
Abstract
Traumatic brain injury (TBI) alters stress responses, which may influence neuroinflammation and behavioral outcome. Sleep disruption (SD) is an understudied post-injury environmental stressor that directly engages stress-immune pathways. Thus, we predicted that maladaptive changes in the hypothalamic-pituitary-adrenal (HPA) axis after TBI compromise the neuroendocrine response to SD and exacerbate neuroinflammation. To test this, we induced lateral fluid percussion TBI or sham injury in female and male C57BL/6 mice aged 8–10 weeks that were then left undisturbed or exposed to 3 days of transient SD.
At 3 days post-injury (DPI) plasma corticosterone (CORT) was reduced in TBI compared with sham mice, indicating altered HPA-mediated stress response to SD. This response was associated with approach-avoid conflict behavior and exaggerated cortical neuroinflammation. Post-injury SD specifically enhanced neutrophil trafficking to the injured brain in conjunction with dysregulated aquaporin-4 (AQP4) polarization. Delayed and persistent effects of post-injury SD were determined 4 days after SD concluded at 7 DPI. SD prolonged anxiety-like behavior regardless of injury and was associated with increased cortical Iba1 labeling in both sham and TBI mice. Strikingly, TBI SD mice displayed an increased number of CD45+ cells near the site of injury, enhanced cortical glial fibrillary acidic protein (GFAP) immunolabeling, and persistent expression of Trem2 and Tlr4 7 DPI compared with TBI mice. These results support the hypothesis that post-injury SD alters stress-immune pathways and inflammatory outcomes after TBI. These data provide new insight to the dynamic interplay between TBI, stress, and inflammation.
Introduction
Traumatic brain injury (TBI) causes widespread neuroinflammation that, if unregulated, results in progressive neurodegeneration. 1,2 Post-injury neuroinflammation is characterized by microglial reactivity, astrogliosis, blood–brain barrier (BBB) disruption, infiltration of peripheral cells, and production of pro- and anti-inflammatory cytokines. 3,4 Data from our group indicate that TBI-induced neuroinflammation sensitizes the brain to the effects of secondary stressors, leading to impaired physical and mental health. 5 For example, when injured animals are exposed to a peripheral immune challenge (e.g., lipopolysaccharide; LPS) at 30 days post-injury, microglia mount an amplified inflammatory reaction that is associated with depressive-like behavior 6 and impaired memory consolidation. 7 We predicted that other environmental stressors, such as sleep disruption (SD), would similarly alter neuroinflammation and influence functional outcome following TBI by engaging the stress-immune axis.
Stressors activate the hypothalamic-pituitary-adrenal (HPA) axis, resulting in the production of stress hormone—cortisol in humans, corticosterone (CORT) in rodents. Studies show that TBI impairs the ability to restore homeostasis in response to acute stressors, indicating HPA axis dysfunction. 8 –11 In the context of HPA axis dysfunction, everyday stressors exert intense “allostatic load” or “wear and tear” on the body. In clinical studies, TBI severity is directly correlated with the magnitude of neuroendocrine deficiency. Specifically, plasma cortisol increases immediately after mild TBI, but decreases immediately after severe TBI. 9 Acute stress-induced CORT facilitates energy expenditure and is immunosuppressive, but blunted CORT responses to chronic stress worsen neuroinflammation and functional outcome. 12 The effect of impaired allostasis after TBI is not well characterized, thus narrowing the interpretation of how this may influence long-term outcome. 13
Sleep is necessary to maintain homeostasis, and circadian rhythms are essential for organizing cellular- and systems-level function throughout the body. Environmental factors such as technology use, 14 hospitalization, 15 and military deployment 16 disturb sleep and circadian rhythms following TBI. These disturbances may act as secondary stressors following injury by altering CORT and HPA axis reactivity. Axons within the suprachiasmatic nucleus (SCN) project to discrete hypothalamic nuclei to control the HPA axis, causing both diurnal and stress-induced release of CORT by the adrenal gland. 17 –19 Because circulating CORT controls HPA activity via negative feedback, sleep-induced changes in CORT release will also affect circadian rhythms. 20 –24 Clinical studies show that SD blunts normal cortisol responses and is related to increased circulating levels of key inflammatory cytokines such as interleukin (IL)-1, IL-6, and tumor necrosis factor alpha (TNFα). 25,26 Further, increased leukocyte hematopoiesis following sleep fragmentation may promote inflammation. 27 These data indicate that SD is a stressor that directly engages the HPA axis and increases inflammation. Thus, SD represents a potential component of post-injury allostatic load that influences recovery.
There is a need to better characterize the neuropathological and functional consequences of post-injury immune stressors. Homeostatic sleep/wake behavior requires coordinated engagement of endocrine and immune pathways. Our studies used daily SD to activate, engage, and interrupt endocrine/immune coordination during acute recovery. This challenged the injured brain to maintain homeostasis. We hypothesized that SD after TBI disrupts neuroendocrine signaling, exacerbates the neuroinflammatory response to brain injury, and impairs behavioral recovery. Moreover, based on previous data, 28 we predicted that the temporal progression of post-injury neuroinflammation would remain altered following a recovery period after SD. Together, these studies aim to spotlight stress-immune communication after TBI as a critical intersection in facilitating optimal recovery.
Methods
Subjects
Equal numbers of male and female C57BL/6 mice aged 8–10 weeks were purchased from Charles River Laboratories (Wilmington, MA) for every study. Mice were group-housed (n = 4/group) by sex in The Ohio State University's Wiseman Hall vivarium and maintained at 21°C under a 12-h light/12-h dark cycle (i.e., lights on 7AM—7PM) with ad libitum access to food and water. Each cohort of male and female mice received either sham injury or lateral fluid percussion TBI and then were left undisturbed or received SD for 3 days before sacrifice at 3 or 7 days post-injury (DPI). Following surgical preparation, each subject was randomized to the TBI or sham group as well as undisturbed or SD group, resulting in four experimental groups: sham, TBI, sham SD, and TBI SD. Attention was given to counterbalance males and females in all procedures and analyses. All conditions were in accordance with the principles set forth by the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of The Ohio State University.
Surgical preparation and lateral fluid percussion injury (FPI)
All surgical procedures were performed as previously described. 29,30 Briefly, mice aged 8–10 weeks were anesthetized with 4% isoflurane gas in an induction chamber for 4 min and then positioned in a stereotaxic frame. Following midline incision, a 3.0-mm craniectomy was trephined midway between bregma and lambda on the right parietal bone, leaving the intact dura mater exposed. A modified portion of a Leur-Loc syringe (3.0-mm inside diameter) was positioned over the craniectomy site and secured with cyanoacrylate adhesive. Following the surgical procedure, mice were placed in their home cages on a heating pad to recover. Twenty-four hours after the surgical procedure, mice were anesthetized with 4% isoflurane in an induction chamber for 4 min and then connected to the fluid percussion injury (FPI) device (Custom Design & Fabrication, Richmond, VA) using the modified portion of a Leur-Loc syringe. To initiate the impact, a pre-positioned hammer was dropped onto the end of the FPI device, delivering a water pulse onto the exposed dura mater to induce a moderate FPI to mice designated to the TBI group. Sham mice were attached to the FPI device, but the hammer was not released, and they did not receive a fluid pulse. The modified syringe and adhesive were removed following FPI or sham injury and the incision was stapled closed. All animals were placed on a heating pad and injury severity was assessed via the self-righting reflex test. After the subjects demonstrated the righting reflex, they were returned to their cages. Mice were sacrificed at either 3 or 7 DPI.
Sleep disruption (SD)
SD was operationally defined as 3 days of minimally invasive mechanical stimulation from 7–11AM beginning the morning after injuries, as adapted from previously published protocols. 31 –33 During SD entire home cages were removed from the standard housing rack and room. In a different room, one to two investigators placed the home cages on a table, removed the plastic cage top, and sat quietly observing sleeping behavior from 7–11AM. Sleeping behavior was operationally defined as sitting quietly with no movement and eyes closed. If sleeping behavior was observed, investigators would gently pet the mouse or nudge it to move. In addition, investigators would remove the cage lid or wire cage rack to stimulate the mice. On 2 DPI, one cage change was performed between 9 and 9:30AM. On 3 DPI, two cage changes were performed at approximately 8:30AM and 10AM. The cage changes were introduced because mechanical stimulation or removing the cage lid/wire rack was not sufficient to interrupt operationally defined sleeping behavior. Mice had ad libitum access to food and water throughout the SD procedures. Published data demonstrate that this protocol of 4 h of SD resulted in increased percent sleep and average sleep bout length for up to 4 h after SD ended. 31 This response suggests that the SD protocol is effective in altering sleep/wake behavior resulting in increased sleep after the disruption ends.
Elevated zero maze
The elevated zero maze was used to examine anxiety-like behavior following exposure to SD or standard housing. The elevated zero maze is equally divided into two open and two closed arms, which were defined as open arm 1, open arm 2, closed arm 1, and closed arm 2. At the beginning of a testing trial, a mouse was placed in open arm 1 next to the entrance to closed arm 1. Thereafter, the mouse could move freely throughout the maze for 5 min. After behavioral testing, each mouse was placed in a clean cage away from cage mates that had not completed behavioral testing. Traces of all feces and urine were removed from the maze before thoroughly cleaning it with 70% alcohol between every testing trial. An aerial view camera and Ethovision tracking software (Noldus) were used to record movement between the open and closed arms. Total time in open and closed arms, as well as total time in each arm, and average time spent in each of the four arms during each minute of the test were primary dependent variables of interest.
Immunohistochemistry and imaging analysis
Mice were euthanized by carbon dioxide asphyxiation. The flow rates and environment of sacrifice were constant across experimental groups and controlled to minimize animal distress. 34 Mice were then transcardially perfused with ice-cold phosphate-buffered saline (PBS; pH 7.4) followed by 4% paraformaldehyde (PFA). Perfused brains were dissected and post-fixed in 4% PFA for 24 h and then cryoprotected in 30% sucrose for 72 h and stored at 4°C. Brains were sectioned at 30 μm using a Leica CM1800 cryostat (Leica Biosystems) and were stored in cryoprotectant (30% ethylene glycol, 30% polyethylene glycol, 40% 0.2 M PBS) at −20°C until labeling. Sections were rinsed with 0.1% Triton X-100 in PBS (PBST) for 10 min and then, for antigen retrieval, placed in a 10-mM sodium citrate buffer (pH 6.0) and heated at 90°C for 10 min. For immunofluorescent labeling, tissue was washed with PBST and blocked (5% normal donkey serum, 0.3% Triton X-100 in PBS) for 2 h at room temperature. Sections were then incubated overnight at 4°C in primary antibody with constant rotation: rabbit anti-mouse Iba1 (1:500; Wako Chemicals), goat anti-mouse GFAP (1:500; Abcam), rat anti-mouse CD45 (1:500; Abcam), or rabbit anti-mouse aquaporin-4 (AQP4) (1:2000; Sigma-Aldrich).
