellular space, exacerbating excitotoxic insult. 249 However, increased extracellular glutamate alone cannot induce the full spectrum of toxic effects observed following CNS injury. 250
Intracellular Ca2+ overload activates apoptotic cellular mechanisms and has been coined the “final common pathway of cell death.” 251 After neurotrauma, there is an immediate rise in extracellular Ca2+ levels due to spillage from injured and inflamed cells, and release from intra-axonal Ca2+ stores. 252 Using 45 Ca autoradiography following a lateral FPI, there is a marked increase in 45 Ca accumulation, even in areas of the brain without gross morphological damage, showing that Ca2+ flux plays a critical role in TBI pathology. 253 Ca2+ rapidly enters neuroglia through a variety of channels and receptors, including AMPA receptors, NMDA receptors, voltage-gated calcium channels (VGCCs), and P2X7 receptors, 254 as well as through membrane pores created by the shearing force during the initial mechanical insult. 255
Increased intracellular Ca2+ is self-propagating, due to Ca2+-induced release of Ca2+ from intracellular stores, such as the endoplasmic reticulum, as well as reversal of the Na+/Ca2+ exchanger, which then pumps additional Ca2+ into the cell. 256 –258 Changes in Ca microdomains are also apparent in optic nerve vulnerable to secondary degeneration, detected using nanoscale secondary ion mass spectroscopy. 259 Following neurotrauma, oligodendrocytes and OPCs are particularly vulnerable to intracellular Ca2+ overload, 40 partially because they contain higher concentrations of P2X7receptors and AMPA receptors than neurons and other glial cells. 260,261 Therefore, more receptors are activated after injury, leading to greater Ca2+ influx when compared with cells that have a lower concentration of these receptors. 262 Further, oligodendrocytes and OPCs do not have the capacity for receptor-mediated desensitization of AMPA receptors when overstimulated, thus increasing their risk of Ca2+-mediated damage. 263 Once Ca2+ has entered cells, it mediates a variety of downstream pathways, including mitochondrial dysfunction and oxidative stress, ultimately resulting in apoptotic cell death and the spread of secondary degeneration, 264 with oligodendrocytes being particularly vulnerable.
Oligodendrocyte susceptibility to oxidative stress
Increased intracellular Ca2+ accumulates in mitochondria, resulting in an increase in mitochondrial and cytoplasmic production of reactive oxygen species (ROS) and reactive nitrogen species (RNS), 130 as shown in Figure 1.
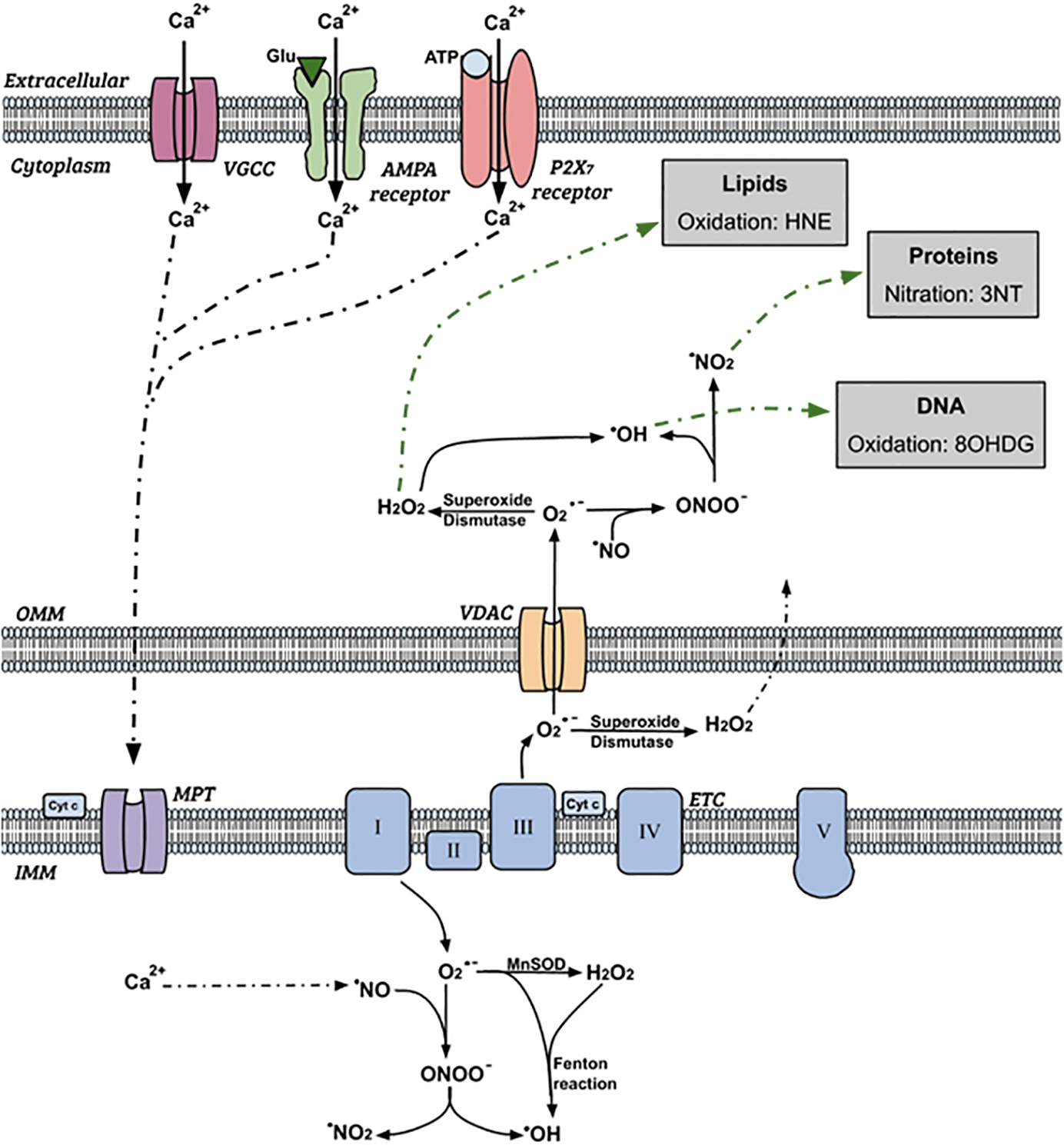
Schematic diagram showing intracellular production and effect of reactive oxygen species during secondary degeneration. Reactions shown are indicative of processes and are not balanced equations. Not all possible reactions are shown., ETC, electron transport chain; MPT, mitochondrial permeability transition; OMM/IMM, outer/inner mitochondrial membrane; VDAC, voltage dependent anion channel. Adapted fromO'Hare Doig et al. 265
Following intracellular Ca2+ influx, Ca2+ accumulates inside the mitochondria via the mitochondrial permeability transition. 266 Mitochondrial protein phosphatase then dephosphorylates cytochrome c oxidase, reducing allosteric ATP-inhibition and increasing the mitochondrial membrane potential. 267 As ATP synthesis increases through the Krebs cycle, electrons are leaked at complexes I and III of the electron transport chain on the inner mitochondrial membrane, resulting in the increased formation of the superoxide anion (O2 •-). 268,269 Superoxide dismutases reduce O2 •- to hydrogen peroxide (H2O2), and the iron-catalyzed Fenton reaction further dismutases H2O2 into hydroxyl radicals (OH•). 270,271 OPC vulnerability is heightened by increased baseline intracellular iron levels, 272 as increased intracellular iron is associated with increased ROS production via iron-facilitated Fenton reactions. 273
Our team has demonstrated that at 1 and 3 days post-injury, there is an increase in H2O2 following a partial optic nerve injury. 265 Nitric oxide (•NO) is enzymatically synthesized by inducible nitric oxide synthase (iNOS), and reacts with O2 •- to produce peroxynitrate (ONOO-). 274 ONOO- can then be further reduced to form OH• and nitrogen dioxide (•NO2). 275 O2•- can travel from the mitochondria into the cytoplasmic space through voltage-dependent anion channels on the outer mitochondrial membrane, resulting in elevated ROS outside of the mitochondria. 276 Further, H2O2 is membrane permeable, and so can diffuse out of the mitochondria into the cytosol. 277 Within the cytoplasm and nucleus, these ROS oxidize DNA and lipid structures, and nitrate proteins, causing cellular damage and apoptosis. 264 OH• also deprotonates lipids and proteins, causing a self-propagating cycle of oxidative damage. 270
Our team has shown that following a partial optic nerve transection, the immunoreactivity of carboxymethyl-lysine (CML), an advanced glycation end-product as a marker of oxidative stress increases significantly post-injury in areas of nerve vulnerable to secondary degeneration, colocalized with Olig1+ oligodendroglial cells. 130 When animals were treated with a combination of ion channel inhibitors aimed to reduce Ca2+ influx through AMPA receptors, P2X7 receptors, and VGCCs, the immunointensity of CML decreased to normal levels, suggesting increased advanced glycation end-products in this model are at least in part a result of Ca2+-dependent mechanisms. 130
