
Abstract
Traumatic brain injury (TBI) can cause persistent cognitive changes and ongoing neurodegeneration in the brain. Accumulating epidemiological and pathological evidence implicates TBI in the development of Alzheimer's disease, the most common cause of dementia. Further, the TBI-induced form of dementia, called chronic traumatic encephalopathy, shares many pathological hallmarks present in multiple different diseases which cause dementia. The inflammatory and neuritic responses to TBI and dementia overlap, indicating that they may share common pathological mechanisms and that TBI may ultimately cause a pathological cascade culminating in the development of dementia. This review explores Australian pre-clinical research investigating the pathological links between TBI and dementia.
Introduction
Traumatic brain injury (TBI) causes neuronal damage and inflammation which may result in both short- and long-term cognitive, behavioral, and motor deficits. Cognitive deficits from mild TBI may resolve within months; however, persistent cognitive disturbances and neurobehavioural symptoms occur in between 15% and 30% of cases. 1 Further, mounting epidemiological, clinical, human brain, and experimental evidence suggests that both single and repetitive TBI can lead to neurodegenerative changes in the brain that increase risk of dementia or dementia-like syndromes. 2 –7 In this regard, TBI has also been linked to the development of chronic traumatic encephalopathy (CTE), which shares many clinical and pathological features with other diseases that lead to dementia. 6,7
There are a number of similarities in the pathological features of TBI and the major diseases that cause dementia, including aggregation of pathological proteins, axon pathology, and inflammation. Australian neurotrauma research groups have made significant contributions to the literature regarding these links. This review discusses the pathological links between TBI and dementia, highlighting recent Australian pre-clinical research. However, to understand the relationship between TBI and dementia and how Australian research groups have contributed to this field, we must first discuss our current understanding of the pathology of TBI and dementia.
The Pathology of Traumatic Brain Injury
TBI results in two main forms of damage: focal and diffuse. Focal brain damage occurs due to a direct impact to the brain and includes intracranial bleeds, lacerations, and cortical and subcortical contusions. 8,9 Diffuse brain injury, however, does not necessarily involve a direct hit to the head, 9 but instead is caused by the straining, shearing, and compression of brain tissue and thus is multi-focal. 10 –14 Here, we will discuss the main forms of TBI-induced pathological changes relevant to enduring cognitive deficits and neurodegenerative conditions.
Diffuse axonal injury (DAI) is the most common pathology observed in diffuse TBI and occurs because of acceleration/deceleration forces on the brain causing shear stress to axons. 14 –16 DAI can cause interrupted axonal transport, resulting in axonal swellings with accumulations of incompletely transported proteins and organelles. 17,18 These transport deficits are hypothesized to be attributed to the stretching of axons, which results in the breakage, disorganization, and disassembly of microtubules, a component of the axonal cytoskeleton. 17,18 Accumulation of amyloid precursor protein (APP) in axonal swellings after TBI has been extensively described and is recognized as a pathological hallmark of axonal injury. 19 –27
Although transport deficits and swellings do not occur in all axons, other important pathological changes induced by TBI can cause axonal dysfunction and/or degeneration. Diffuse brain injury can lead to compaction of neurofilaments (NFs), another component of the axonal cytoskeleton. 28,29 Further, TBI can also perturb the axonal ionic balance, including elevated intra-axonal sodium and calcium levels. 30,31 Elevated intra-axonal calcium levels activate proteases, such as calpain, resulting in damage to the axonal cytoskeleton and ion channels. 30,32,33 Additionally, TBI-induced oxidative stress and lipid peroxidation can cause cytoskeletal degradation and mitochondrial dysfunction in the axon, as reviewed in a previous work. 34 Enduring axonal pathology has been observed years after TBI, 35 and it is suggested to be one of the underlying causes of chronic and progressive cognitive impairment after TBI. 36,37 Further, it may provide an important mechanistic link between TBI and dementia.