Next, tissue was rinsed in PBST and incubated in corresponding fluorochrome-conjugated secondary antibody for 1 h (1:1000; Alexa Fluor 594, 488, or 647). Sections were then mounted and cover-slipped using Fluoromount-G (Invitrogen). For diaminobenzidine (DAB) labeling, tissue was placed in 0.3% H2O2 in PBS for 30 min after antigen retrieval and then was blocked (5% normal goat serum, 0.3% Triton X-100 in PBS) with Mouse IgG Blocking Reagent (1:100; Vector Laboratories) for 2 h. Sections were then incubated overnight at 4°C with mouse anti-mouse AT180 (1:200; Thermo Fisher) with constant rotation. Sections were washed with PBST and incubated with biotinylated anti-mouse secondary antibody (1:200, Vector Laboratories) for 1 h. Next, sections were washed with PBST and incubated according to instructions for the ABC Kit for DAB staining (Vector Laboratories). After a PBST wash, sections were placed in DAB solution (Vector Laboratories) for 45 sec with an additional 15 sec for each subsequent tissue set. DAB reaction was stopped with PBS and sections were washed a second time in PBS. Sections were then mounted and dehydrated (1 min 70% ethanol, 1 min 90% ethanol, 2 × 2 min 100% ethanol, 2 × 4 min xylenes) and cover-slipped using Permount (Thermo Fisher).
For Iba1 and GFAP, slides were imaged using an EVOS FL Auto 2 Imaging System (Thermo Fisher) at 20 × magnification. For each animal, 2–4 images per area of interest were taken and percent-area was quantified using the National Institutes of Health's ImageJ software. For AQP4 polarization, slides were imaged using a Leica SP8 upright confocal microscope at 20 × magnification. Sequential optical sections were captured using the Leica Application Suite X imaging software. Sequential optical sections were analyzed using custom in-house software. AQP4 labeling intensity was calculated at each point along cross-sections of large penetrating cortical vessels. AQP4 polarization was then calculated as the ratio of perivascular (<10 μm from vessel) to parenchymal (20–25 μm from vessel) AQP4 immunofluorescence. Dysfunction is indicated by a lower polarization because AQP4 should be primarily sequestered to the perivascular astrocytic end feet. 35
For lesion analysis, 30-μm sections were selected at 10 evenly spaced coordinates covering the entire area under the craniectomy (+0.02 mm through −3.16 mm relative to bregma). Sections were stained with DAPI to visualize cytoarchitecture and 10 × tile-scan images were taken of each section on an EVOS cell imaging system. Lesion area was measured for each section using ImageJ. Lesion volume was extrapolated by averaging adjacent sections and multiplying by the distance between sections. Investigators were blinded throughout analysis. Brain regions were identified using the Allen Mouse Brain Atlas. For CD45, 10 × tile-scan images were taken of each section using the aforementioned EVOS system. Images were then rotated to center on the injury site and cropped to select a 3.65-mm × 2.01-mm region for quantification using ImageJ. The number of CD45+ cells for each section was counted and averaged for quantification. For AT180, slides were imaged using the aforementioned EVOS system for bright field at 40 × . For each animal, 2–4 images per ipsilateral temporal cortex were taken and AT180+ cells were counted and averaged for each animal.
Quantitative polymerase chain reaction (qPCR)
Mice were euthanized by carbon dioxide asphyxiation and the brains were immediately removed. The ipsilateral cortex was micro-dissected and snap frozen in liquid nitrogen (−196°C) and stored at −80°C. RNA was isolated from cortical homogenate using Tri-zol Reagent (Sigma Aldrich) per manufacturer's protocols. Next, RNA (2.4 ng) was diluted in 40 μL of RNase-free water and converted to complementary DNA (cDNA) using the High Capacity cDNA Reverse Transcription Kit (HiCap RT PCR kit; Applied Biosystems). Resulting cDNA was diluted 1:10 in TaqMan Advanced Fast Master Mix (Thermo Fisher) and gene expression was quantified by real-time quantitative polymerase chain reaction (qPCR) using validated probes from TaqMan Gene Expression (Applied Biosystems) on a QuantStudio3 PCR machine (Thermo Fisher)for 40 amplification cycles. Samples that amplified in more than 38 cycles were excluded. Genes of interest were normalized to Gapdh and expressed as fold-change from control (ΔΔCt). Gene targets included: H2-Eb1 (Mm00439221_m1); Aqp4 (Mm00802131_m1); Gfap (Mm01253033_m1); Vim (Mm01333430_m1); Itgam (Mm00434455_m1); S100b (Hs00902901_m1); Il1b (Mm00434228_m1); Ccl2 (Mm00441242_m1); Il6 (Mm00446190_m1); Ccr2 (Mm99999051_gH); Trem2 (Mm04209424_g1); Cxcr2 (Mm99999112_s1); Cxcl1 (Mm04207460_m1); Cx3cr1 (Mm00438354_m1); Tlr4 (Mm00445273_m1); Cd68 (Mm03047340_m1); and Tnf (Mm00443258_m1).
Corticosterone (CORT) enzyme-linked immunosorbent assay (ELISA)
Blood samples were collected in 1.5-mL Eppendorf tubes with 20 μL of 50 mM ethylenediaminetetraacetic acid (EDTA) as an anti-coagulant. Blood was collected via submandibular bleeding immediately after SD or after remaining undisturbed at 11AM, and then was immediately placed on ice. After collection, blood was spun at 6000 rcf for 15 min, after which plasma was extracted and stored at −80°C as previously described. 36 Plasma concentration of CORT was evaluated using the Corticosterone EIA Kit (catalog #ADI-900-097; Enzo) according to the manufacturer's instructions.
Collecting blood for CORT measurement is a sensitive and time-dependent procedure. Cheek bleed samples were taken in the same room as SD occurred to minimize movement stress. Order of bleed collection was randomized between days, ensuring no group condition or sex was sampled in the same order. Exclusion criteria included any animal that took longer than 3 min from initial investigator contact to successful sample collection and any animal that had to be scruffed more than once. After blood collection, each mouse was placed in a new clean cage away from littermates that had not yet undergone collection and then was returned to its home cage once collection was completed for that cage. Blood collection was done at 11AM to quantify the CORT response immediately following the stressor of SD. Blood was collected at 11AM 2 DPI in animals aging to 3 DPI. Blood was collected at 11AM at 1 and 3 DPI in mice aging to 7 DPI. The 2 DPI time-point was excluded for the 7 DPI experiments to reduce the number of cheek bleeds required over testing days.
Flow cytometry
Mice were euthanized by carbon dioxide asphyxiation, after which blood was collected via cardiac puncture into Eppendorf tubes with 20 μL 50 mM EDTA and placed on ice. Brains were then immediately collected and put into PBS on ice. Blood was collected at 3 DPI and processed for flow cytometric analysis as previously described. 37 Briefly, red blood cells were lysed and the remaining cells were centrifuged. Supernatant was removed and cell pellets were resuspended in antibody solution: B220 (APC-Cy7), Ly6C (PE-Cy7), CD3 (PE), CD11b (APC), and Ly6G (FITC) for 45 min at 4°C. All antibodies were diluted 1:50 and from Fisher Scientific. Compensation beads (Abcam) were also incubated for 45 min at 4°C for each antibody listed.
Brain tissue was homogenized and pelleted. After supernatants were separated, the cell pellets were resuspended in 70% isotonic Percoll (GE Healthcare). A Percoll density gradient was then applied in three layers of 50%, 35%, and 0% (PBS) isotonic Percoll and then centrifuged. The fat layer was removed from all tubes and the microglial layer between the 70% and 50% layers was separated and washed to remove any remaining Percoll. Cells were pelleted and supernatant was removed. Cells were then incubated in antibody solution as described above: Ly6G (FITC), Ly6C (PE), CD45 (Percp), and CD11b (APC). After antibody incubation, brain, blood, and compensation beads were washed and counting beads (123 count beads; eBiosciences) were added. Surface expression was determined using a FACS Caliberflow cytometer (Becton-Dickinson) and analysis was completed using FlowJo7 software (Tree Star).
Experimental study design
The primary objective of this study was to characterize the immediate and delayed effects of transient post-injury SD on behavioral and immunological outcomes after TBI. This resulted in a 2 (sham, TBI) × 2 (undisturbed, SD) factorial design. To determine post-injury SD effects on CORT responses, submandibular cheek bleeds were taken at 2 DPI (n = 6–8 per group). Immediate behavioral effects of post-injury SD were determined through elevated zero maze at 3 DPI immediately before sacrifice (n = 10–12 per group). Flow cytometry was used to measure immune cell populations (n = 3–4 per group) in both blood and brain. Inflammatory responses were also characterized by immunohistochemistry (n = 4–6 per group) and messenger RNA (mRNA) expression (n = 4–6 per group). To determine delayed and persistent effects of post-injury SD, separate cohorts of male and female mice were divided as previously described and following 3 days of SD remained undisturbed for 4 additional days until sacrifice at 7 DPI. To determine post-injury SD effects on CORT responses, submandibular cheek bleeds were taken at 1 and 3 DPI (n = 6–8 per group). Delayed behavioral effects of post-injury SD were determined through elevated zero maze at 7 DPI immediately before sacrifice (n = 10–12 per group). Inflammatory responses were characterized by immunohistochemistry (n = 4–6 per group) and mRNA expression (n = 4–6 per group). An independent investigator blinded to experimental groups performed subsequent data analysis.
Statistical analysis
All statistical analysis was completed with GraphPad Prism. A mixed model factorial analysis of variance (ANOVA) with correction for repeated measures was used to evaluate group differences in elevated zero maze performance and weight change. A mixed model ANOVA was selected because subject numbers varied between experimental groups. 38,39 For all other analyses, a two-way ANOVA was used with correction for multiple comparisons and with injury (TBI, sham) and sleep (no SD, SD) as independent variables. Main effects of injury and SD, as well as interaction effects, were considered. The Holm-Sidak procedure was used for multiple comparisons following statistically significant main effects. Male and female mice were pooled for all analyses as no sex × injury × SD effects were identified. Statistical significance was determined as p < 0.05. All data are presented as mean ± standard error of the mean (SEM).