In a normal, uninjured system, ROS and RNS are detoxified by antioxidants, such as glutathione peroxidase (GPx1), heme oxygenase-1 (HO-1), and manganese superoxide dismutase (MnSOD), preventing a buildup of free radicals within cells. 78 Cells respond to increased production of ROS and RNS by increasing production of antioxidants; with research from our team showing a significant increase in MnSOD immunoreactivity in the optic nerve as early as 5 min after partial transection, colocalized within astrocytes. 278,279 By 3 days following injury, there is a significant increase in GPx1, often colocalized within CC1+ oligodendrocytes, with a significant increase in HO-1 by 7 days post-injury, predominantly colocalized inionized calcium-binding adaptor molecule (Iba1) positive microglia. 265 Following two repeated closed-head weight-drop mTBIs in rats, MnSOD immunoreactivity increases 11 days post-injury. 280 When a combination of ion channel inhibitors to limit Ca2+ entry via AMPA receptors, P2X7 receptors, and VGCC is used following repeated mTBI, MnSOD immunoreactivity decreased, indicating modulating Ca2+ influx is key to limiting this antioxidant response. 280 At both 1 and 14 days post-injury following a spinal cord contusion in rats, the level of MnSOD is significantly increased, with a similar increase in copper zinc superoxide dismutase (CuZnSOD) and GPx1 at 14 days post-injury. 281
When the rate of ROS and RNS generation becomes greater than the rate at which antioxidants can counterbalance, cells are unable to prevent the damaging effects of these free radicals and undergo oxidative and nitrosative stress. 249 OPCs have lower concentrations of endogenous antioxidants, such as GPx1 and MnSOD and are thus particularly vulnerable. 272,282 Glutathione prevents cell apoptosis by conjugating 4-hydroxynonenal (HNE), a by-product of lipid peroxidation, and suppressing the toxic activity of 12-lipoxygenase. 283,284 Therefore, lower levels of glutathione are correlated with an increased risk of oxidative damage to lipids. 285 Further, when OPCs are engineered to overexpress the antioxidant MnSOD, there is an observable decrease in apoptosis. 286
Mitochondrial oxidative stress also induces endoplasmic reticulum (ER) stress. 287 ER stress results in unfolded proteins, 288 and if the stress persists for an extended period, it can result in cell death. 289 Further, in periods of stress, the ER releases large quantities of Ca2+, which in turn increase the production of ROS/RNS in the mitochondria. 287 Our research has shown that mitochondria in both axons and glia develop subtle structural changes following injury, with increased autophagic profiles demonstrating susceptibility of mitochondria to secondary degenerative processes. 290
Oligodendrocyte vulnerability to oxidative stress is further exacerbated by it role in maintaining the myelin sheath. 282 The maintenance of myelin requires large amounts of energy, and involves catalyzing the production of lipids. 291 One by-product of these reactions is ROS; therefore, there is an intracellular accumulation of ROS within oligodendrocytes, amplifying oxidative stress mechanisms. 282
Lipid peroxidation following injury
Lipid peroxidation, the process where ROS “steal” electrons from a cells membranous lipid bilayer, is an important downstream mechanism of oxidative stress. 292 ROS attack the carbon to carbon double bonds of lipids, in particular of polyunsaturated fatty acids, as well as glycolipids, phospholipids, and to a lesser extent cholesterol. 293 This attack on lipids results in the formation of toxic aldehydes, such as HNE. 294 Further, iron plays a key role in ROS-mediated lipid peroxidation and enhances cytokine-mediated mitochondrial and membrane damage. 81,295 Therefore, during secondary degeneration, OPCs not only produce more ROS but have greater ROS-mediated cell damage than other neuroglia due to their increased intracellular iron levels. 272 HNE mediates cell damage by binding to and disrupting membrane transport systems, including impairing glutamate reuptake in astrocytes. 296,297 Therefore, the production of HNE by lipid peroxidation causes a positive feedback for glutamate excitotoxicity and exacerbates the cycle of cell damage. 298
Our team has shown overall increased levels of HNE from 3 days after partial injury to the optic nerve, 265 with a significant increase in HNE within the OPC and mature oligodendrocyte populations at this time. 43 When a combination of three ion channel inhibitors targeting AMPA receptors, P2X7 receptors, and VGCCs was delivered acutely following the injury, OPCs were preserved when HNE decreased. The protection was associated with improvements in visual function, suggesting that limiting lipid peroxidation may aid in maintaining the number of OPCs following injury. 42 Following a clip-compression-induced SCI, there is a biphasic increase in lipid peroxidation as shown by malondialdehyde assays, with the first peak at 4 h post-injury, and the second increase at 24 h to 5 days post-injury. 299 In a model of repeated mTBI, our team has also shown increased lipid peroxidation in cortical neurons, demonstrating that oligodendroglia are not the only cell type vulnerable to lipid peroxidation. 300 Further, following TBI in humans, circulating plasma levels of HNE increase compared with control patients. 301
Protein nitration following injury
Nitrosative stress also has a neurodestructive effect, mediating further lipid peroxidation, as well as protein nitration. 302,303 Protein nitration occurs when a nitro group is added into the structure of a protein, 304 and primarily occurs as the reaction between •NO or •NO2 and the amino acid tyrosine, to form 3-nitrotyrosine (3-NT). 305 Immunointensity of 3-NT, a protein nitration indicator, is significantly increased in a diffuse distribution 3 days following partial injury to the optic nerve. 42 Further analysis using NanoSIMS technology shows that 3-NT is increased at 3 days within mature oligodendrocytes, NG2+ cells, Olig2+ cells, βIII tubulin+ axons, and paranodes, with no structure selectively vulnerable to damage. 43 In addition, following a moderate diffuse closed-head injury, 3-NT immunoreactivity increased at 48 h post-injury. 306 This suggests that the nitrosative pathways also contribute to the cellular damage observed in secondary degeneration.