TBI also initiates inflammatory cascades, which can further exacerbate the original injury. Inflammatory cascades are mediated by microglia, the resident immune cells of the brain. Over the past decades, evidence has come to light regarding the shared inflammatory pathways initiated by TBI and those involved in other neurological diseases, such as Alzheimer's dementia, as reviewed in an earlier work. 38
The Pathology of Dementia
Multiple neurodegenerative diseases, which have overlapping pathology, cause the global decline in brain function referred to clinically as dementia. This review will primarily focus on the most prevalent cause of dementia, Alzheimer's disease (AD), and its relationship to CTE. 6 AD is characterized by four main pathological hallmarks: extracellular aggregates of insoluble amyloid-beta (Aβ) peptides known as plaques, neurofibrillary tangles (NFTs) and neuropil threads, both composed of hyperphosphorylated tau, and Aβ plaque-associated dystrophic neurites. 39 –41
As AD progresses, there is a characteristic pattern of pathology development, which differs between NFTs and plaques. Plaques first appear at low density in the isocortex, then, in conjunction with increased plaque density in the isocortex plaques, also develop in the primary and sensory cortices. 39 Finally, plaque deposition occurs in all cortical areas and the hippocampus. 39 In contrast, immunohistochemical analysis of NFTs and neuropil thread deposition reveals that changes appear first in the transentorhinal region of the temporal lobe, lateral to the hippocampus, before spreading to the entorhinal cortex, hippocampus, and association neocortex. This pattern of pathology led to Braak staging of AD pathology, with stages I–II affecting the transentorhinal area; in stages III–IV, pathology moves into the limbic regions, with the neocortical regions exhibiting pathology in stages V–VI. 42 It should be noted that medial temporal lobe pathology, associated with stages I and II, is a very common neuropathological finding in older cognitively intact individuals.
The Aβ peptide, of which plaques are comprised, is derived from the proteolytic cleavage of the APP protein. Two proteolytic pathways exist by which APP can be processed: a non-amyloidogenic pathway and the Aβ-producing amyloidogenic pathway. 43 Increased Aβ production through the amyloidogenic pathway is hypothesized to underlie, at least in part, the series of neurodegenerative changes associated with AD. 44 Aβ plaques may be characterized morphologically into three main types—diffuse, fibrillar, or dense-cored—and can be classified as neuritic by the presence of dystrophic neurites or non-neuritic if dystrophic neurites are absent. 40
The tau protein, which abnormally accumulates in a fibrillar form in NFTs, neuropil threads, and dystrophic neurites in AD, is a microtubule-associated protein that promotes assembly of tubulin into microtubules and aids microtubule stability. 45,46 In the human brain, there are six isoforms of tau, generated by the alternative splicing of its encoding gene, microtubule-associated protein tau, with these isoforms being categorized into two main groups, 3-repeat tau (3R-tau) and 4-repeat tau (4R-tau), depending on the number of microtubule-binding domains they contain. 47 Some diseases result in the pathological accumulation of just one isoform of tau, such as 3R-tau in Pick's disease; however, both AD and CTE have filamentous accumulations of 3R- and 4R-tau. 48 The microtubule-stabilizing role of tau is regulated by its phosphorylation; abnormal hyperphosphorylation of tau in AD results in a loss of function and increases the propensity for tau to self-aggregate, resulting in the insoluble accumulation of tau in NFTs, neuropil threads, and dystrophic neurites. 45
Dystrophic neurites in AD are neuronal processes, likely of axonal origin, with abnormal shapes and accumulations of cytoskeletal and cytoskeletal-associated proteins, which are specifically associated with Aβ plaques, as reviewed elsewhere. 49 Our laboratory and others have demonstrated that dystrophic neurites can be immunoreactive for accumulations of numerous proteins, including both phosphorylated and dephosphorylated NFs, α-internexin, APP, beta-secretase 1 (BACE1), growth-associated protein 43 (GAP43), ubiquitin, synaptophysin, chromogranin A, lysosomal-associated membrane protein 1, and/or tau. 40,41,50 –64
CTE is a neurodegenerative condition caused by TBI, which shares many clinical and pathological features with AD. 6,7 Pathologically, CTE is characterized by brain atrophy, astrocytic tau tangles, and TAR DNA-binding protein-43 accumulation, as well as the pathological hallmarks of AD, tau-NFTs, neuropil threads, and, in some cases, Aβ plaques. 6,65 –67 CTE is defined pathologically as a tauopathy, with the abnormal tau accumulations (NFTs and dystrophic neurites) occurring primarily in the perivascular regions and at the depths of the cortical sulci. 68 It is important to note, however, that the focus of studies to date have been on abnormalities in tau, and it is not yet clear how these may relate to other neuronal cytoskeletal alterations.