Results
Post-injury SD attenuates plasma CORT and elicits approach-avoid conflict 3 DPI
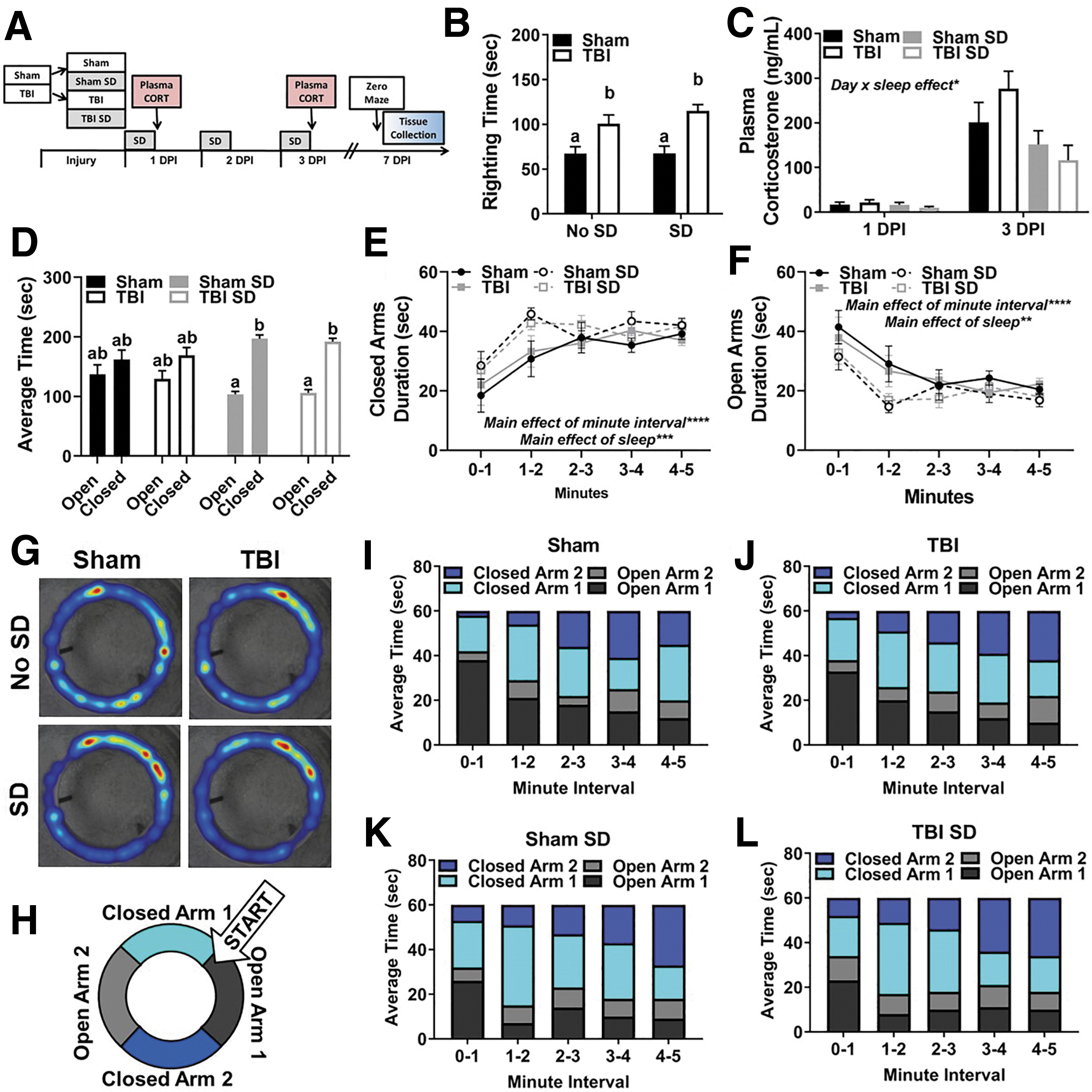
To examine the immediate effects of post-injury SD, C57BL/6 mice aged 8–10 weeks received lateral fluid percussion TBI or sham injury. Afterward, half of the mice in each group remained undisturbed and the other half received 3 days of SD from 7–11AM (Fig. 1A). Blood and brain tissue collection occurred 3 DPI immediately after one test trial in the elevated zero maze. As expected, TBI induced a longer righting time to regain consciousness than sham injury, F(1, 68) = 62.36, p < 0.001. Notably, at the time of injury SD had not been performed. Thus, Figure 1B depicts a posteriori analysis of righting times between no SD and SD groups to ensure that righting time (as an indicator of injury severity) did not confound subsequent data analysis and interpretation. Indeed, TBI mice that were subsequently exposed to SD (M = 164.11, SEM = 15.17) displayed a similar righting time to TBI mice that remained undisturbed (M = 161.50, SEM = 18.03). Daily body weights were collected throughout the experiment beginning on surgery day, and 1–3 DPI. Sham and TBI mice in control housing lost weight 1 DPI compared with sham and TBI mice exposed to SD resulting in an interaction effect of day and injury group, F(3, 350) = 30.65, p < 0.001.; however, these differences resolved between 2 and 3 DPI and likely occurred because undisturbed mice slept sooner after injury. 40
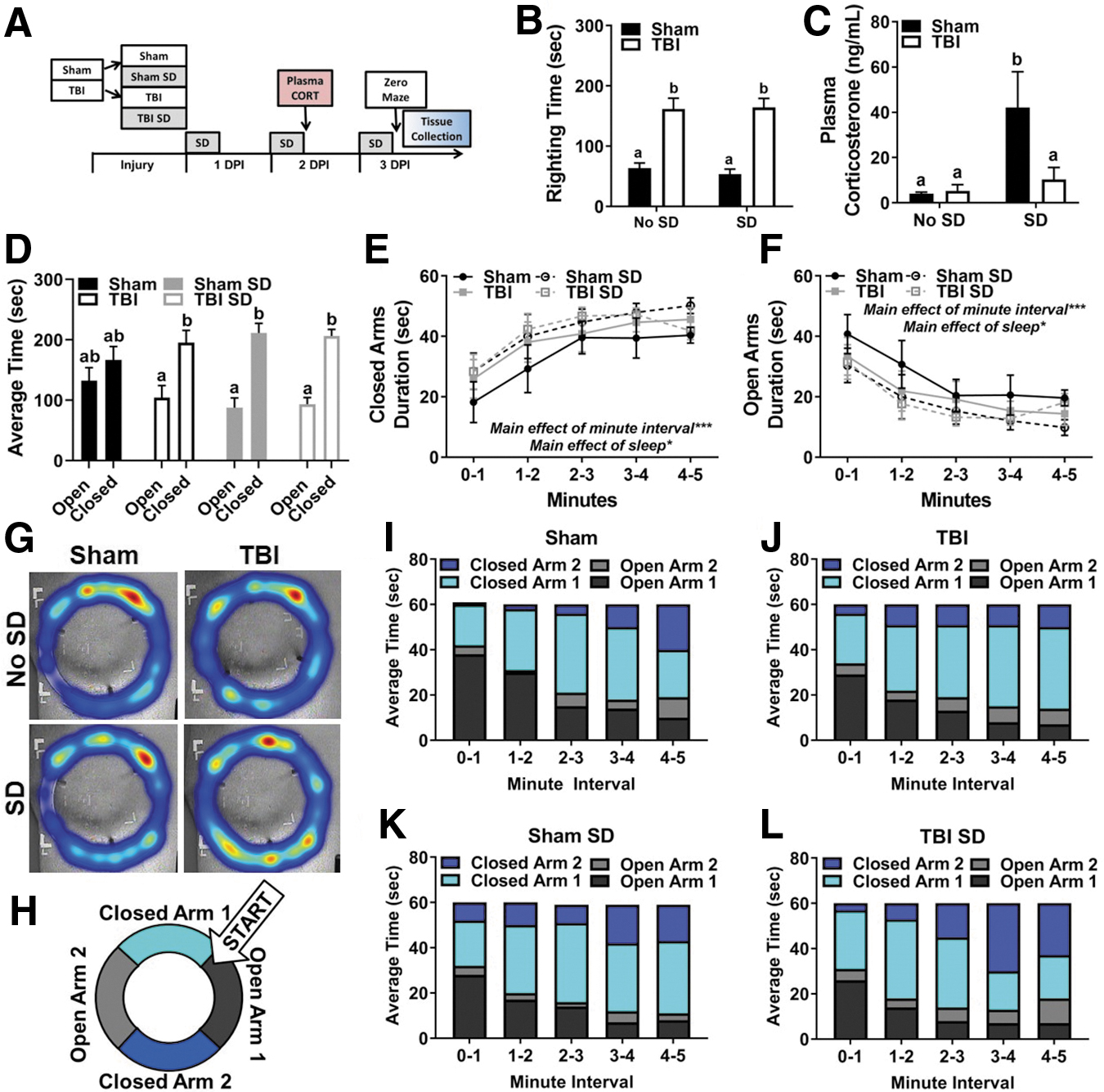
Post-injury SD attenuates plasma CORT and elicits approach-avoid conflict 3 DPI.
To evaluate the peripheral stress response to post-injury SD, blood was collected via submandibular bleed at 11AM 2 DPI. Blood was also collected from undisturbed sham and TBI animals for comparison. This time-point was selected to avoid the potential influence of stress-induced behavioral changes following blood collection at 3 DPI. Notably, sham surgical effects could influence peripheral CORT at acute time-points. Thus, all animals were given the same surgical preparation and anesthetic treatment to account for surgical effects in addition to injury effects. Also, no anesthetic was used prior to submandibular bleeding. Nonetheless, TBI mice displayed a blunted blood plasma CORT response to SD at 11AM 2 DPI compared with sham mice exposed to SD, injury effect F(1, 26) = 7.152, p < 0.05; interaction effect F(1, 26) = 4.198, p = 0.05 (Fig. 1C). These results are in line with previous studies demonstrating impaired glucocorticoid negative feedback in TBI animals. 10,41
At approximately 12PM 3 DPI, mice completed one testing trial in the elevated zero maze to evaluate anxiety-like behavior. Blood and brain tissue were collected upon trial completion. No significant difference in total distance traveled was detected between experimental groups, indicating that motor deficits did not confound movement throughout the maze. A mixed model ANOVA revealed a main effect of arm (i.e., open vs. closed) F(1, 20) = 27.45, p < 0.05, an arm × sleep interaction effect, F(1, 20) = 7.43, p < 0.05, as well as an arm × sleep × injury effect, F(1, 16) = 8.25, p < 0.05.
Post hoc comparisons confirmed that TBI, sham SD, and TBI SD mice preferred spending time in the closed arms compared with the open arms in the elevated zero maze. Sham mice spent a similar amount of time exploring open and closed arms (Fig. 1D). To examine arm preference over time, a mixed-model ANOVA was performed. This analysis revealed a main effect of minute interval, F(4, 190) = 10.18, p < 0.05 and sleep, F(4, 190) = 5.81, p < 0.05, in duration of time spent in closed arms (Fig. 1E). Similar effects were identified in duration of time spent in open arms, main effect of minute interval, F(4, 190) = 9.60, p < 0.05 and sleep, F(4, 190) = 5.81, p < 0.05 (Fig. 1F). Representative heat maps from each experimental group provide a summary of where mice spent their time during the testing trial and further suggest that there may be group-dependent preferences in where the mice explore (Fig. 1G). Next, the average time spent in each arm of the elevated zero maze was calculated, resulting in four zones of interest: open arm 1, open arm 2, closed arm 1, and closed arm 2 (Fig. 1H).