Oxidative DNA damage following injury
DNA is highly vulnerable to oxidative damage, and there are a variety of mechanisms through which free radicals mediate DNA damage during oxidative stress, one of which is nucleobase modification. 249 These DNA lesions can have extremely detrimental effects, by either altering the genetic code or by acting as a block during DNA replication. 307 DNA is highly vulnerable to oxidative damage, as ROS, particularly •OH, can mediate hydroxylation of DNA nucleic acid bases. 249 This process converts deoxyguanosine to 8-hydroxy-2-deoxyguanosine (8OHDG), through a spontaneous transversion mutation, altering DNA structure. 308 Our team has shown that by 24 h following partial optic nerve injury, 8OHDG is significantly increased in the ventral optic nerve white matter, and this is associated with apoptotic cell death. 265,309 However, in our closed-head weight-drop model of repeated mTBI, there is no increase in 8OHDG immunoreactivity in a range of brain regions, with either 1, 2, or 3 injuries at 3 months following injury, 310 or with two injuries at 11 days post-injury. 280 More work is needed to assess the acute time course of 8OHDG immunointensity in this model.
Going on to define the cellular specificity of the changes, we have shown that oligodendroglial cells are significantly more likely to succumb to oxidative 8OHDG DNA damage following neurotrauma compared with other neuroglial cells and structures. 43 Three days following the optic nerve injury, both OPCs and oligodendrocytes had increased levels of 8OHDG DNA damage. 43 Newly derived oligodendrocytes exhibited significantly more DNA damage than pre-existing oligodendrocytes, suggesting newly differentiated cells are more vulnerable to oxidative damage. 43 The DNA-damaged oligodendrocytes were more likely than non-DNA-damaged oligodendrocytes to become apoptotic. 43 However, newly derived oligodendrocytes were less likely to be apoptotic than pre-existing oligodendrocytes. 43 Thus, despite their increased susceptibility to DNA damage, newly derived oligodendrocytes are less vulnerable to apoptotic cell death than their pre-existing oligodendrocyte counterparts. 43 Interestingly, by 28 days after injury, these newly derived oligodendrocytes are still less likely to be apoptotic, but they also have decreased expression of MyRF. 43 This suggests that newly derived DNA damaged oligodendrocytes are more likely to survive following injury but have a reduced myelination capacity. 43 Therefore the ability for remyelination is compromised following oxidative damage due to neurotrauma and may lead to poor repair of damaged myelin. 43
Changes in numbers of oligodendroglia following injury
It has been widely reported, including in Australian research, that following experimentally induced TBI and SCI there is a loss of oligodendrocytes. 40,41,311 –314 However, some studies have not detected oligodendrocyte death, 120 perhaps indicating differential responses of these cells depending on injury severity or mode.
We have shown that following partial injury to the optic nerve white matter, 43 using Olig2 together with NG2 as markers for OPCs, the number of OPCs remains similar to control at 1 and 3 days after injury. 315 However, OPC densities significantly decline from 7 days to 3 months, which is not attributable to optic nerve swelling. 315 At 3 days post-injury, OPCs proliferate more than controls. 315 At 1 and 3 days post-injury, the density of CC1+/Olig2+ immature oligodendrocytes decreases; however, by3 days there is an increase in the density of CC1+/Olig2- mature oligodendrocytes, indicating differentiation of a subset of surviving immature oligodendrocytes. 315 By labeling dying oligodendroglia with the cell death marker TUNEL, it was found that an increase in TUNEL+ staining occurred across the oligodendroglial lineage by 7 days following injury, with a significant increase in OPC cell death sustained at 1 month post-injury. 315 This suggests that despite acute proliferation, there is an overall chronic depletion in the OPC population. 43,315
Similarly, following a contusion SCI, numbers of BrdU+/NG2+ OPCs increase, with proliferation of OPCs tripling in the first week following injury. 313 However, within the first 7 days following injury, the total number of OPCs decreases, demonstrating an overall reduction of this cellular population despite the proliferative response. 313
Change in the OPC population may be slightly different following TBI. Using an FPI model, the number of OPCs increases in white matter tracts from 2 days to 21 days following injury, 41 quantified based upon use of Olig2 or Tcf4 individually to indicate OPCs. Further, although BrdU+/PLP+ newly derived oligodendrocytes are rare in the corpus callosum, they are significantly higher in number following a CCI than sham injury. 120 Finally, in another study utilizing a CCI, immunoreactivity of the OPC marker NG2, quantified by optical density measurements, peaks at 1 to 4 days following injury in the corpus callosum. 316 Taken together these studies suggest that OPC populations increase in response to injury in white matter following TBI. In the first few days following a moderate to severe TBI in humans, oligodendrocytes are lost with a concomitant increase in the number of OPCs. 317 Despite these findings, it is possible that variability exists in number of oligodendroglia depending on the type of white matter injury.