The pattern of abnormal tau accumulation in CTE has been proposed to be distinct from other tauopathies, with NFTs present perivascularly and focally in the cerebral cortex, preferentially in neocortical layers II and III of deep sulci, as opposed to AD where they mainly occur in layers III and V, as reviewed earlier 48 ; however, NFTs and Aβ plaques are immunohistochemically identical to those observed in AD. 6 Additionally, CTE frequently occurs in conjunction with other neurodegenerative disorders, including AD. 66 The overlap between CTE and AD suggests a common underlying pathogenesis between these neurodegenerative conditions.
The Axonal Response in Traumatic Brain Injury and Alzheimer's Disease
The axonal pathologies in TBI and AD are strikingly similar. Further, the axonal response to injury shares many of the neurochemical and morphological signatures of dystrophic neurites in AD. Australian laboratories have demonstrated that focal transection of axons in mature culture preparations 69 and acute models of penetrative brain injury in rodents 70 result in axons undergoing an initial reactive phase involving the accumulation of NFs in bulb- and ring-like structures. Further, they have shown that this reactive phase of injured axons is morphologically identical to early plaque-associated dystrophic neurite pathology in AD, which also exhibits NF-labeled ring- and bulb-like structures. 40,55
Australian TBI research has also focused on the neurochemical changes that occur in injured axons. Using cortical neuronal cultures and in vivo rodent models of penetrative brain injury, they have demonstrated that injured axons accumulate the neuritic outgrowth marker, GAP43, as well as NFs in sprouting axons. 69 –73 This neurochemical response of axons to TBI is comparable to that of neurites in AD where GAP43-positive dystrophic neurites as well as fine-caliber sprouting GAP43-immunolabeled neurites have been demonstrated in the hippocampus. 74 Further, as mentioned above, NFs also accumulate in plaque-associated dystrophic neurites in AD. 40,41,50 –64 Thus, the plaque and periplaque environment in AD may induce abnormal stimulation of a regenerative response similar to that observed post-TBI in the surrounding neuropil.
Because of the striking array of similarities between dystrophic neurites and traumatically injured neurites, we and others have hypothesized that Aβ plaques may represent chronic local lesions within the neuropil that structurally damage neighboring dendrites, synapses, and axons. 49,75,76 This hypothesis is supported by data from Australia and abroad showing that the denser, fibrillar plaques in AD more frequently associate with dystrophic neurites compared to diffuse plaques, and that the density of axons and dendrites is decreased within fibrillar plaques compared to the surrounding neuropil. 40,75,77 –81 Thus, Aβ plaques in AD may cause physical damage to axons in the neuropil as they grow in size and density, similar to the damage caused by the stretching and shearing forces of TBI.
As mentioned previously, the axonal accumulation of APP is a pathological hallmark of TBI and is thought to result from disruption of axonal transport, 17,18 while dystrophic neurites in transgenic AD mice and human AD also accumulate APP. 52,74,82 –86 However, Australian and international neurotrauma research has demonstrated TBI-induced APP upregulation, suggesting that it may have a neuroprotective role in the injured brain. 26,87 –89 The neuroprotective role of APP after TBI will be discussed in more detail below.
Similar to neurites post-TBI, 35,90 evidence suggests that coaccumulation of APP and BACE1 in dystrophic neurites may result in production and release of Aβ and the potentiation of plaque seeding and growth. 83,84 Our laboratory and other international laboratories have demonstrated coaccumulation of APP and Aβ in injured axons following experimental models of diffuse TBI in AD transgenic mice and in human TBI cases. 2,35,90 –94 Our data indicate that diffuse TBI, but not focal penetrative TBI, before onset of plaque deposition in transgenic AD mice, results in increased plaque burden after injury. 94,95 Further, research from another Australian laboratory examined Aβ distribution in brains using [ 18 F]-AV45 tracer positron emission tomography (PET) in a human cohort with and without a history of TBI. They demonstrated that people who had a history of TBI had a pattern of mainly increased Aβ tracer uptake in comparison to controls. 96 These data further support the idea that TBI induced diffuse axonal injury and subsequent intra-axonal accumulations of APP and Aβ, may seed plaque development and be an important mechanistic link between TBI and the development of AD (see Fig. 1).