Time binned contingency tables visualize where mice spent their time throughout the 5-min test trial (Fig. 1I–L). Throughout the test trial, all mice moved from open to closed arms; however, TBI SD mice spent the most time in closed arm 2 compared with all other groups as the testing trial progressed from 3 to 5 min. Although this response cannot be described as typical disinhibitory behavior (i.e., spending more time in open arms), the data suggest that TBI SD mice explore the entire maze but maintain reservation to stay within the closed arms, indicating conflict between approach-avoid exploration. 42
Post-injury SD facilitates brain infiltration of neutrophils 3 DPI
Flow cytometry was used to assess the peripheral effect of post-injury SD in the blood 3 DPI. Immediately after behavioral testing, mice were anesthetized and blood was collected via cardiac puncture. Figure 2A shows representative bivariate dot plots of Ly6C and Ly6G labeling in the blood. TBI significantly increased the number of circulating Ly6Chigh monocytes, (main effect of injury) F(1, 10) = 7.95, p < 0.05, and neutrophils, (main effect of injury) F(1, 10) = 15.73, p < 0.05 in circulation (Fig. 2B,C). No statistically significant differences in the number of circulating Ly6Clow monocytes or lymphocytes were detected. Moreover, SD did not enhance the number of circulating leukocytes in sham or TBI mice compared with controls.
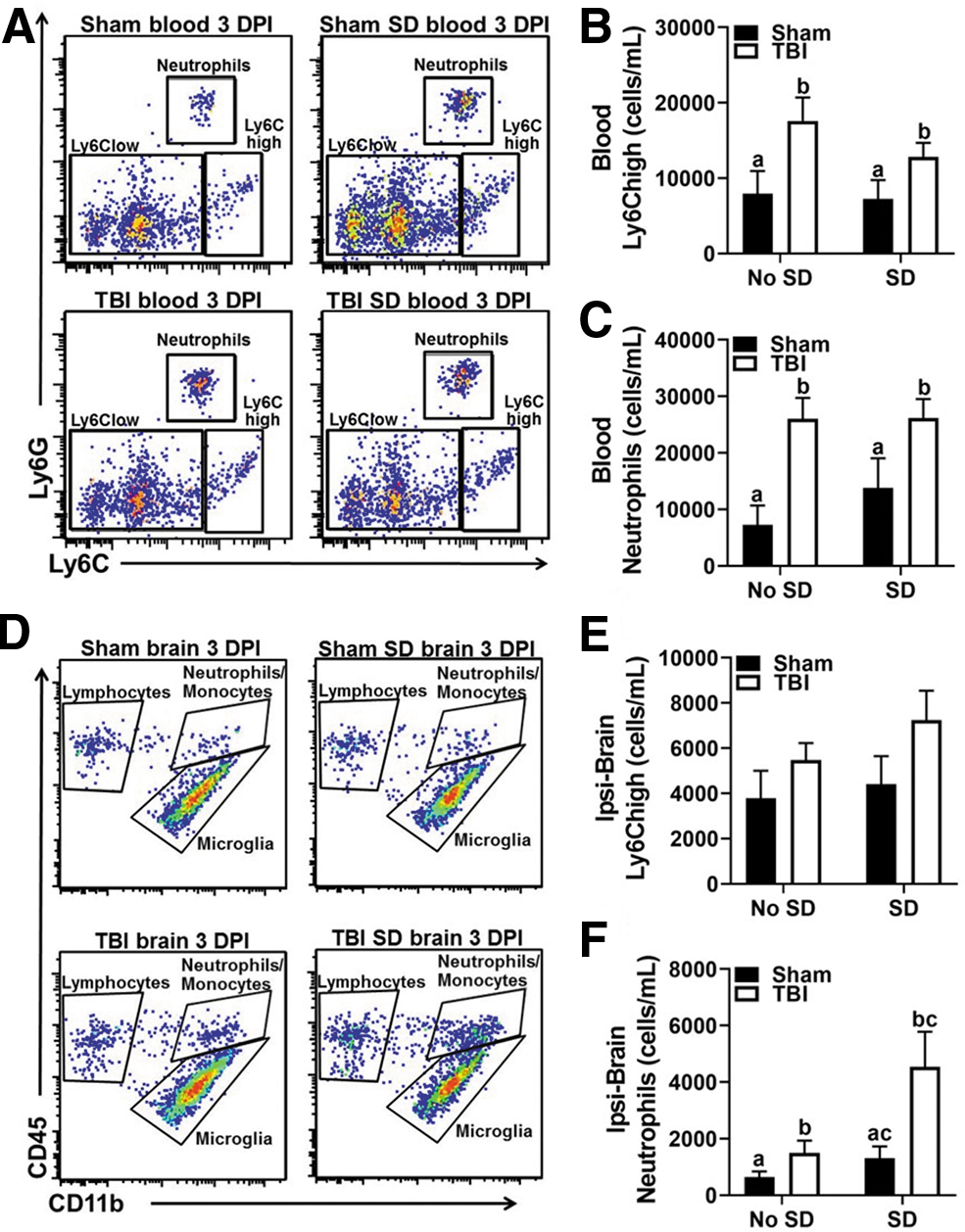
Post-injury SD facilitates brain infiltration of neutrophils 3 DPI.
Next, the numbers of brain-infiltrating leukocytes, as well as brain microglia, were determined using flow cytometry. The brain was bisected into ipsilateral and contralateral hemispheres to better identify injury-induced effects. Figure 2D shows representative bivariate dot plots of CD45 and CD11b labeling in the ipsilateral brain 3 DPI. TBI mice displayed increased numbers of Ly6Chigh monocytes in the ipsilateral hemisphere; however, a main effect of injury was not statistically significant (p = 0.08; Fig. 2E). Strikingly, the number of brain neutrophils was highest in TBI SD mice compared with all other groups in the ipsilateral hemisphere; main effect of injury, F(1, 10) = 6.45, p < 0.05; main effect of sleep, F(1, 10) = 5.29, p < 0.05 (Fig. 2F). No significant differences in the number of microglia or infiltrating Ly6Clow monocytes and lymphocytes were detected between groups. Together, these results suggest that post-injury SD facilitates infiltration of neutrophils into the ipsilateral hemisphere of the brain.
Post-injury SD exacerbates cortical gliosis 3 DPI
Although the number of brain resident microglia did not differ between experimental groups, we predicted that TBI would induce a distinct spatial distribution of reactive microglia/macrophages. Separate cohorts of mice were undisturbed or exposed to 3 days of SD from 7 to 11AM. At approximately 12PM 3 DPI, brain tissue was collected and cryoprotected for subsequent immunohistochemical analysis. Coronal brain sections were labeled with Iba1 antibody and percent-area was quantified in the primary somatosensory cortex and retrosplenial cortex of the ipsilateral and contralateral hemispheres, which corresponds to lateral and medial to the injury site, respectively. Figures 3A and 3C display representative images of Iba1 labeling in the lateral and medial ipsilateral cortex of sham and TBI mice with and without SD. Insets are included to better visualize the morphological changes in Iba1+ cells. As expected, Iba1+ cells in TBI mice appear de-ramified with swollen cell bodies and short, thickened processes. Two-way ANOVA revealed a main effect of sleep, F(1, 16) = 16.63, p < 0.05, and injury, F(1, 16) = 72.61, p < 0.05, in percent-area covered by Iba1 labeling in the lateral cortex.
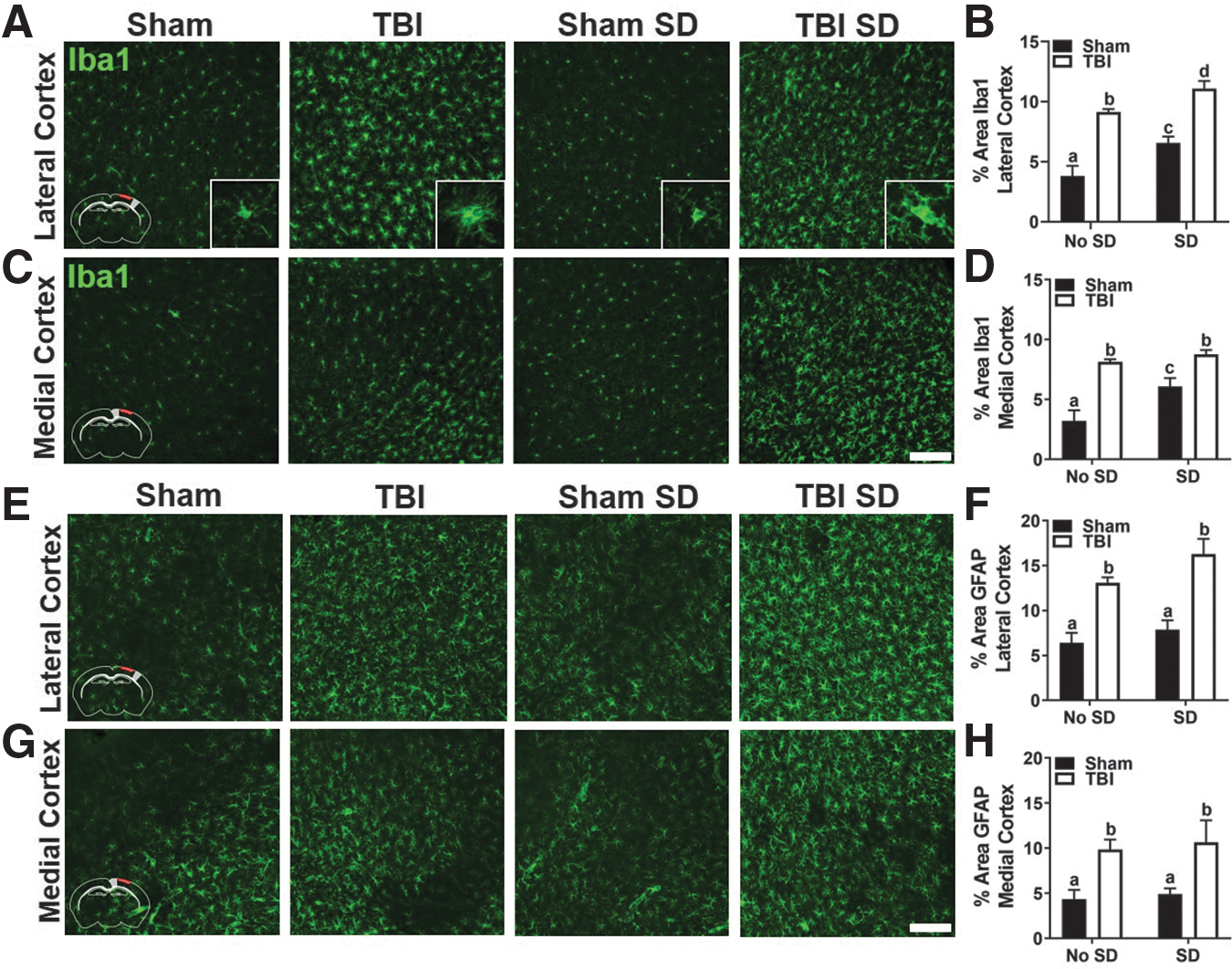
Post-injury SD exacerbates cortical gliosis 3 DPI.