Axonal loss and neuronal damage following injury
Neuronal loss both within and around the injury site is a hallmark of white matter injury. 318 In TBI, neural degeneration has been observed within minutes following injury, 319 featuring the degenerative cascade of TAI. 33 Certain areas of white matter are more vulnerable to TAI, such as the corpus callosum and the brainstem. 171,320 Staal and Vickers found that in vitro unmyelinated axons are more vulnerable to stretch injury that myelinated axons. 321 Further, unmyelinated fibers in the corpus callosum are more selectively vulnerable to damage than myelinated axons in vivo. 322 Following injury, increased intracellular Ca2+ within neurons results in damage to the axonal cytoskeleton and impairments to axonal transport via activation of degenerative enzymes and cysteine proteases such as caspase-3 and the Ca2+-dependent neutral protease calpain. 323 Caspase-3 cleaves proteins associated with DNA repair, contributing to the DNA damage observed following injury. 324 The damage cascade results in accumulation of axonal transport proteins within the axon, DNA fragmentation, and axonal swelling, with affected neurons eventually undergoing either apoptotic or necrotic cell death, impairing function. 325 In addition, DNA damage from ROS enhances post-traumatic neuronal death following injury. 326
The partial optic nerve transection model has allowed specific insight into the changes to axons and their parent somata vulnerable to secondary degeneration in a white matter tract. We have shown that at 24 h after injury, retinal ganglion cell somata show increased c-jun expression, associated with pro-apoptotic mechanisms. 278 By 3 days after injury, poly (ADP-ribose) polymerase (PARP) is upregulated in axons in the ventral optic nerve, implicating necrosis as an additional pathway to cellular demise. 327 There is also decreased axonal stability at 1 day following PT, shown through decreased acetylated tubulin expression. 42 Further, there are no changes in the ratio of Ca2+-independent phosphorylated Tau/panTau at [S 262 ], whereas there is increased Ca2+-dependent phosphorylated Tau isoforms at this time. 42
When treated with a combination of ion channel inhibitors that reduce Ca2+ influx through AMPA receptors, P2X7 receptors, and VGCCs, the ratio of phosphorylation of both [S 396 ] and [T 205 ] relative to total Tau decreases significantly at 3 day post-injury compared with injured untreated controls. 42 The ion channel inhibitor combination also ameliorates acetylated tubulin immunoreactivity. 42 Conversely, NogoA immunoreactivity decreases in the ventral optic nerve with injury but increases when animals were treated with the ion channel inhibitor combination. However, changes to NogoA did not appear to be a driver of therapeutic benefit following ion channel inhibitor administration, and the functional significance of changes to NogoA remain unclear. At 3 months following the optic nerve injury, β-III tubulin immunointensity is decreased, indicative of loss of axonal integrity and axonal death. 130 Indeed, by 3 months after injury, the number of axons in the ventral optic nerve has decreased by 30.7%. 34 The ion channel inhibitor combination protects β-III tubulin immunoreactivity 3 months post-injury, associated with preservation of myelin and improvements in visual function. 130 There is also a significant decrease in retinal ganglion cell densities in the central retina both 1 and 3 months after these injuries, which suggests retrograde damage to cell bodies along axons. 328 Taken together the data indicate that loss of neurons is associated with Ca2+ dysregulation, oxidative stress mechanisms, and dysmyelination, and likely contributes to a decrease in function. 42,130
These degenerative changes to neurons and their axons are not limited to the optic nerve injury model. Following mTBI with TAI, degenerating axons are apparent with electron microscopy at 3 days through to 6 weeks post-injury, with swollen mitochondria, either abnormally high or low cytoplasmic density, and vesicle accumulation. 120 Unmyelinated axon density decreases in the corpus callosum following a moderate FPI, with diameter decreasing from 3 to 7 days post-injury and recovering at 15 days. 329 In a TAI study utilizing compound action potentials evoked in the corpus callosum, it was found that the amplitude of myelinated axons was decreased up to 3 days post-FPI but recovered to control levels by 7 days. Whereas the unmyelinated axonal amplitude did not recover, therefore a differential vulnerability was observed. 322 Following a single-impact open-head weight-drop TBI, axons are damaged, with a concomitant decrease in conduction velocities across the corpus callosum. 330 However, there is no increase in TUNEL+ neurons in white matter following a FPI, which suggests neurons are less vulnerable to apoptotic cell death than oligodendroglia. 331
Tau hyperphosphorylation is a driver of neurodegeneration as it results in a disruption of microtubules and may result in interrupted axonal transport and therefore decreased synaptic transmission. 332 The Corrigan team at the University of Adelaide (Collins-Praino and colleagues 333 ) have shown that regardless of whether rats receive a single moderate to severe TBI or three mTBIs, the level of AT180 tau phosphorylation has a biphasic pattern, with an initial spike at 24 h following injury, which returns to baseline before increasing again at 3 months post-injury. Further, levels of protein phosphatase 2Ac (PP2Ac) decrease at both 24 h and 3 months following injury, hypothesized to demonstrate a loss of PP2A phosphatase activity. 333 Overall, this indicates that tauopathy and tau phosphorylation may be a significant feature of neurodegeneration following both repeated mTBIs and more moderate to severe single TBIs.