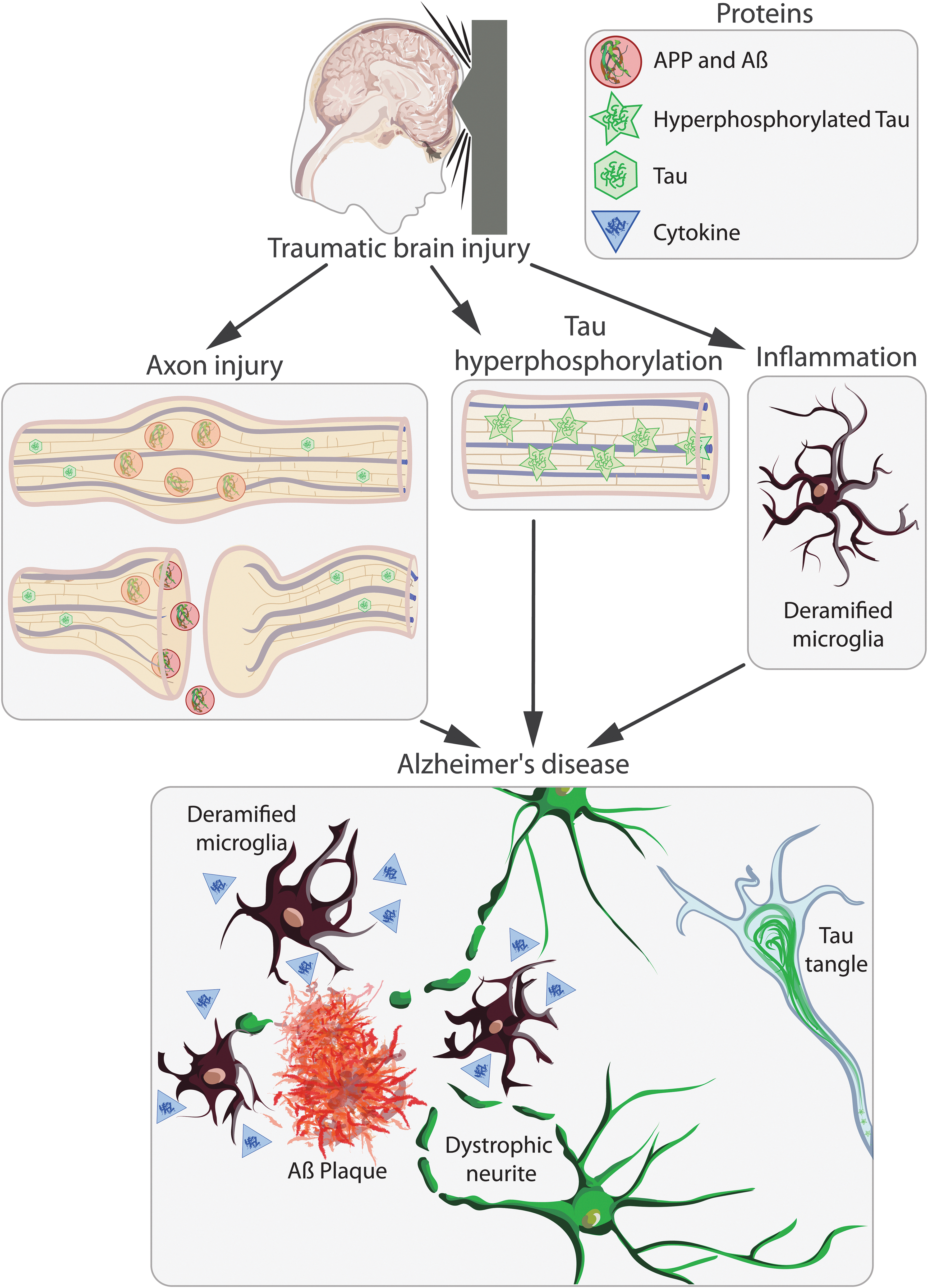
Schematic representation of the injury cascades initiated by traumatic brain injury (TBI). TBI causes axonal injury and tau hyperphosphorylation and elicits an inflammatory response, which may initiate a pathological cascade resulting in the development of Alzheimer's disease. Axonal injury can result in interrupted axonal transport, leading to the accumulation of APP, which may be cleaved to form Aβ and then released into the extracellular space by exocytosis or after lysis of the damaged axon. This extracellular Aβ may then aggregate forming Aβ plaques. Aβ plaques cause damage to neurites by either physically damaging them or by the release of toxic diffusible forms of Aβ. Tau hyperphosphorylation after TBI may also lead to the development of interneuronal neurofibrillary tangle formation, an important pathology in multiple different forms of dementia. TBI induces an inflammatory response in the brain, including microglial activation and the release of anti- and proinflammatory cytokines. This inflammatory response may persist chronically after TBI and may either prime the brain for or initiate a neurodegenerative cascade, resulting in the development of Alzheimer's disease. Aβ, amyloid-beta; APP, amyloid precursor protein. Color image is available online.