Post hoc comparisons showed a significant difference between all groups (Fig. 3B), suggesting that SD exacerbates the microglial/macrophage response to TBI. An interaction effect was identified in the medial cortex, F(1, 16) = 4.58, p < 0.05, indicating that the effect of SD was unique in sham and TBI mice. Nonetheless, post hoc comparisons showed no significant difference between TBI and TBI SD mice in percent-area covered by Iba1 labeling in the medial ipsilateral cortex (Fig. 3D). Interestingly, there was an interaction effect of Iba1 labeling in the medial cortex of the contralateral hemisphere, F(1, 16) = 5.16, p < 0.05 (data not shown), suggesting region-specific response to post-injury SD. Next, coronal brain sections were labeled with GFAP antibody to define the astrocytic response to injury and SD. Figures 3E and 3G show representative images of GFAP labeling in the lateral and medial ipsilateral cortex of sham and TBI mice with and without SD. Two-way ANOVA revealed a main effect of injury in both the lateral cortex, F(1, 19) = 39.71, p < 0.05 (Fig. 3F), and medial cortex, F(1, 19) = 13.67, p < 0.05 (Fig. 3H) of the ipsilateral hemisphere. No significant differences in percent-area covered by GFAP labeling were found in the contralateral hemisphere (data not shown). Together, these data highlight the vulnerability of the ipsilateral primary somatosensory cortex to glial reactivity following TBI and indicate that transient post-injury SD enhances the microglial/macrophage response to TBI.
Post-injury SD disrupts AQP4 polarization and elevates mRNA expression of inflammatory mediators in the cortex 3 DPI
To further define the astrocytic response to post-injury SD, we examined the proportion of perivascular AQP4 over parenchymal AQP4, defined as AQP4 polarization. At baseline, AQP4 is primarily expressed in the perivascular end feet of astrocytes and maintains a critical role in water transport. Deletion of the Aqp4 gene in mice dramatically decreases perivascular exchange of cerebral spinal fluid and interstitial fluid, making it a key player in the glymphatic system. 43,44 Following ischemic, inflammatory, or traumatic injury, AQP4 polarization decreases as AQP4 shifts from the perivascular astrocytic end feet to the parenchymal astrocytic soma. 45 This dysfunction in polarization may be critical in mediating outcome and further compromised by post-injury SD. 46 It is important to note that previous studies have shown that an anesthetic such as isoflurane may influence glymphatic clearance through altering AQP4. 47
To account for this, all animals received the same anesthetic dosage to minimize confounding influences as previously described. 48 Coronal brain sections were co-labeled with GFAP and AQP4 antibodies and AQP4 polarization was quantified in the primary somatosensory cortex of the ipsilateral and contralateral hemispheres. Figure 4A displays representative images of AQP4 in the ipsilateral cortex of sham, TBI, sham SD, and TBI SD mice. An inset is provided to visualize co-labeled GFAP. Two-way ANOVA revealed a significant interaction effect in AQP4 polarization in the ipsilateral cortex 3 DPI, F(1, 20) = 7.95, p < 0.05 (Fig. 4B). Notably, AQP4 polarization was uniquely altered in sham and TBI mice following post-injury SD. For example, the AQP4 labeling in TBI is diffuse and extends away from the vasculature with a hazy appearance (Fig. 4A). A similar pattern, although not as distinct, can be seen in the sham SD representative image. AQP4 labeling in sham and TBI SD mice is localized to the vasculature and does not extend into the parenchyma (Fig. 4A). No significant differences in AQP4 polarization were detected in the contralateral cortex (data not shown).
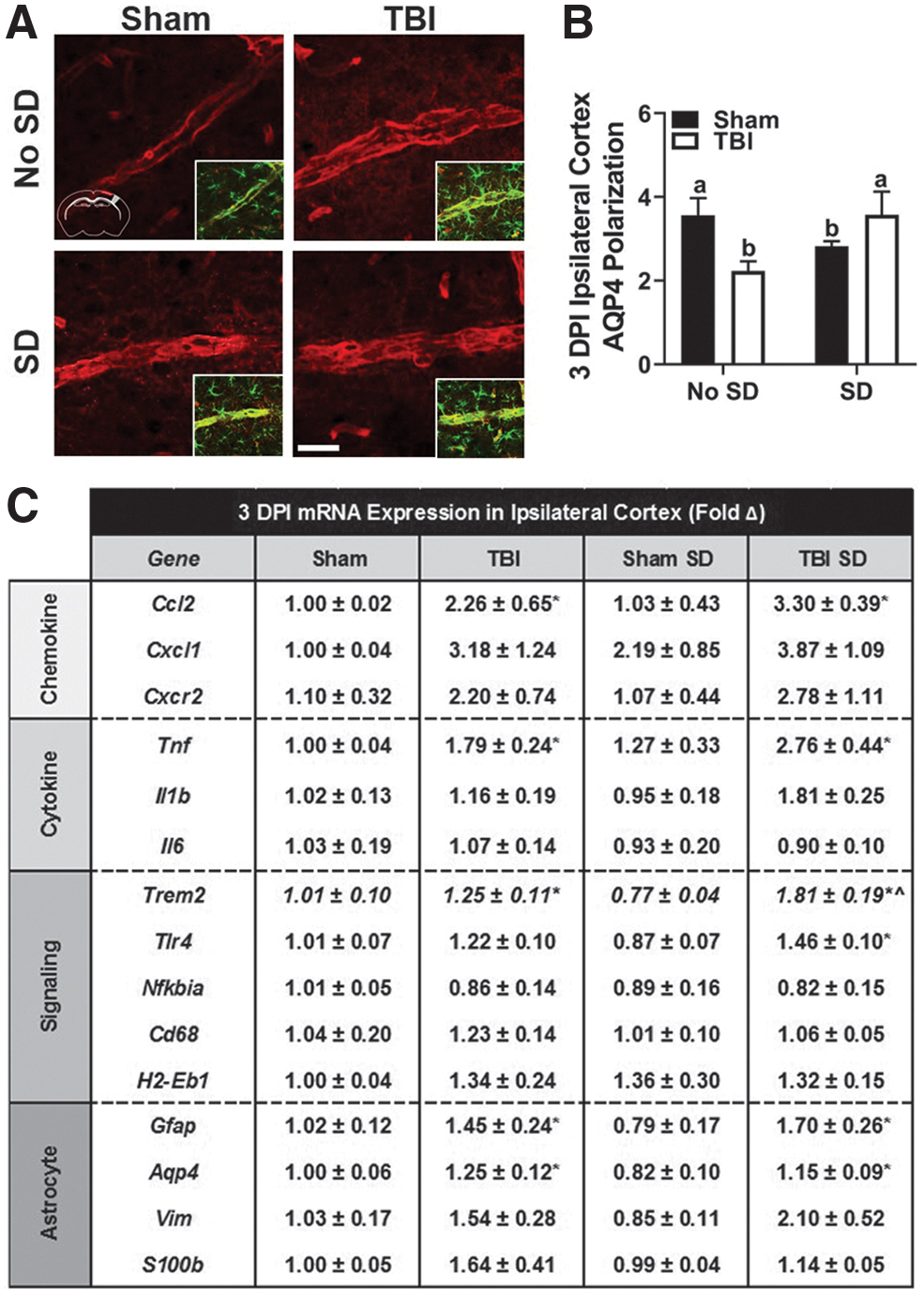
Post-injury SD disrupts AQP4 polarization and elevates mRNA expression of inflammatory mediators in the cortex 3DPI.
Next, the mRNA expression of genes associated with chemokines (Ccl2, Cxcl1, Cxcr2), cytokines (Tnf, Il1b, Il6), cell signaling (Trem2, Tlr4, Nfkbia, Cd68, H2-Eb1), and astrocytes (Gfap, Aqp4, Vim, S100b) was determined (Fig. 4C). Cortical Tnf expression significantly increased with SD, t(14) = 2.83, p < 0.05, and is consistent with previous literature implicating TNFα as a regulator of sleep/wake behavior. 49 TBI SD mice displayed increased expression of many genes compared with all other groups (i.e., Ccl2, Cxcl1, Cxcr2, Tnf, Il1b, Trem2, Tlr4, Gfap, Vim); a main effect of injury was identified via two-way ANOVA in only a subset of genes (i.e., Ccl2, Tnf, Trem2, Gfap, Aqp4, p < 0.05). An interaction effect was detected in expression of Trem2, F(1, 13) = 5.66, p < 0.05. Post hoc comparisons confirmed that Trem2 expression was higher in TBI mice compared with sham mice; however, this interaction effect was likely driven by the reduction in Trem2 expression in sham SD mice. Taken together, these data confirm that TBI induces a widespread inflammatory response in the injured cortex. Although this conserved response is selectively enhanced in genes regulating microglial (or macrophage) reactivity, astrocytes are also affected. Consequently, the glymphatic system may be altered.
Three days of post-injury SD attenuated plasma CORT and increased anxiety-like behavior independent of TBI 7 DPI
To examine the delayed effects of post-injury SD, C57BL/6 mice aged 8–10 weeks received lateral fluid percussion TBI or sham injury. Afterward, half of the mice in each group remained undisturbed and the other half received 3 days of SD from 7–11AM. All animals then remained undisturbed for 4 days. Brain tissue collection occurred 7 DPI immediately after one test trial in the elevated zero maze. Also, because behavioral testing was delayed to 7 DPI, blood collection for plasma CORT measurement occurred at 11AM 1 and 3 DPI. These time-points were selected to complement the 2 DPI data from the first set of experiments and also to avoid repeated, daily blood collection in TBI and sham mice (Fig. 5A). A posteriori analysis of righting time confirmed that there were no differences with SD exposure, and as expected TBI induced a longer righting time than sham injury, main effect of injury, F(1, 41) = 9.31, p < 0.05 (Fig. 5B). No significant differences in body weight were detected between groups through 7 DPI, which is in contrast to what we saw in the first set of experiments. Generally though, mice exposed to SD lost less weight than undisturbed mice. A mixed model ANOVA with repeated measures was used to evaluate group differences in plasma CORT at 11AM on 1 and 3 DPI. A significant main effect of DPI, F(1, 14) = 33.0, p < 0.05, and SD, F(1, 14) = 9.76, p < 0.05 was found. In addition, a DPI × SD interaction effect was found, F(1, 14) = 7.42, p < 0.05.
Three days of post-injury SD attenuated plasma CORT and increased anxiety-like behavior independent of TBI 7 DPI.