Axonal loss is also a hallmark of SCI. By 1 week following a contusion SCI, only 13% of axons remained in the degenerated core of the dorsal column at the injury site. 334 Axons swelled, with the axoplasm becoming dense, followed by myelin detaching from the axon. 334 Axons subsequently degenerated, leaving a microcyst inside a slowly disappearing myelin sheath. 334
The amyloid-β (Aβ) peptide is derived from amyloid precursor protein (APP), with the aggregation of Aβ believed to play a role in Alzheimer's disease pathogenesis. 335 Aggregation of Aβ is evident following severe TBI in humans, 336 so it is not surprising that there is an epidemiological association between TBI and Alzheimer's disease later in life. 6 The Vickers team have recently shown (Collins and associates 337 ) that 30 days following a lateral FPI in the APP/PS1 transgenic model of Alzheimer's disease in mice, the aggregation response of Aβ is dependent on the state of amyloidosis at time of injury, with pre-plaque mice showing increased Aβ deposition in the cortex, and mice with established amyloidosis showing decreased total Aβ deposition. More work is needed to determine the Aβ response in white matter following injury. The functions of APP are manifold, including roles in neuronal and synaptic processes. 338 The Vink team has shown (McAteer and co-workers 339 ) that following repeated mTBI, there are increases in APP immunoreactivity at 12 weeks post-injury. However, APP derivatives can mediate both neurotoxic and neuroprotective effects. Indeed, the team in South Australia (Corrigan and colleagues 340 ) has gone on to show that if APP is knocked out in mice, there is increased cortical and hippocampal cell loss, axonal injury, and decreased behavioral deficits following diffuse mTBI, suggesting upregulation of APP plays a neuroprotective role. 340 Exogenous treatment with the APP derivative sAPPα prevents deficits in APP knockout mice after injury. 341 Together with Roberto Cappai, the team have shown (Plummer and associates 342 ) that this neuroprotective effect of APP is mediated by residues 96-110 in the heparin-binding site in the growth-factor like domain (D1) of sAPPα, with APP96-110 having a neuroprotective effect following focal CCI. Intravenous administration of APP96-110 30 min after a moderate to severe diffuse impact-acceleration TBI results in reduced motor deficits, and decreased axonal injury and neuroinflammation in the corpus callosum at 3 days following injury. 343 Further, when APP96-110 is administered intravenously 5 h post-injury, there are improvements in motor deficits and axonal injury in the corpus callosum. 343 Therefore, APP96-110 may be a potential therapeutic option to limit axonal injury in white matter following TBI.
Dysmyelination and myelin structural changes
The myelin sheath is predominantly lipid-based, which means it is highly vulnerable to oxidative stress and lipid peroxidation, 344 resulting in widespread dysmyelination following white matter injury. 15,345,346 Dysmyelination is a destructive process whereby the myelin sheath becomes abnormal in structure, associated with a loss of function due to reduced conduction of neural signals. 347 A hallmark feature of dysmyelination is myelin decompaction. Myelin decompaction occurs when the myelin lamellae separate and the paranodal loops detach and split, associated with increased intracellular Ca2+, as shown in Figure 2. 348

Characteristics of dysmyelination and myelin decompaction. Following CNS injury, myelin structures succumb to oxidative stress and lipid peroxidation. This results in paranodal splitting, myelin retraction and decompaction, Ca2+ influx, and cytoskeletal alterations to the nodal complex. Diffusion of nodal components results in impaired saltatory conduction along nerves, resulting in axonal injury and/or loss. As irregular myelin repair is attempted by newly myelinating oligodendrocytes, the number of atypical nodal complexes increases. Red circles represent nodal and paranodal components. CNS, central nervous system. Figure created with
Oxidative stress disrupts both the integral myelin sheath proteins, and the proteins between the surfaces of the myelin lamellae. 351,352 We have shown that from 5 min following partial optic nerve injury, generalized increases in MBP immunofluorescence are observed in the ventral optic nerve, indicative of myelin degradation and increased immunoreactivity of MBP. 278 However, by 3 months following injury, MBP immunoreactivity is decreased, suggesting long-term myelin loss. 34 In a rat model of TBI, MBP becomes proteolyzed in a calpain-mediated manner, contributing to lost integrity of the myelin sheath and dysmyelination. 353 MBP has also been found to be elevated in the CSF following severe TBI in humans. 354
Luxol fast blue (LFB) staining is a frequently used indicator of myelin integrity. Following a CCI, LFB staining reveals lesions within the corpus callosum from 1 day to 3 months after injury. 355 At 1 year following a parasagittal FPI in rats, abnormal LFB staining is observed in the corpus callosum, with a decrease in LFB staining in the ipsilateral cerebral peduncle compared with sham animals, indicating loss of myelin. 356 At 24 h and 2 days following a contusion SCI, LFB staining is considerably decreased in the dorsal column at the middle of the injury site, with more granular staining visible in the ventrolateral white matter; by 10 weeks post-injury, a thick rim of LFB staining is apparent surrounding the injury site. 334
Given the optic nerve is a highly myelinated white matter tract, the partial transection model has proved useful for studying changes to myelin structure following injury. We have used electron microscopy to demonstrate a significant increase in the proportion of axons with decompacted myelin in the ventral optic nerve at 3 months after injury, 357 associated with lipid peroxidation and oxidative stress as described above. 358 At 6 months, loosening of the myelin sheath is widespread throughout the nerve, with myelin thickness significantly increased at this time-point, likely due to the loosening. 34 The number of intraperiodic lines surrounding myelinated axons also increases at 6 months post-injury, suggesting oligodendrocytes may be wrapping more layers of myelin around axons in an attempt at repair. 34
In SCI, the necrotic core of the injury is surrounded by a rim of demyelinated axons. 359 After a contusive SCI, the number of demyelinated axons peaks at 1 day following injury, and starts to resolve by 7–14 days, before then increasing again up to 450 days post-injury. 360 Following a chronic clip compression injury of the rat thoracic spinal cord, the myelin is thinner in the injured dorsal column, associated with reduced action potential amplitudes in affected axons. 361
Following mTBI with TAI, electron microscopy has revealed an increase in double-layered myelin, 120 which can occur as myelin degenerates around injured axons. 362 We have shown that in normally myelinated axons myelin thickness in the corpus callosum is not significantly altered with up to three repeated mTBIs. 310 However, more work is needed to quantify dysmyelination, myelin decompaction, and loosening at various time-points following TBI at the electron microscopy level.
