
Abstract
Despite multiple prior pharmacological trials in traumatic brain injury (TBI), the search for an effective, safe, and practical treatment of these patients remains ongoing. Given the ease of delivery and rapid absorption into the systemic circulation, inhalational gases that have neuroprotective properties will be an invaluable resource in the clinical management of TBI patients. In this review, we perform a systematic review of both pre-clinical and clinical reports describing inhalational gas therapy in the setting of TBI. Hyperbaric oxygen, which has been investigated for many years, and some of the newest developments are reviewed. Also, promising new therapies such as hydrogen gas, hydrogen sulfide gas, and nitric oxide are discussed. Moreover, novel therapies such as xenon and argon gases and delivery methods using microbubbles are explored.
Introduction
Traumatic brain injury (TBI) results in death and disability in millions of individuals throughout the world annually. Although various pharmacological agents have been trialed and many of them are still under development, an exciting field of therapeutic intervention is the use of inhalational gases that have various neuroprotective effects. Given the ease of delivery and lack of discomfort in mild TBI patients, as well as direct control of exact concentration of delivered gas in intubated severe TBI patients, inhalational gases that have neuroprotective effects have advantages as a medical intervention. This review will discuss four major inhalational gases shown to have neuroprotective effect in traumatic brain injury: oxygen (O2), hydrogen (H2), hydrogen sulfide (H2S), and nitric oxide (NO). Additionally, new advances in the field of targeted gas delivery will also be discussed. Thus far, the majority of evidence in support of their neuroprotective role is in the pre-clinical setting. Although clinical trials using hyperbaric oxygen are under way, there is a lack of robust clinical data on majority of other gases. Further validation of their therapeutic efficacy in clinical trials is much needed to advance their utility and impact the current management algorithm for patients with TBI.
To perform a comprehensive review of inhalational gases that have been used in either TBI patients or animal models of TBI, a literature search was conducted in September of 2020 using PubMed database. Search terminology was performed with the phrases (‘nitric oxide’ [MeSH Major Topic] or ‘inhalational gas’ [MeSH Major Topic] or ‘hyperbaric oxygen’ [MeSH Major Topic] or ‘hydrogen sulfide’[MeSH Major Topic] or ‘xenon’[MeSH Major Topic] or ‘hydrogen gas’[MeSH Major Topic]) AND (‘traumatic brain injury’[MeSH Major Topic] or ‘TBI’ [Title/Abstract] or ‘traumatic brain injury’ [Title/Abstract]). Appropriate articles under these search terms were included. Additional searches of articles that were referred by these articles were also included. This resulted in approximately 150 unique articles that reported the use of various inhalational gases or related chemical compounds used for treatment of TBI or other associated conditions that were relevant to the discussion in this review.
Normobaric Hyperoxia
In severe TBI, multiple mechanisms may contribute to decreased oxygen supply to the brain tissue such as increased intracranial pressure, systemic hypotension, and acute respiratory distress syndrome. 1 -3 As TBI can result in cerebral ischemia and hypoxia, 4 optimization of arterial oxygen level has been suggested as a potential strategy to mitigate secondary insult. 5
Supplemental oxygen is given readily in the intensive care unit, but the effect of potential hyperoxia on the outcome of TBI patients has always been a question among neurointensivists and neurosurgeons. Prior studies that looked at normobaric hyperoxia showed mixed results but the level of evidence was not very strong, as most were retrospective studies. 6 –9 Thus far, there has not been any randomized prospective study looking at the effect of normobaric hyperoxia.
In a study of a large cohort of severe TBI patients, 6 hyperoxia with arterial partial pressure of oxygen (PaO2) > 300 mm Hg or hypoxia with PaO2 < 60 mm Hg was not associated with any difference in Glasgow Outcome Scale-Extended (GOSE) at 6, 12, and 24 months. However, a more focused approach looking at several PaO2 cutoff values showed that mild levels of hyperoxia were associated with better outcomes. 7 In this study, PaO2 value >150 and >200 mm Hg (but not >250, > 300, or >350 mm Hg) within 24 h of hospital admission were associated with higher GOSE at 6 months, as well as improved neuropsychological outcome on a battery of eight tests. However, other retrospective studies of large cohort of patients showed that higher mortality 8 and lower discharge GCS 9 may result from higher PaO2 levels.