In summary, Holm-Sidak's multiple comparisons revealed no between-group differences at 1 DPI or 3 DPI. TBI, sham, and sham SD mice displayed a statistically significant increase in plasma CORT from 1 DPI to 3 DPI; however, plasma CORT remained unchanged in TBI SD mice from 1 DPI to 3 DPI. TBI SD mice displayed the lowest amount of plasma CORT at both 1 and 3 DPI compared with all other experimental groups (Fig. 5C). This is consistent with what was observed at 2 DPI (Fig. 1D) and suggests that glucocorticoid negative feedback is impaired in TBI SD mice. Importantly though, these data also suggest that there are between-day differences in response to post-injury stress as well as repeated cheek bleeds. For example, the sham SD mice do not display the same elevated plasma CORT at 1 or 3 DPI compared with all other groups that was observed at 2 DPI. In addition, undisturbed TBI and sham mice show a striking increase in plasma CORT at 3DPI. Together, these results suggest that repeated environmental stressors such as sleep disruption and even handling can elicit a CORT response that changes from day to day.
At approximately 12PM 7 DPI, all mice completed one testing trial in the elevated zero maze to evaluate anxiety-like behavior. Brain tissue was collected upon trial completion. No significant difference in total distance traveled was detected between experimental groups indicating that injury- or SD-induced motor deficits did not confound movement throughout the maze. A mixed model ANOVA revealed a main effect of arm (i.e., open vs. closed), F(1, 82) = 66.52, p < 0.05, and an arm × sleep interaction effect, F(1, 82) = 15.10, p < 0.05. Post hoc comparisons confirmed that mice exposed to SD preferred spending time in the closed arms compared with the open arms (Fig. 5D).
To examine arm preference over time, a mixed-model ANOVA was performed. This analysis revealed a main effect of minute interval, F(4, 55) = 13.15, p < 0.05 and sleep, F(1, 55) = 12.49, p < 0.05, in duration of time spent in closed arms (Fig. 5E). Similar effects were identified in duration of time spent in open arms, main effect of minute interval, F(4, 55) = 13.67, p < 0.05 and sleep, F(1, 55) = 12.0, p < 0.05 (Fig. 5F). Representative heat maps from each experimental group provide a summary of where mice spent their time during the testing trial and further suggest that there may be group-dependent preferences in where the mice explore (Fig. 5G). Next, the average time spent in each arm of the elevated zero maze was calculated resulting in four zones of interest within the maze: open arm 1, open arm 2, closed arm 1, and closed arm 2 (Fig. 5H). Time-binned contingency tables visualize where mice spent their time throughout the 5-min test trial (Fig. 5I–L). Both sham SD and TBI SD mice spent more time in closed arm 1 compared with undisturbed mice during the 1- to 2-min interval of maze. This initial anxiety-like behavior improved over the next 2- to 3-min interval though. The distribution of average time in each arm was similar between experimental groups during minutes 3 to 5. Together, these data show that there are persistent but subtle behavioral changes associated with 3 days of post-injury SD.
Three days of post-injury SD did not affect lesion volume but increased the number of CD45+ cells in the penumbra of TBI mice 7 DPI
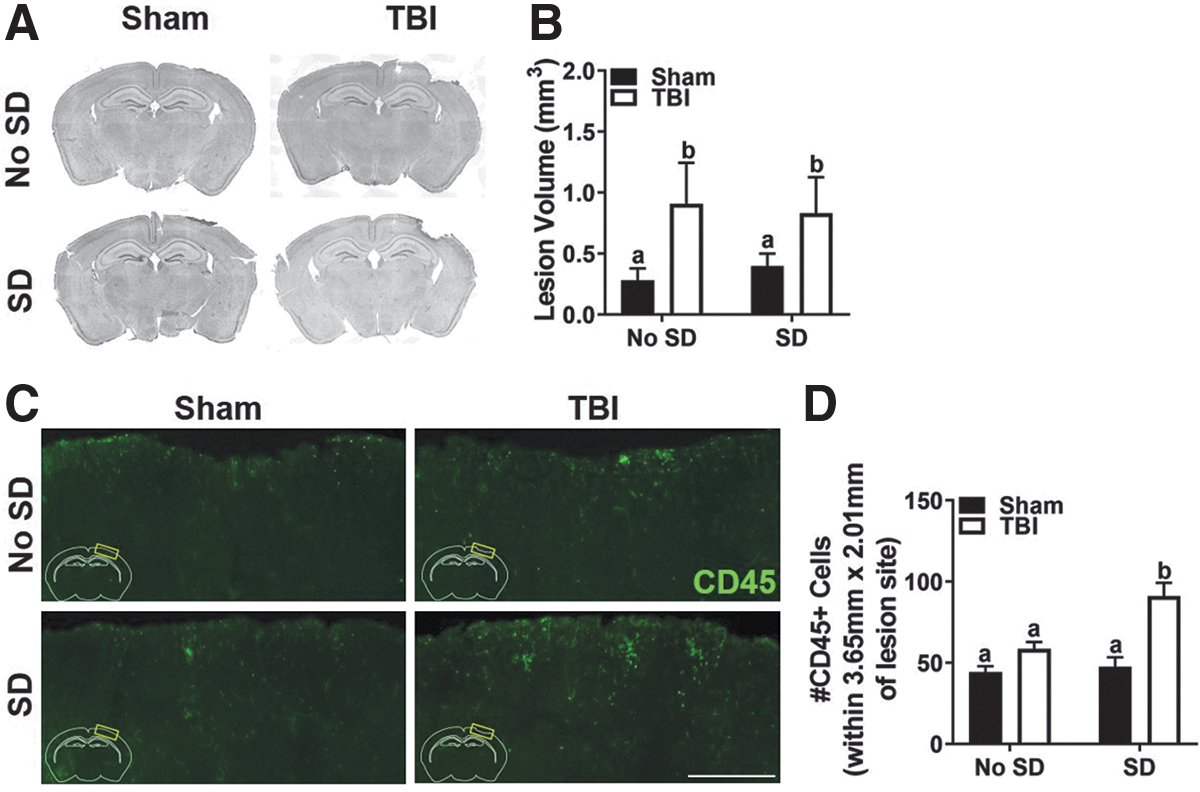
A quantitative lesion analysis was performed to determine if post-injury SD influenced progressive tissue loss following TBI. Representative photomicrographs of coronal brain sections at bregma −1.70 mm reveal a small cortical lesion associated with tissue loss in TBI mice 7 DPI (Fig. 6A). Coronal brain sections were selected from predetermined spatial coordinates spanning the length of the craniectomy to quantify the lesion volume in sham and TBI mice. As expected, TBI resulted in a larger lesion area than sham injury, which peaked between bregma −1.34 and −1.70 mm. Two-way ANOVA confirmed that average lesion volume was highest in TBI mice with no effect of SD, F(1, 26) = 4.92, p < 0.05 (Fig. 6B).
Three days of post-injury SD did not affect lesion volume but increased the number of CD45+ cells in the penumbra of TBI mice 7 DPI.
Flow cytometric data from 3 DPI indicated that post-injury SD increased the number of brain Ly6Chigh monocytes and neutrophils in TBI mice. To identify the spatial presence of infiltrating cells and persistence of the infiltrating cells, brain tissue was labeled with CD45 antibody. As expected, CD45+ cells were identified in close spatial proximity to the site of injury in both sham and TBI mice (Fig. 6C). Two-way ANOVA confirmed a main effect of injury, F(1, 24) = 27.54, p < 0.05, sleep, F(1, 24) = 10.55, p < 0.05, as well as an interaction effect, F(1, 24) = 7.00, p < 0.05. Post hoc analysis showed that TBI SD mice had significantly more CD45+ cells than any other group near the site of injury (Fig. 6D, p < 0.05). This indicates that the exacerbated population of peripheral cells in the TBI SD mice persists near the lesion, even once SD has ceased. Flow cytometry was used to assess the peripheral effect of post-injury SD in the blood 7 DPI. No statistically significant differences in the number of circulating Ly6Chigh monocytes or neutrophils were detected though (data not shown). These data suggest that post-TBI SD does not influence tissue loss at the lesion area; however, it does facilitate persistent inflammation through peripheral immune cells even after SD has ceased.
TBI-induced gliosis persists 7 DPI and 3 days of SD enhanced GFAP labeling in bilateral primary somatosensory cortex
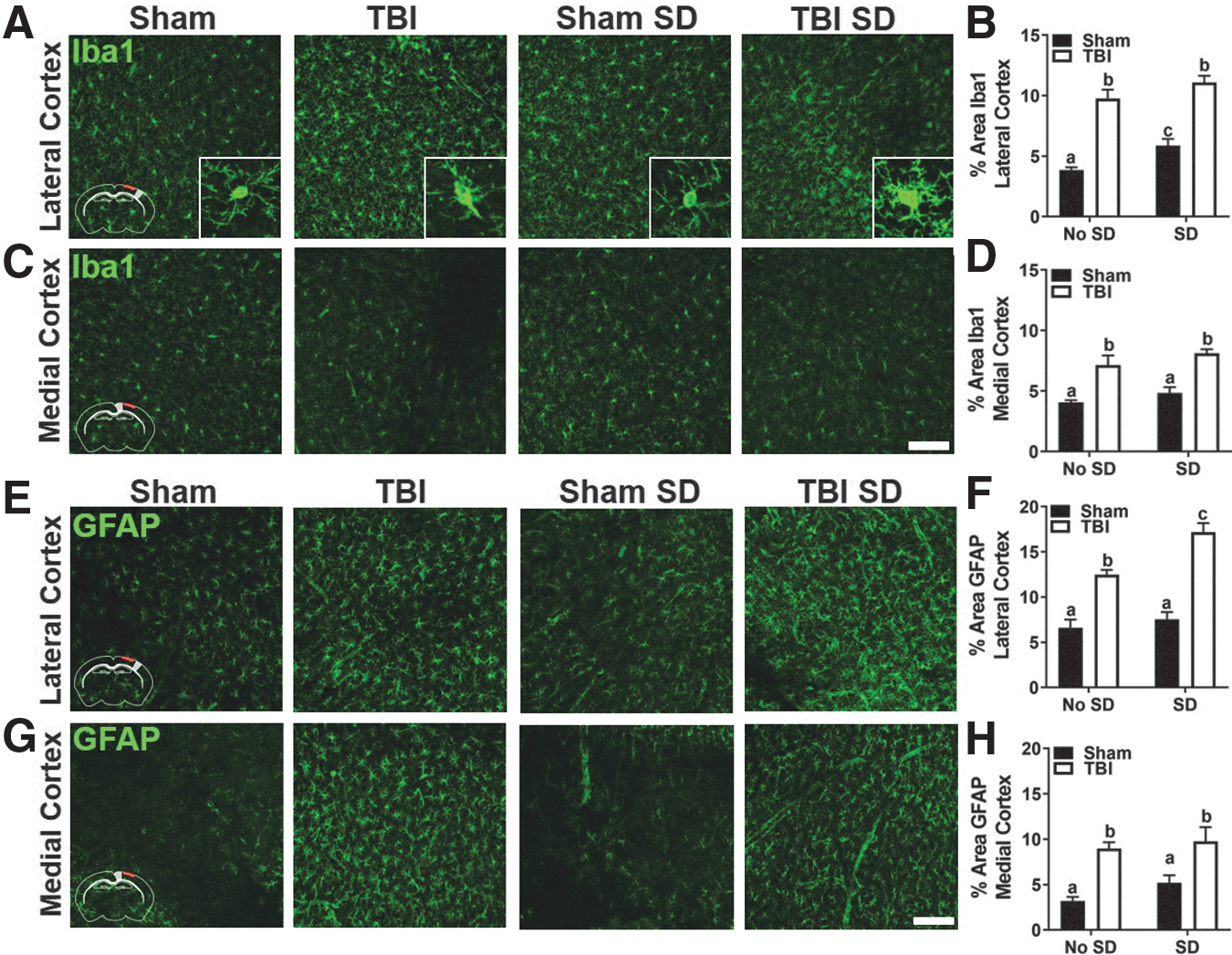
Microglial responses were determined by Iba1 labeling as previously described. Figures 7A and 7C display representative images of Iba1 labeling in the lateral and medial ipsilateral cortex of sham and TBI mice with and without SD. Insets show the morphological changes in Iba1+ cells. Iba1+ cells remain deramified in TBI mice compared with shams at 7 DPI. Two-way ANOVA revealed a main effect of sleep, F(1, 28) = 9.22, p < 0.05, and injury, F(1, 28) = 101.7, p < 0.05, in percent-area covered by Iba1 labeling in the lateral cortex. Post hoc comparisons showed a significant difference between all but the TBI groups (Fig. 7B). A main effect of injury was found in percent-area labeled by Iba1 in the medial cortex, F(1, 28) = 38.0, p < 0.05, (Fig. 7D). Together, these data show that cortical microglial/macrophage reactivity is persistent after TBI. Also, these data hint at a possible microglia/macrophage response to transient SD alone. No significant differences in Iba1 labeling were found in the contralateral hemisphere (data not shown).