The Role of Amyloid Precursor Protein in Traumatic Brain Injury and Dementia
Australian neurotrauma research groups have contributed substantially to the literature on the expression and role of APP after TBI. As mentioned previously, intra-axonal accumulation of APP is a key pathological hallmark of axon pathology in TBI. 19 –27 However, Australian neurotrauma researchers 89,97 and others 26,87,88 have also demonstrated an increase in neuronal APP staining after TBI. Further, using an ovine left temporal TBI model, an Australian research group have shown that this increase in neuronal APP staining is caused predominantly by enhanced APP messenger RNA expression, 89 rather than accumulation of existing neuronal APP. This has led them and others to hypothesize that after TBI, APP is upregulated to perform neuroprotective and -trophic roles.
As mentioned previously, APP may be cleaved by an amyloidogenic pathway producing Aβ or a non-amyloidogenic pathway. The non-amyloidogenic pathway produces a soluble α form of APP (sAPPα). Australian neurotrauma research has demonstrated that sAPPα is neuroprotective after experimental TBI in both mice and rats. First, they demonstrated that intracerebroventricular injection of sAPPα 30 min after a severe impact acceleration model of TBI in rats resulted in improved motor and cognitive outcomes. 98 Additionally, they demonstrated that rats treated with sAPPα had a reduced number of apoptotic neuronal nuclei in the cortex and CA3 region of the hippocampus and lower levels of axonal injury in the corpus callosum. 98
Using an APP knockout mouse model (APP–/–), they subsequently demonstrated that endogenous APP is also neuroprotective after experimental TBI. 99 APP–/– mice were shown to have worse motor and cognitive impairment after moderate controlled cortical impact (CCI) injury and larger cortical lesion volume and hippocampal damage. Intracerebroventricular injection of sAPPα 15 min after CCI injury improved motor and cognitive function in APP–/– mice and reduced the amount of cortical and hippocampal damage. This indicates that the neuroprotective activity of APP is most likely mediated by sAPPα. 99 Using similar research methodology, this Australian laboratory identified that the heparin-binding sites of sAPPα, in particular residues 96–110 of the D1 domain, confer sAPPα's neuroprotective functions after TBI. 100 –102
Together, these results demonstrate that the upregulation of APP after TBI may be a neuroprotective response of the brain to trauma. However, these increased levels of APP after injury may also provide the substrate for enhanced Aβ production, seeding future plaque development. In support of increased Aβ genesis after TBI, an Australian meta-analysis of 19 experimental TBI studies using animal models found that cerebral Aβ levels are increased from 24 h up to 1 month post-injury. 103 Thus, an alternate therapeutic avenue to harness sAPPα's neuroprotective role in TBI without subsequently increasing Aβ production, and an area of research in the AD field, is therapeutically targeting APP cleavage toward the non-amyloidogenic pathway.