Whether normobaric hyperoxia can result in neuroprotection or not is controversial. 7 In a small, focused study that looked at the effects of increasing fraction of inspired oxygen (FiO2) supplied to the severe TBI patients who are on ventilators (from 35% to 60% then 100% over a period of 6 h in 14 patients), microdialysate lactate levels decreased significantly. 10 Additionally, 3-month clinical outcome was better among patients with better brain tissue oxygenation. Another prospective study on 52 severe TBI patients treated for 24 h with 100% oxygen resulted in lower intracranial pressure (ICP) and lower lactate/pyruvate ratio. 11 Aside from the lack of large-scale, prospective randomized study, the lack of clarity on the effects of normobaric hyperoxia after TBI in these prior randomized studies may also be due to the fact that the optimal level of oxygenation is difficult to determine in severe TBI. 5 As cerebral metabolic rate is reduced in severe TBI, oxygen extraction is reduced. Targeting high levels of arterial oxygen than required by cerebral metabolism may theoretically result in oxygen toxicity due to increased risk of oxidative stress.
Hyperbaric Oxygen
Perhaps the oldest and most rigorously tested inhalational gas for various purposes is hyperbaric oxygen therapy (HBOT). It is defined as inhalation of pure oxygen in a pressurized chamber that is greater than 1 atmospheric pressure. During more than 50 years of clinical use, HBOT has been primarily used for treatment of carbon monoxide poisoning and air embolism. In addition, HBOT has also been used empirically in the treatment of cerebral edema, spinal cord injury, stroke, and other ischemia-reperfusion injuries. 12 -14 The mechanisms of its benefit are thought to be multi-fold: by increasing PaO2, there is a subsequent increase in oxygen supply to the brain tissue. There is also a potential secondary effect of reducing tissue edema, thereby attenuating intracranial pressure increase and reducing secondary ischemia and resulting neuroinflammation. 15
The pre-clinical evidence of benefits of HBOT is extensive, as summarized in Table 1. In a weight drop model of TBI, 15 –18 fluid percussion injury, 19,20 blast injury, 21 and controlled cortical impact, 22 improvements in cell survival and functional outcome were demonstrated. In other forms of trauma, such as stab injury 23 and dynamic cortical deformation 24 -26 improved cell survival, reduced inflammatory, and apoptotic changes were shown. In line with these findings, early clinical trials of HBOT in severe TBI patients also showed various improvements in oxidative metabolism, cerebral blood flow (CBF), reduced ICP, and functional outcomes as reported in Table 2. 27 -29
TBI, traumatic brain injury; ATA, atmosphere absolute; LOC, loss of consciousness; HIF, hypoxia-inducible factor; N/A, not applicable; ATP, adenosine triphosphate; BBB, blood–brain barrier; MRS, magnetic resonance spectroscopy; TNF, tumor necrosis factor; IL, interleukin.
TBI, traumatic brain injury; N/A, not applicable; ATA, atmosphere absolute; PCS, post-concussion syndrome; SPECT, single photon emission computed tomography; GOS, Glasgow Outcome Score; ICP, intracranial pressure; CSF, cerebrospinal fluid; CMRO2, cerebral metabolic rate of O2; DSC, dynamic susceptibility contrast-enhanced magnetic resonance imaging; DTI, diffusion tensor imaging; GCS, Glasgow Coma Score; GOS, Glasgow Outcome Scale; HBOT, hyperbaric oxygen therapy; QOL, quality of life.
The beneficial effect of HBOT may be dependent on the timing of the therapy. As reduced oxygenation in TBI occurs acutely after injury, comparison of initiation times for HBOT demonstrates a therapeutic window of 6 h after TBI, but not at 60 days after TBI in rats. 20 Markers of cell death and injury in the hippocampus and performance on Morris water maze improved only when intervention was performed prior to 6 h. Another detailed study looking at the therapeutic window for HBOT showed that if multiple sessions of HBOT are given, initiation of HBOT even at 48 h after TBI can be therapeutic. 18 However, HBOT at more delayed initiation times may also improve memory and histological measures of injury. 15
In mice subjected to weight drop model of TBI, HBOT administration for 4 days initiated either immediately (3 h following injury) or at delayed times (7 days following injury) resulted in significant improvement in novel object recognition and Y-maze test, as well as attenuated astrogliosis and losses in neurons and myelin. 15 Thus, therapeutic window may be longer than the initial few hours, as cerebral oxygenation study in TBI patients also showed that a large proportion of patients have hypoxic episodes later than 24 h. 30 In this earlier study by Gopinath and colleagues looking at severe TBI patients who had jugular venous oxygen saturation monitored continuously, 36.8% of desaturation episodes were within 24 h, but the rest of the episodes were in subsequent days with 12-16% of the episodes each day during Days 2-5.