TBI-induced gliosis persists 7 DPI; 3 days of SD enhanced GFAP labeling in bilateral primary somatosensory cortex.
Next, coronal brain sections were labeled with GFAP antibody to define the astrocytic response to injury and SD as previously described. Figures 7E and 7G display representative images of GFAP labeling in the lateral and medial ipsilateral cortex of sham and TBI mice with and without SD. Two-way ANOVA revealed a main effect of sleep, F(1, 28) = 11.59, p < 0.05, injury, F(1, 28) = 86.48, p < 0.05, as well as an interaction effect, F(1, 28) = 5.18, p < 0.05, in GFAP labeling in the lateral cortex. Post hoc comparisons confirmed that all but the sham groups were significantly different from one another (Fig. 7F). A main effect of injury, F(1, 28) = 28.18, p < 0.05 was observed in GFAP labeling in the medial cortex. The GFAP response to injury was also apparent in the lateral and medial contralateral cortex, but failed to reach statistical significance (Fig. 7H). These data further suggest that the primary somatosensory cortex is particularly vulnerable to persistent glial reactivity following post-injury SD in TBI mice. Moreover, these data indicate that astrogliosis follows microgliosis and is exaggerated in the injured and uninjured hemisphere of TBI mice. Thus, the delayed neuroinflammatory effects are not limited to areas of direct cortical impact following TBI.
AQP4 polarization normalizes but cortical inflammation persists in conjunction with elevated tau phosphorylation in TBI mice 7 DPI
Persistent astrogliosis following post-injury SD may be associated with chronic glymphatic disruption. To evaluate lingering alterations in AQP4 polarization following post-injury SD, coronal brain sections were co-labeled with GFAP and AQP4 antibodies. AQP4 polarization was quantified in the primary somatosensory cortex in the ipsilateral and contralateral cortices as previously described. No significant differences in AQP4 polarization were detected in the ipsilateral or contralateral cortex (data not shown). These data suggest that dysfunction in AQP4 polarization following TBI or SD is transient and normalizes over time.
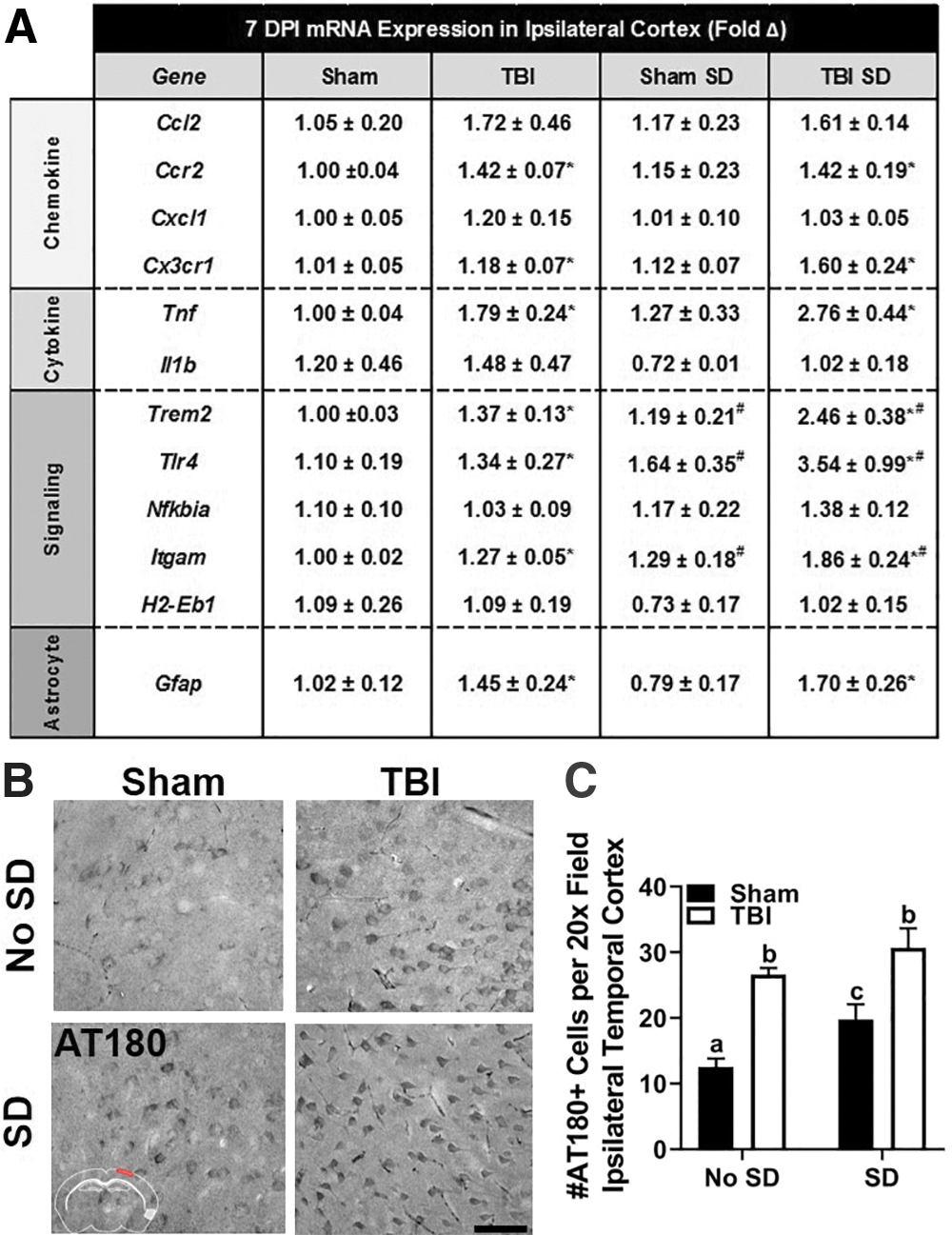
The mRNA expression of a variety of genes associated with chemokines, cell signaling, and astrocytes was determined in the ipsilateral cortex (Fig. 8A). Similar to 3 DPI, TBI SD mice displayed increased expression of many genes compared with all other groups (e.g., Trem2, Tlr4, Cx3cr1, Nfkbia, Itgam, Gfap) at 7 DPI; however, a main effect of injury was identified by two-way ANOVA in only a subset of genes (i.e., Trem2, Tlr4, Cx3cr1, Itgam, Gfap; p < 0.05). A main effect of sleep was identified in mRNA expression of Trem2, Tlr4, Itgam, and Gfap (p < 0.05). No interaction effects were present. Notably, in the central nervous system (CNS), TREM2 is enriched in microglia. Microglia are the primary innate immune cells in the brain equipped with pattern recognition receptors, including all toll-like receptor subtypes, to survey and protect parenchymal cells from injury. 50 Moreover, accumulating evidence shows that TREM2-DAP12 signaling attenuates TLR4-mediated production of inflammatory cytokines, including TNFα. 51 Taken together, changes in mRNA expression of these key microglial receptors suggest that the TREM2-DAP12-TLR4 pathway may be particularly sensitive to post-injury SD and alter microglial responsiveness to TBI. This in turn may influence the astrocytic responses once SD has ended. These results further demonstrate a persistent change in the glial response to TBI after post-injury SD; however, the pathological consequence of this altered immune response remains unclear.
Cortical inflammation persists in conjunction with elevated tau phosphorylation in TBI mice 7 DPI.
Abnormal tau phosphorylation is a characteristic feature of many neurodegenerative diseases, including Alzheimer's disease (AD). Given that TBI is a risk factor for AD and that neuroinflammation is associated with tau pathology in both TBI and AD, we sought to determine if post-injury SD facilitated tau phosphorylation in TBI mice. Coronal brain sections were labeled with AT180 antibody to detect tau phosphorylation at Thr231. We selected three brain regions of interest in the ipsilateral hemisphere: the primary somatosensory cortex (i.e., lateral cortex), the hippocampus, and the perirhinal/dorsolateral entorhinal cortex (i.e., temporal cortex). Figure 8B displays representative images of AT180 labeling in temporal cortex of sham and TBI mice with or without SD. No significant differences in the number of AT180+ cells were identified in the lateral cortex or hippocampus; however, a main effect of sleep, F(1, 25) = 8.45, p < 0.05 and injury, F(1, 25) = 41.38, p < 0.05 was detected in the temporal cortex. Although TBI SD mice had the highest number of AT180+ cells, post hoc comparisons showed no differences between the two TBI groups (Fig. 8C). These results are consistent with previously published reports of increased tau phosphorylation in the temporal cortex after lateral FPI 29 but suggest that post-injury SD alone increases tau phosphorylation. Notably, tau phosphorylation itself is not necessarily pathological and thus additional studies are required to define the temporal pathological response to post-injury SD.