The Role of Tau in Traumatic Brain Injury and Alzheimer's Disease
Tau hyperphosphorylation is a key pathological feature of both AD and CTE. Changes in tau phosphorylation have been demonstrated after TBI 104 –107 and has been a major recent focus in Australian neurotrauma research. Shultz and colleagues have demonstrated that fluid percussion injury (FPI) in rats acutely reduces expression and activity of components of protein phosphatase 2A (PP2A), a major protein phosphatase in the brain, while increasing expression of phosphorylated tau and the ratio of phosphorylated tau to total tau. 106 Similarly, a significant increase in tau phosphorylation was also observed in post-mortem human brain tissue from acute TBI patients. 106 Long-term increases in tau phosphorylation, as well as brain atrophy, corpus callosum damage, cognitive impairment, enhanced anxiety-like behavior, and sensorimotor impairments, were also demonstrated in rats at 12 weeks post-FPI. 106 This increase in tau phosphorylation, as well as the cognitive and motor impairments and brain damage, were attenuated when rats were treated with sodium selenate, a potent activator of PP2A. 106 Although brain damage and behavioral impairments still remained after sodium selenate treatment, these results indicate that enhanced tau phosphorylation may contribute to the neurodegenerative process after TBI and be an important pathological link between TBI and development of dementias such as CTE.
These results have been supported by other Australian studies examining the effects of single and repetitive brain injuries in rats using the FPI model and a modified impact acceleration model. 104,105,107 As observed for single TBI, repetitive mild FPI in rats induced a decrease in PP2A and an increase in tau phosphorylation in the brain. Further, brain atrophy and cognitive and sensorimotor deficits were also reported, with sodium selenate treatment shown to reduce these TBI-induced pathophysiological effects. 107 Although a decrease in the chief tau, phosphatase, has been shown after both single and repetitive TBI in rats, 104,106,107 Collins-Praino and colleagues have shown that there are minimal changes in tau-related protein kinases such as glycogen synthase kinase 3 beta. This suggests that the increase in tau phosphorylation after TBI is attributable to reductions in tau dephosphorylation by phosphatases, rather than increases in tau phosphorylation by kinases. In addition to an increase in levels of tau phosphorylation after single and repetitive experimental TBI in rats, there is also a significant increase in the number of neurons positive for hyperphosphorylated tau. 104,105
These data from experimental models of TBI in rodents are further supported by an Australian study by Mohamed and colleagues using the [ 18 F]AV145 PET imaging to examine tau accumulations in the brains of non-demented control and TBI cases. This study demonstrated that TBI cases had increased tau accumulation in neocortical regions of the brain compared to controls. Distribution of this widespread tau accumulation was similar to that of AD as well as atypical of AD, but similar to CTE. 108
From these studies, it is evident that TBI in rodents increases the hyperphosphorylation of tau in the brain, and this may be the result of TBI-induced changes in PP2A expression and activity. These changes in PP2A activity may be an important pathogenic mechanism linking TBI to AD. Numerous studies have demonstrated that PP2A levels and activity are decreased in the AD brain. 109 –111 Further, PP2A activity is negatively correlated to tau phosphorylation levels at most phosphorylation sites in the human brain. 112 Similar to the results observed for TBI studies, treatment with sodium selenate or selenomethionine in AD mouse models has been shown to increase PP2A expression, reduce tau expression and hyperphosphorylation, prevent NFT formation, improve cognitive and motor function, and reduce glial activation and synaptic protein loss. 113,114
Taken together, these data indicate that tau hyperphosphorylation is an important pathology in AD and TBI and that changes in expression and activity of PP2A may be the driver of tau pathology. Recent Australian early clinical trials in human AD patients have demonstrated that sodium selenate supplementation is well tolerated and results in elevated selenium concentration in the cerebrospinal fluid (CSF), 115,116 and pilot data are indicative of reduced cognitive deterioration, as measured by the Mini-Mental Status Examination, over the 24-week trial period. 115 Therefore, enhancing selenium levels in the CSF may be an important therapeutic intervention for TBI and may prevent the secondary neurodegenerative cascade which may lead to conditions such as CTE and AD.