Some clinical studies on patients with post-concussion syndrome show benefit up to several years after injury. 31,32 As shown in Table 2, it is possible that enhanced oxygenation even after resolution of acute injury can still improve neurological function. Also, in another study of clinical trial of HBOT assessed by diffusion tensor imaging (DTI) and dynamic susceptibility contrast-enhanced (DSC) MRI, 15 subjects were administered 60 daily sessions. 33 The imaging analysis showed increased fractional anisotropy and decreased mean diffusivity, which signify preserved axonal integrity, as well as increased blood flow and volume. Cognitive testing showed that subjects exposed to HBOT had improvement in major cognitive functions including memory, executive function, and information processing.
Despite these findings, the true benefit of HBOT in post-concussion syndrome has been debated, as two prospective randomized studies showed no benefit. 34,35 As these studies used rigorous control groups with 1.2-1.3 atmosphere absolute (ATA) of room air, which could theoretically increase plasma oxygen by 30%, the difference between control group and experimental group in these studies may also have been difficult to detect. 36 There is no arteriolar oxygen level that has been reported in these studies for confirmation, but future clinical trials could benefit from confirmation of blood oxygen content difference between the groups to ensure an adequate treatment effect.
Also, despite the reports showing therapeutic potential, there are a number of complications of HBOT that should be considered, including middle ear barotrauma, upper respiratory infection, and availability of equipment and specialized staff. 31 In terms of side effects of the 16 subjects with concussion who were treated with HBOT, 31 five (31%) experienced mild reversible middle ear barotrauma. Additionally, four of the subjects (25%) also experienced upper respiratory infection, possibly due to the mild immunosuppression that has been previously reported. 37 Others have reported seizures in TBI patients treated with HBOT at 9%, but this may be due to their severe TBI and higher dose of oxygen at 2.0 ATA for 90 min. 38 Among severe TBI patients who have risk of seizure and hemodynamic instability, treatment inside a hyperbaric chamber may pose a greater risk than other patients. Thus, a closer monitoring and access to rapid treatment should be in place before HBOT is initiated.
Neurotoxic effects can result when HBOT is applied at high pressures around 4.9 ATA. 39 This is a major concern in HBOT, as high levels of oxygen can generate reactive oxygen species (ROS), as major source of oxidative stress. However, at 2.5 ATA for 75 min, there was no increase in lipid peroxidation in pre-clinical studies of global cerebral ischemia. 40 Despite these concerns for generation of ROS, HBOT protocols in TBI studies have durations ranging in hours. During this short period of time, antioxidant chemicals stored within cells (superoxide dismutase, catalase, glutathione dependent peroxidase, vitamin C) may be able to prevent damage from ROS. 41 However, there is much evidence that TBI alone reduces antioxidant reserve and potential added effect of oxidative stress from HBOT must be considered. Cerebrospinal fluid of severe pediatric TBI patients showed reduction of ascorbate and glutathione. 42 In the post-TBI setting where antioxidant supply is largely depleted, there is a higher risk of oxidative stress from HBOT and a more thorough characterization of oxidative stress at different durations of HBOT exposure is needed in the future.
Additional insights can be gained from some reports that looked at the effects of atmospheric pressure change to neural tissue in TBI. The effect of post-TBI exposure to hypobaric conditions, for example during flight transport following military blast, have been explored previously. 43 Although hyperbaric conditions were shown to increase oxygen delivery to brain tissue and reduce intracranial pressure, 44 hypobaric exposure for 6 h in rats after TBI showed that there was significantly worse cognitive function, hippocampal neuronal loss, and glial activation at 7 days post-injury. 45 The mechanism of deleterious effects of hypobaric condition is decreased CBF and brain tissue oxygenation, as shown by intracranial monitor in Yorkshire swine that underwent hypobaric condition for 4 h. 46 However, based on these few studies, it is difficult to conclude whether the deleterious effect of hypobaric condition is due to limited oxygen delivery, or any other unknown effect of atmospheric decompression and compression of nervous tissue. Further mechanistic studies are needed on the effect of high and low atmospheric pressure asides from oxygen delivery in the setting of TBI.