Dysmyelination severely disrupts the myelin node/paranode complex. The nodal sodium channels and the paranodal protein, Caspr, diffuse along the axon, increasing the size of the node and paranode respectively. 363 Our team has shown that at 1 day following partial optic nerve injury, there is a significant increase in both the paranodal gap length and the proportion of atypical nodal complexes, and these changes are maintained at 3 months after injury. 364 After closed-skull impact, paranodes were either shortened or absent with a concomitant increase in heminodes. 330 Following closed-head weight-drop mTBI, the length of the paranode, the node of Ranvier, and the proportion of atypical nodal complexes in the corpus callosum increases. 280 Interestingly, inhibiting VGCCs, P2X7 receptors, and AMPA receptors results in improvements in both the length of the node as well as the proportion of atypical nodal complexes in both the optic nerve and mTBI models, indicating increased intracellular Ca2+ is a key mediator of myelin integrity at the node of Ranvier. 42,280,365
Dysmyelination after injury is associated with higher axonal susceptibility to excitotoxic insult, 366 evidenced by a significant decrease in axonal density associated with dysmyelination both 1 and 3 months after partial optic nerve injury. 357 It is therefore not surprising that dysmyelination is associated with a loss of visual tracking behavior after partial optic nerve injury at both acute and chronic timepoints. 42,130 Abnormal myelination at a single internode can be sufficient to block neural signal transduction for an entire axon. 344 White matter damage has also been associated with functional deficits following experimental models of TBI, with deficits dependent on the location and severity of the injury. 367,368 Therefore, limiting the extent of myelin damage, both by attenuating dysmyelination and promoting successful remyelination following injury, may result in improved functional outcomes.
Inflammation and gliosis
Myelin degradation is both a cause and consequence of inflammation, as shown in animal models of multiple sclerosis where myelin debris regulates microglial activation. 369 Along with infiltrating macrophages, activated microglia secrete numerous cytokines, chemokines, ROS, and growth factors, affecting nearby cells. 370 Proinflammatory chemokines further recruit immune cells to the injury site, exacerbating damage, 371,372 with depletion of microglia following a moderate FPI resulting in a therapeutic effect on neuronal loss early post-injury. 373 However, microglia have pleiotropic responses to injury, and can also mediate anti-inflammatory responses depending on environmental cues and exposure to specific cytokines. 374 In SCI, it has recently been discovered that microglia form a “microglial scar,” without which functional outcomes after injury are worsened. 375 From 3 days after partial transection to the optic nerve, we have shown that Iba1+ microglia and ED1+ macrophages become activated and their numbers increase in ventral nerve vulnerable to secondary degeneration and remotely in the brain. 278,376
The Corrigan team have shown (Collins-Praino and associates 377 ) that following a single diffuse impact-acceleration mTBI, there is an acute increase in Iba1+ cells in the hippocampus at 6 days, which resolves chronically by 3 months post-injury. The finding built upon earlier demonstrations of a significant increase in cortical microglial activation at 24 h following three impact acceleration diffuse mTBIs, and a significant increase following both one and three injuries at 12 weeks. 339 Meanwhile, we have shown in a closed-head weight-drop model of repeated mTBI that microglia in the corpus callosum remain hypertrophic after two injuries at 3 months, 310 but only after one injury in cortex, suggesting repeated mTBI may cause more chronic microglial activation in white matter tracts. From 3 days following mTBI with TAI, there is an increase in microglial activation in the corpus callosum, persisting through to 6 weeks post-injury. 120,378
Further, following a closed-skull impact TBI with TAI, astrogliosis also significantly increases up to 6 weeks post-injury in the corpus callosum where axons are damaged. 120 To form a barrier around the injury site, reactive astrocytes become hypertrophic, their processes become interlaced, and these cells show an increased expression of glial fibrillary acidic protein (GFAP). 379 We have shown that by 24 h after partial optic nerve transection, astrocytes display a hypertrophic morphology, with a significant increase in both the width of astrocytic processes and number of minor astrocytic processes. 278 Neurotrauma can activate at least two different types of astrocyte reactivity, A1 and A2, 380 nomenclature that mimics the M1/M2 macrophage/microglia terminology. 381 However, as with macrophages and microglia, 381 it is likely that astrocyte reactivity is a spectrum of various phenotypes, rather than distinct reactive states. 380 In particular, A1 astrocytes have been associated with neuroinflammation, whereas A2 astrocytes have been associated with the production of reparative trophic factors. 380
Following injury, it appears that there may be a mixture of these phenotypes present. Ablation of proliferating reactive astrocytes following a moderate TBI resulted in inflammation and neural degeneration, 382 suggesting the importance of astrocytes in the reparative response. Further, following white matter ischemic injury, astrocytes promote OPC differentiation and oligodendrocyte generation by secreting BDNF, a mechanism that may occur within traumatic injuries. 383 However, reactive astrocytes also release inhibitory growth factors, cytokines, ROS, and glutamate that exacerbate injury and may have a detrimental effect on overall function. 384 –387 Recent data have shown that within the cortex, the astrocytic marker GFAP and A1 astrocyte phenotype marker C3 are upregulated from 8 h following both rat FPI and mouse CCI, with the microglial marker Iba1 increased at 24 h following injury. A peak was reached in both models 72 h after injury, suggesting astrocytes may become reactive prior to microglial activation. 388 However, it is important to note that not all GFAP+ astrocytes were C3+, suggesting there may be a mix of reactive astrocyte phenotypes present following injury. 388
These mechanisms may also be at play within the white matter following injury. In LFP TBI, reactive astrocytes are found 1 day post-injury in the subcortical white matter tracts, continuing at least until 1 month post-injury. 389 .At both 24 h and 12 months following a repeated closed-head injury, astrogliosis increases, but when only a single injury is administered, there is no increase in astrocyte reactivity at 24 h and only a mild elevation at 12 months. 122 However, at 24 months post-injury, a single mTBI produced mild astrogliosis, which was more severe following repeated injuries. 121 However, in our closed-head weight-drop model of repeated mTBI in rat, we find no astrogliosis with either one, two, or three injuries 3 months post-injury in the corpus callosum. 310 However, more research is needed to determine whether microglia and astrocytes have beneficial or detrimental effects following CNS injury.