Discussion
This study aimed to define neuroendocrine and neuroinflammatory effects of post-injury SD 3 DPI and, after a period of recovery, 7 DPI. To do this, we used mechanical stimulation to compromise the normal pattern of sleep/wake transitions acutely after TBI. SD was an environmental stressor that engaged and challenged the HPA axis, a critical intersection of stress-immune pathways. We then looked at plasma CORT as an indicator of stress, behavioral changes, and common markers of inflammation immediately following 3 days of SD and after a period of recovery from SD. These data provide insight to the immediate and delayed effects of environmental SD, which could enhance the effects of TBI-induced sleep disturbances and compromise post-injury recovery.
We show novel evidence that the plasma CORT response to SD was reduced in TBI mice compared with sham mice 2 DPI. This recapitulates previous data showing that the CORT response to restraint stress and forced swim are blunted in TBI mice compared with shams. 10,41 Importantly, both restraint stress and forced swim induce sleep disturbances and neuroinflammation in a glucocorticoid-dependent manner. 13 Because glucocorticoids are classically anti-inflammatory, we predicted that a blunted CORT response may alter behavioral recovery and exacerbate neuroinflammation. TBI, sham SD, and TBI SD mice preferred exploring closed arms in the zero maze 3 DPI confirming that both TBI and SD influence anxiety-like behavior. Although TBI SD mice preferred closed arms, they explored the entire maze more than any other group and spent the most time in closed arm 2 during the last 2 min of the trial. This behavior is indicative of approach-avoid conflict and suggests TBI SD mice may take longer to discriminate novel stimuli. 42 As a result, exploration is increased while a preference for the closed arm is maintained.
Another notable finding was that SD increased neutrophil infiltration to the ipsilateral hemisphere of TBI mice 3 DPI. Neutrophils migrate to the injured brain (see review by Liu and colleagues 52 ) and can perpetuate inflammation through activation of other immune cells. 53 Indeed, post-injury SD increased percent-area of cortical Iba1 labeling in both sham and TBI mice 3 DPI; however, this response was exaggerated in the lateral cortex of TBI SD mice. In addition, post-injury SD altered microglial reactivity in the medial cortex of the contralateral hemisphere, indicating global changes in neuroinflammation. Astrogliosis, indicated by GFAP percent-area, was enhanced by TBI lateral to the site of injury 3 DPI, but was unaffected by SD. These data suggest that Iba1+ cells are uniquely vulnerable to the effects of SD, potentially through interaction with infiltrating granulocytes. This effect is exacerbated after TBI in a brain region dependent manner.
Reactive astrocytes have dysregulated expression of AQP4, a water channel important in BBB maintenance 54 and glymphatic function. 48 Dysfunction in either or both of these systems may increase presence of immune cells in the brain. As expected, TBI reduced AQP4 polarization in the ipsilateral cortex of TBI mice compared with sham mice. 45 This same effect was not observed after post-injury SD; in fact, AQP4 polarization was decreased in sham SD mice compared with TBI SD mice. This response highlights the independent effects of SD on BBB maintenance and/or glymphatic function and further suggests that TBI alters this response. Further analysis is needed to identify specific effects of post-injury SD on AQP4 polarization.
TBI increased mRNA expression of inflammatory markers Ccl2, Tnf, Trem2, Gfap, and Aqp4 in the ipsilateral cortex regardless of SD. TBI SD mice showed the highest expression of Trem2, which has genetic variants associated with AD. Trem2 was decreased in sham SD mice compared with undisturbed sham mice, indicating a unique post-injury SD effect on Trem2 expression. TREM2 deficiency alters the macrophage response to TBI and improves long-term outcome. 55 However, mouse models of AD have shown that TREM2 deficiency promotes AD pathogenesis. 56 –58 These results showcase the dynamic role TREM2 plays in neuroinflammation. Our data suggest that TREM2 may also has an important role in stress-immune interactions after SD.
We predicted acute post-injury SD would cause lasting behavioral and inflammatory impairment after TBI. Thus, we defined delayed effects of 3 days of post-injury SD at 7 DPI. On average, TBI SD mice had the lowest plasma CORT at 11AM 1 and 3 DPI, which is consistent with what was observed at 2 DPI. Interestingly, both sham and TBI mice had increased plasma CORT 3 DPI compared with sham SD and TBI SD mice. Increased plasma CORT may reflect initiation of the stress response in control mice because they were “disturbed” by handling to collect cheek blood. In contrast, reduced plasma CORT in SD mice may reflect habituation to handling (during the 4 h SD) and/or compromised HPA axis function. Generally, these results suggest that temporal plasma CORT response to cheek bleed and post-injury SD changes daily. Additional studies are needed to define the temporal CORT response as a single daily time-point 4 h after initiation of SD (i.e., 11AM) may not be sufficient to identify the peak CORT response.
At 7 DPI, all mice completed a single trial in the elevated zero maze. Undisturbed mice explored open and closed arms, whereas SD mice maintained a preference for closed arms. Time-binned analysis revealed that SD mice spent more time in closed arm 1 during the first 2 min of testing; however, this preference declined thereafter. Contrary to 3 DPI, all experimental groups explored the entire maze and spent a similar amount of time in closed arm 2. These data suggest that even transient SD interferes with behavioral performance for several days in both sham and TBI mice. We predict that the neuroinflammatory response to SD (i.e., increased Iba1) may be critical in perpetuating behavioral changes in sham and TBI mice.
Post-injury SD did not result in lesion area or volume changes in TBI mice. Notably, post-injury SD increased the number of CD45+ cells near the lesion in TBI mice compared with controls, which was not due to increased number of circulating monocytes or neutrophils. This mirrors data from 3 DPI where TBI SD mice showed a significant population of CD45+neutrophils in the ipsilateral hemisphere. It is possible that post-injury SD facilitated peripheral cell entry into the brain. Another possibility is that post-injury SD prevented clearance or removal of infiltrating peripheral cells from the brain, as sleep is shown to increase clearance. 59 Additional flow cytometry experiments on brain tissue are needed to identify specific cell populations. Nonetheless, these data show that peripheral cell infiltration and/or clearance from the injured brain is altered even after post-injury SD ends.
Microglial/macrophage reactivity increased in the ipsilateral cortex of TBI mice compared with shams 7 DPI but was not exaggerated by SD. Importantly, sham SD mice continued to display increased cortical Iba1 percent-area compared with sham mice. This suggests that Iba1+ cells are sensitive to SD and may be key players in behavioral changes. At 7 DPI, TBI SD mice showed the most astrogliosis in the lateral cortex of both hemispheres. Astrocytes are heavily implicated in sleep homeostasis 60 –62 and have distinct gene expression profiles during sleep, wake, and SD. 63 These data suggest that astrocytes have a delayed and exaggerated response to post-injury SD that may contribute to inflammation and long-term consequences of TBI. We hypothesized that astrogliosis contributed to BBB or glymphatic impairment in TBI SD mice resulting in more peripheral cell infiltration to the brain. Surprisingly though, no differences in AQP4 polarization were detected between groups 7 DPI. Further analysis of BBB and glymphatic function may provide insight to the persistence of CD45+ cells in the parenchyma following post-injury SD.
We next determined the delayed effects of post-injury SD on inflammatory gene expression. Several genes were elevated in TBI mice, such as Trem2, Tlr4, Tnf, Ccr2, and Gfap. Interestingly, Tnf was highest in TBI SD mice. TNFα is a potent neutrophil chemoattractant (see review by Liu and colleagues 52 ), which could explain the number of brain neutrophils at 3 DPI as well as the CD45+ cells in TBI SD mice at 7 DPI. Expression of Itgam, Tlr4, and Trem2 was exacerbated in both sham SD and TBI SD mice. Given that TREM2 negatively regulates TNFα and TLR4 gene transcription, 64 an increase in both Trem2 and Tlr4 indicates a disruption of this pathway that may influence gliosis. 65,66 Further analysis is needed to confirm the specific molecular mechanisms mediating outcome.
Next, we examined tau phosphorylation, a hallmark of neurodegenerative tauopathies associated with neuroinflammation. 67,68 Tau phosphorylation was detected near the lesion and hippocampus; however, a main effect of injury was only identified in the ipsilateral temporal cortex. In this same region, tau phosphorylation increased in sham SD mice compared with controls. This suggests that microtubule stabilization is influenced by SD as well as TBI. We hypothesize that altered cortical Iba1 reactivity in sham SD and TBI SD mice promotes tau phosphorylation; however more data are needed to confirm this relationship.
In conclusion, we provide novel data showing that post-injury SD has both immediate and delayed effects on behavior and inflammation. For example, Iba1+ cells appear to be particularly vulnerable to the effects of post-injury SD because both sham and TBI mice display increased cortical reactivity 3 DPI that correlates with persistent behavioral changes and tau phosphorylation 7 DPI. Surprisingly, only TBI mice displayed a delayed astrocytic response to SD that correlated with transcriptional changes in Trem2 and Tlr4 7 DPI.
One limitation of this study is a lack of physiological or quantitative measurement of sleep. For future studies, one aim is to include a detailed analysis of sleep/wake behavior to best define the effects of post-injury SD. The summation of injury-induced sleep/wake deficits and environmental SD may result in a unique sleep/wake pattern that influences stress-immune pathways. Together, these results indicate that even transient interruption of the HPA axis via SD compromises post-injury recovery and neuroinflammation. Further understanding of the mechanisms contributing to altered stress-immune interactions after TBI is needed and could help to improve management of chronic recovery.
Footnotes
Acknowledgments
We thank the Center for Brain and Spinal Cord Repair at The Ohio State University for use of the fluid percussion device. We thank Jackie Fontana, Harsha Pulluru, and Andrew Perl for technical assistance.
Authors Contributions
J.E.K. performed SD, behavioral analysis, immunohistochemistry, qPCR analysis, and wrote the manuscript; Z.M.T. performed SD, immunohistochemistry, flow cytometry, and wrote the manuscript; K.G.W. provided training in tissue processing, expertise in data analysis and interpretation, and edited the manuscript; R.R.A. performed SD, behavioral analysis, and immunohistochemistry; J.A.V. performed sham and fluid percussion injury, SD, and qPCR analysis; S.M.O. performed immunohistochemistry and analysis for AQP4 polarization; J.E.D. and C.E.B. performed qPCR and lesion analysis; J.F.S. provided funding, expertise in data analysis and interpretation, and edited the manuscript; J.P.G. provided funding, expertise in data analysis and interpretation, and edited the manuscript; O.K.C. provided funding, designed, performed, analyzed all experiments, and wrote the manuscript.
Funding Information
This work was supported by National Institute of Neurological Disorders and Stroke (NINDS) Grant R01NS109585 (to O.K.C.), NINDS Grant R56NS090311 (to J.P.G.), a College of Medicine Dean's Discovery Grant (to J.P.G.), funding from The Ohio State University Center for Brain and Spinal Cord Repair (to J.P.G.), a Chronic Brain Injury Discovery Grant (to O.K.C. and J.F.S.), National Institute of Dental and Craniofacial Research Training Grant T32DE014320 (to K.G.W. and S.M.O.) and a P30 Core Grant (NINDS, P30NS045758).
Author Disclosure Statement
No competing financial interests exist.