However, much of the literature on HBOT have flawed methodology and do not describe factors that can lead to placebo effect in their study subjects. 47 For example, there is paucity of description on social experience, environment that the patient was treated in, and the expectation of the patient and staff in terms of group assignment. Some of the prior studies often did not randomize patients or perform blinded analysis, introducing high risk for bias. 48 Additionally, given wide range of protocols throughout these prior studies, an optimal parameter regarding the pressure and frequency of treatment remains unclear.
To address some of the controversies about the efficacies of HBOT, a new multi-center phase II clinical trial supported by Strategies to Innovate Emergency Care Clinical Trials Network (SIREN) across 15 centers in the United States have been recently initiated. 49 Multiple arms with various pressure and duration of HBOT will be used to determine if HBOT has efficacy in severe TBI in this phase II clinical trial named Hyperbaric Oxygen Brain Injury Treatment (HOBIT).
Hydrogen
As ROS play an important role in the pathophysiology of TBI, free radical scavengers and antioxidants have been extensively studied as one of the critical strategies in treatment of TBI. One of the major oxidative stress scavengers of recent interest is molecular hydrogen (H2) which is in a gaseous state. Given its small size, it can freely penetrate cellular membranes and reduce damaging effects of ROS. 50 It also has limited side effect profile and its safety has been demonstrated even in prolonged exposures of mice for 72 h, 51 as well as human subjects in a 14-day treatment. 52 Mixture of inhalational gases with H2 content up to 56% has been previously demonstrated as safe among deep sea divers. 53 The additional advantage of H2 use is that it can also be administered in a dissolved state in saline or water with potentially similar therapeutic effect. 54 This would allow the potential option of delivery via injection or oral intake.
The key factor to consider in reducing oxidative species is that there are also ROS that have important physiological roles at low concentrations such as superoxide anions (O2∙-) or hydrogen peroxide (H2O2). These agents function in signal transduction of major cellular functions such as growth, differentiation, and apoptosis. 55,56 In cell free and cell culture experiments, H2 was shown to specifically reduce hydroxyl radicals (OH∙) but did not react with ROS that have major physiological roles such as H2O2, NO∙, O2∙-, and ROS inside mitochondria. 57 Moreover, in these experiments it reduced peroxynitrite, another highly damaging agent after hydroxyl radical. 57
Hydroxyl radicals (OH∙) may be one of the most potent ROS, reacting with nucleic acids, proteins, and lipids. It directly kills erythrocytes, damages cell membranes as well as nuclear DNA. 12 As H2 can specifically reduce OH∙, the therapeutic efficacy for H2 has been investigated in the setting of high oxidative stress conditions such as reperfusion injury. 57,58 Other organs with reperfusion injury such as the heart 59 and liver 60 have also been studied, with a therapeutic benefit of H2 demonstrated. As oxidative stress is a major driver of pathophysiology in neurodegenerative diseases, intracranial hemorrhage, and TBI, H2 has also been investigated in these settings. 61 –64
In an ischemia-reperfusion injury model of mice, H2 administration improved survival rate of animals and reduced hippocampal neuronal injury by reduction of oxidative stress. 58 Similarly in a study of 7-day-old neonatal rats that underwent common carotid artery ligation and hypoxic injury, intraperitoneal injection of H2 in saline resulted in reduced caspase activity, inflammation, and cognitive performance. 54 Intracerebral hemorrhage model of rats also showed that H2 administration reduced cerebral edema and functional impairment. 62 As expected with the time course of oxidative stress after neural injury, there was a window of therapeutic effect when administered at 24 h but not at 72 h. In addition to free radical scavenging effects, there may be additional effects of H2 which are indirectly related to its function as an antioxidant. Anti-inflammatory effects of H2 also have been demonstrated. 65,66
Although there have been many reports on neurodegenerative and cerebrovascular pathologies in which H2 was shown to be protective, TBI studies have not extensively explored H2 as a therapeutic agent shown in Table 3. There are three rodent studies that showed H2 given through hydrogen rich water 63 and inhalation at 2% 64,67 to be neuroprotective. The H2 administration resulted in reduced cerebral edema and infarct volume, as well as protection of cognitive function in rats. 64,67 Histological analysis of hippocampal sections, cytokine levels in brain tissue, and magnetic resonance spectroscopy showed reduction of inflammatory markers. 63
TBI, traumatic brain injury; IP, intraperitoneal; TNF, tumor necrosis factor; HGMBI1, High mobility group box 1; MRS, magnetic resonance spectroscopy.