Remyelination
Remyelination occurs when the normal myelin sheath is restored to dysmyelinated axons, allowing saltatory conduction, promoting axonal survival and improving function. 390 Once a mature oligodendrocyte has myelinated a series of axons, it is traditionally thought to be relatively unable to remyelinate additional axons. 391 Instead, new oligodendrocytes must be generated for remyelination, with OPCs as the main source of remyelinating oligodendrocytes. 349 Schwann cells may also contribute to remyelination, as OPCs possess the ability to differentiate into Schwann cells in the context of CNS demyelination. 392
Recent work using single-cell transcriptomics as well as a 14 C-based birth dating techniques in human multiple sclerosis tissue has revealed that mature oligodendrocytes may in fact possess the capacity to remyelinate additional axonal segments. 393 –395 However, the ability of mature oligodendrocytes to provide myelin to demyelinated axons has not yet been investigated following neurotrauma. Nevertheless, reduced generation of new myelinating oligodendrocytes from OPCs following injury is associated with decreased function following injury. 313 Further, transplantation of OPCs following SCI results in remyelination and functional recovery. 396 Therefore, preserving and encouraging proliferation and differentiation of OPCs after injury is likely to be associated with improved functional outcomes after neurotrauma.
OPCs rely on factors released by microglia and astrocytes to switch from a quiescent to a regenerative phenotype. 73 Whereas normal astrocytes create a facilitatory environment for OPC differentiation, once activated, astrogliosis can inhibit differentiation and maturation of OPCs. 397 However, blocking astrocyte scar formation inhibits axonal regeneration following a severe crush SCI in mice, suggesting that astrocytes also play a crucial role in recovery following injury. 398 The effect of microglial cells on OPC differentiation depends on the balance of pro-inflammatory or anti-inflammatory phenotypes, with a switch from pro-inflammatory toward anti-inflammatory coinciding with commencement of remyelination. 399 –401 Macrophages may also play a role in oligodendrogenesis following injury, as depletion of these cells delayed remyelination and OPC recruitment after lysolecithin-induced spinal cord demyelination. 402 Myelin proteins inhibit OPC differentiation, therefore MBP degradation may act as a further barrier to OPC differentiation as it is released into the extracellular space from damaged sheaths. 403,404 However, microglia are in part responsible for the clearance of myelin debris, thus regulating oligodendrogenesis through myelin phagocytosis. 405 Further, neural activity from the pre-motor cortex enhances OPC proliferation, differentiation into oligodendrocytes, and myelination. 93 Collaborative Australian research has shown that under normal conditions, increased neural activity leads to axons being myelinated with thicker myelin than axons with less neural activity, 406 which may also be relevant to remyelination following white matter injury.
In demyelinating disease, remyelinated fibers are often characterized by thinner myelin sheaths than would be expected based on axonal diameter. 407 However, Australian researcher Kaylene Young and colleagues discovered that myelination is an ongoing adult phenomenon, with evidence for myelin remodeling by adult-born oligodendrocytes, which made shorter and thinner myelin, but more internodes of myelin than early-born oligodendrocytes. 52 This suggests that thinner myelin typical of remyelination may be a characteristic of adult myelination, rather than remyelination itself.
Our team has shown that the number of intraperiodic lines increase chronically following partial optic nerve injury, suggesting oligodendrocytes may be forming new layers of myelin; however, this was not associated with improvement in visual function. 34 However, unlike in demyelinating disease, thicker myelin sheaths were observed, which was posited to be due to loosening of the myelin lamellae. 34 Following a contusion SCI, newly formed oligodendrocytes and Schwann cells remyelinated axons, and these myelin internodes were significantly shorter than in control mice, 408 and elongated over 6 months post-injury. 408 At most time-points tested, myelin thickness was not different in injured animals compared with controls. 408 Therefore, long-term following SCI, remyelinated sheaths were neither thinner nor shorter than myelin in control mice. 408
Following an open-head weight-drop TBI, conduction velocity along axons in the corpus callosum was partially restored by 2 weeks post-injury, alongside concomitant increased synthesis of myelin membranes, suggesting remyelination had occurred. 330 Following mTBI with TAI, myelin thickness was reduced at 1, 2, and 6 weeks post-injury, following an earlier increase in the number of newly generated oligodendrocytes at 3 days post-injury. 120 However, more work is needed to determine the extent and pattern of remyelination in various models of white matter injury.
Conclusion
White matter injury is a key pathology that contributes to functional loss following neurotrauma of all etiologies. Evolving technologies are revealing the subtleties of structural changes in white matter tracts and the underlying causative biochemical and cellular disruptions (Fig. 3). Models including partial optic nerve transection are allowing an examination of contributing cellular dynamics in tissue exclusively vulnerable to secondary degeneration (summarized in Table 1). The next step will be to use this information to design intervention strategies to minimize the oxidative damage associated with dysmyelination, preserve myelin, and promote remyelination where required, and in so doing preserve axonal and behavioral function.