The Role of Inflammation in Traumatic Brain Injury and Alzheimer's Disease
Research internationally as well as in Australia has shown that TBI initiates a cascade of inflammatory events which begin at the time of injury and can take months to years to fully resolve. 38,117 –119 Proinflammatory cytokines, such as interleukin (IL)-1, tumor necrsosis factor, and IL-6 and chemokines (C-C motif chemokine ligand 2, IL-8/C-X-C motif chemokine ligand 2), as well as other mediators, including cell adhesion molecules, prostaglandins, complement, and damage-associated molecular patterns, are the first responders to TBI, stimulating further the upregulation of other factors which generate a multi-faceted inflammatory response. This response is required for repair; however, if not tightly controlled, it will exacerbate injury.
Neutrophil recruitment mostly occurs within the first 24 h post-injury. Clinical studies have reported that the absolute number and frequency of neutrophils are significantly increased after TBI, with one study reporting a 4.5-fold increase as early as 3 h. 120 Research from Australia has shown that in experimental models of focal TBI, mice deficient in C-X-C motif chemokine receptor 2, the mouse homologue of human IL-8, have reduced neutrophil infiltration, decreased lesion volume, and altered cytokine profiles. 121 These data suggest that neutrophils may exacerbate secondary brain injury, potentially through production of reactive oxygen species.
European groups have reported data from experimental models of AD which indicate that neutrophil accumulation occurs in all stages of disease, suggesting that neutrophil trafficking may cause blood–brain barrier dysfunction and inflammation. 122 When neutrophil accumulation was depleted or blocked by lymphocyte function-associated antigen 1 integrin, the clinical and neuropathological hallmarks of AD were reduced. Clinically, changes in neutrophil survival, migration, and phagocytic activity have been reported in patients with AD. 123 These alterations were most evident in patients with AD dementia compared to AD mild cognitive impairment. Further, patients with dementia of other types had neutrophils that exhibited normal survival, migration, and phagocytic activity, indicating AD-specific pathology.
Microglia are traditionally described as the immune cells of the brain. However, with advances in technology, additional dynamic roles of microglia, such as synaptic maintenance, have been identified. Microglia are distributed widely throughout the brain, such that they can quickly detect disturbances to homeostasis. After TBI, microglia play a dynamic role, scavenging debris and assisting in controlling the inflammatory response. Australian researchers have documented that, morphologically, microglia change from a ramified surveillance structure to an activated morphology, 124 –126 where the production of cytokines and chemokines change. 119,121,127,128
Evidence suggests that acute and chronic microglial activation contributes to circuit pathology after diffuse TBI in rats. 124 Moreover, the role microglia play in TBI may vary with age, including changes in morphology and cell-surface marker expression. With age, microglia become dystrophic and undergo cytoplasmic fragmentation, with cell processes appearing stripped of fine ramifications. 129 In this state, microglia elicit a more proinflammatory profile. This suggests that the age at which TBI is acquired may drastically affect the microglial responses and, potentially, the outcome. Experimentally, Australian researchers have shown that lipopolysaccharide administration for 5 days after a mild diffuse TBI in rats produces an exaggerated proinflammatory response at 24 h, whereas at 3 months it caused persistent microglial activation and behavioral deficits. 130
In AD, it has long been hypothesized that microglia primarily respond to Aβ misprocessing and/or plaque load. However, evidence from experimental models is beginning to challenge this view, given that microglial activation begins before overt plaque formation. Microglia are reported to migrate to the plaque and phagocytose Aβ 131 ; over time, these microglia become enlarged and are no longer able to process Aβ. 132 Early activation of microglia is postulated to be beneficial; however, once the activation rolls into the chronic phase, this prolonged activation has been demonstrated to exacerbate AD pathology, as reviewed in an earlier work, 132 resulting in accumulation of Aβ and sustained proinflammatory cytokine signaling, which damages neurons. 133 –135 Sustained activation of microglia results in decreased efficiency for binding and phagocytosing Aβ, 136,137 without affecting microglial capacity for producing proinflammatory cytokines. 135
Our Australian research group has shown that environmental enrichment changes microglial morphology and increases CD68 immunoreactivity in a mouse model of amyloidosis (APP/PS1 [presenilin 1]). 138 Additionally, we have reported that when APP/PS1 mice were subjected to mild lateral FPI, CD68 immunoreactivity did not change. 94 Moreover, CD68-positive microglia throughout the white matter tracts were evident 30 days after FPI, suggesting chronic inflammatory processes and axonal pathology. 94 These data show the potential to harness microglia as a target for therapeutics both in AD and TBI.