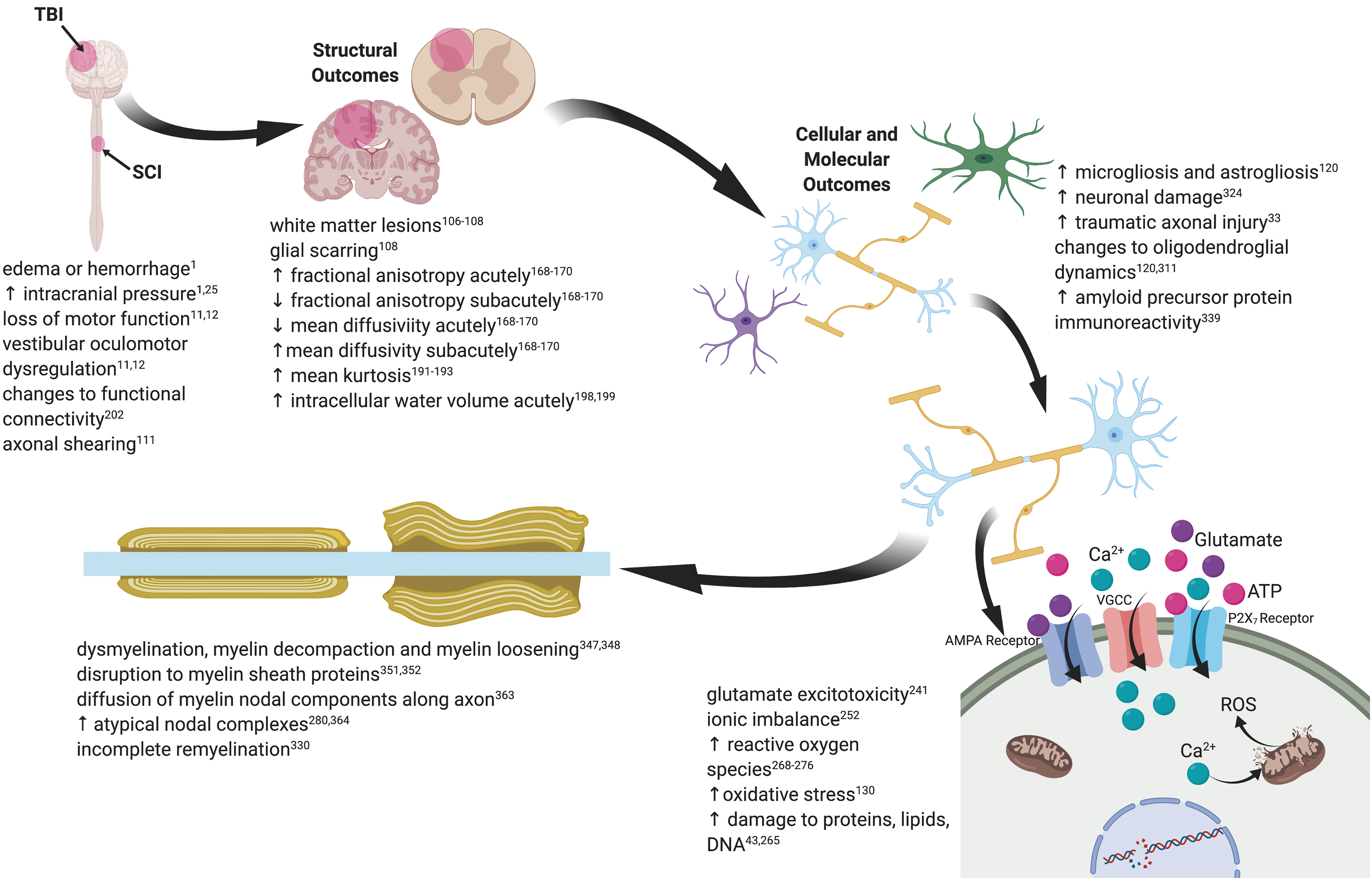
Schematic illustration of structural and molecular mechanisms of white matter damage in central nervous system injury. Note: Traumatic brain injuries represented are non-penetrating. Figure created with BioRender.com.
Summary of Cellular and Pathophysiological Changes Reported by the Fitzgerald Team in Rat Models of CNS Injury


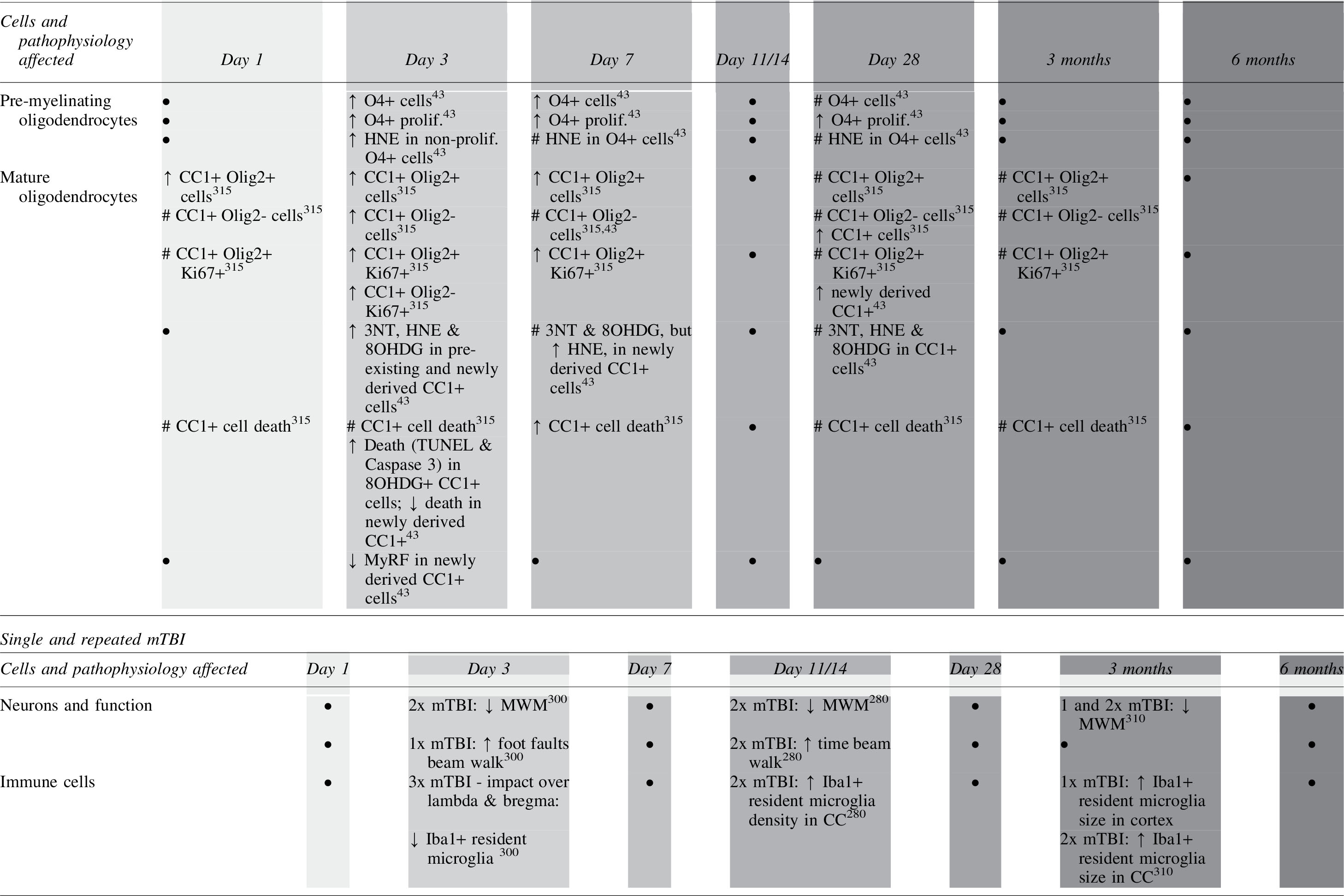

Footnotes
Funding Information
The work was supported by the National Health and Medical Research Council of Australia (APP1160691).
Author Disclosure Statement
No competing financial interests exist.