Microglial inflammatory processes appear to have a dual function: Microglia form the first line of defense when there are disturbances to brain homeostasis; however, uncontrolled or chronic activation may be directly toxic to neurons attributable to release of inflammatory cytokines, nitric oxide, and superoxide. 139 Autopsy studies of individuals with recent brain injury have shown increased cerebral Aβ deposits 1–3 weeks post-injury, with increased levels of IL-1ß being associated with the increased APP production and Aβ load. 140,141 IL-1ß has been shown to increase other cytokines, such as IL-6, which has been shown to stimulate cyclin-dependent kinase 5 activation, a kinase which hyperphosphorylates tau. 142 Therefore, the inflammatory response may initiate pathological cascades, resulting in accumulation of AD-associated pathology. The inflammatory response to TBI is similar to that reported in AD and leads to the question, Does this inflammatory response to TBI drive the development of AD?
Cognitive Impairment after Traumatic Brain Injury and Its Link to Dementia
Another mechanism by which TBI may increase the risk of subsequent dementia is by affecting cognition. Australian neurotrauma research groups have investigated TBI-induced cognitive changes using both experimental animal models and human cohort studies. Eramudugolla and colleagues examined the cognitive outcomes of TBI over 8 years in a cohort of people 20–64 years of age. They found that a history of TBI was associated with long-term cognitive decline and impairment across the life span in adults. 143 This is in contrast, however, to research from another Australian group who have demonstrated that adults who recovered to a healthy level of cognitive function, after a mild TBI with loss of consciousness, did not have deficits in cognitive function, 5 years or more after the injury. The data did, however, indicate that the effect of TBI on the cognitive domain, episodic memory, may be modulated by the duration of TBI-induced unconsciousness and the age at which the TBI occurred. 144
Differences in the findings of these studies can be attributed to a number of experimental factors, including sample size, the way TBI is defined, the time after TBI examined, and cognitive testing performed, among others. However, looking at literature from all sources, TBI is associated with long-term deficits in cognition. 1
Australian research using rodents have also shown subtle changes in cognitive function after experimental brain injury. One study demonstrated that moderate/severe TBI in rats caused significant increases in depressive-like behavior from 1 to 3 months post-injury and impaired cognitive flexibility at 3 months post-injury, despite learning and memory remaining unaffected. 145 Further, this same group later demonstrated that at 12 months post-injury, a single moderate/severe, but not a single mild or repetitive mild, TBI in rats also caused significant impairment in cognitive flexibility without causing significant impairments in depressive-like behavior, spatial learning, or memory. 146 Together, these data indicate that TBI may cause long-term, subtle changes in cognition; however, the implications of these changes in terms of dementia risk are currently unknown.
From the literature, it is evident that TBI can cause long-term deficits in cognition. TBI-induced changes in cognition may reduce the amount of pathology the brain can withstand, therefore accelerating the onset of cognitive impairment. These cognitive changes may be a mechanism by which TBI leads to or accelerates the onset of dementia.
Conclusion
TBI is a recognized risk-factor for development of dementia, including AD. Aβ and tau are hypothesized to be closely linked to neurodegeneration in AD and are evident in both rodent TBI models and patients who have had a TBI. Overall, the pathological changes of TBI and AD are similar, indicating that they may share underlying disease mechanisms. Further, it could be postulated that TBI may either initiate and/or accelerate the onset of dementia. While this review has mainly focused on how TBI-induced axonal injury, APP processing, tau phosphorylation, inflammation, and cognitive changes may underlie the relationship between TBI and the development of dementia, other important mechanisms, such as metabolic, vascular, and synaptic changes, are also likely to be involved.
Footnotes
Acknowledgment
Figure provided by Mr. Graeme McCormack.
Funding Information
A.E.K. is supported by an NHMRC Boosting Dementia Research Leadership Fellowship (APP1136913).
Author Disclosure Statement
No competing financial interests exist.