Although efficacy remains to be determined, the potential use of H2 as a therapy in TBI would be versatile (it can be used in either inhaled form or injected form dissolved in liquid). It functions via oxygen radical scavenging agent, and it has the most therapeutic effect when administered early after injury. It is one of the newly discovered therapies in TBI that can be further explored with much potential for future studies.
Hydrogen Sulfide
Hydrogen sulfide is endogenously expressed in areas such as the ileum, portal vein, and aorta, and have various effects in many systems, such as both pro- and anti-inflammatory effects, vasodilatory effects, anti-apoptotic effects. 68,69 Its role in various processes is incompletely understood, but it is an agent that warrants further investigation given its numerous implications for cytoprotection. As it can be delivered by inhalation 70 or hydrogen sulfide donor, 71,72 it also has many versatile potential applications for clinical use similar to hydrogen gas.
Neuroprotective effect of hydrogen sulfide has recently been explored in several animal studies of TBI, but the body of data is much smaller than that of other gases as shown in Table 4. It has previously been considered a toxic substance but was shown to have protective effects in stroke 73,74 and neurodegenerative states. 75 Much of the neuroprotective effect is considered to involve anti-apoptotic and anti-inflammatory function. 76 Additionally, it is known to attenuate excitotoxicity in models of ischemic brain injury 77 and Parkinson's disease 78 by functioning as an N-methyl-D-aspartate (NMDA) receptor antagonist.
TBI, traumatic brain injury; NaHS, sodium hydrosulfide (H2S donor); IP, intraperitoneal; N/A, not applicable.
Intraperitoneal injection of H2S by a donor: sodium hydrosulfide (NaHS) has also been demonstrated as a feasible delivery method. Rats subjected to controlled cortical impact had significant attenuation of spatial memory deficits when pretreated with NaHS injection. 71 Another report using this model of injury showed reduced lesion volume, cerebral edema, and improved blood–brain barrier function as well as motor behavioral function. 72 In a mouse weight drop model of TBI, H2S was also administered via intraperitoneal injection of NaHS. 79 This ameliorated the surge in markers of apoptosis and autophagy, as well as edema and cognitive deficits. Although autophagy has been reported to be both harmful, 80 as well as protective, 81,82 in TBI, the authors of this report interpreted this finding as H2S preventing oxidative stress to the degree that it reduced an autophagic burden of the injured tissue. Similar to H2 use, hydrogen sulfide has the advantage of usage as gas or liquid delivery forms. Its mechanism of action is more complex however, as it does not function as free radical scavenger but affect various pathways as outlined.
One of the proposed mechanisms of H2S having neuroprotective effects is by reducing metabolic rate, as inhaled H2S was shown to induce hypothermia and reduced metabolism in rodents. 83 However, when the same effect was explored in a mechanically ventilated piglets, there was a lack of hypometabolic effects. 84 Similarly, in a pig model of hemorrhagic shock, 85 there was no difference in hemodynamics, temperature, and metabolic rate of pigs exposed to H2S compared with control pigs. As increasing the dose was associated with lung toxicity without further reduction in metabolism in this study, the efficacy of H2S in TBI will need to be thoroughly assessed in clinical trials.
Nitric Oxide
Unlike many of the other inhaled gases in this report, nitric oxide (NO) has a much more complicated effect as it is used in various physiological processes. Whether nitric oxide can be therapeutic or cytotoxic in the setting of injury has been a subject of debate for many years. This is due to the multiple roles it can serve as an oxidant or antioxidant, and its byproducts that can have various different influences on the vascular dynamics and oxidative stress of the cells. Although NO has scavenging function of free radicals, its byproduct can induce stress pathways.
Nitric oxide functions as a neurotransmitter, essential for dilation of blood vessels. 86,87 However, in the presence of O2∙- it is converted into a strong oxidant peroxynitrite (ONOO∙-) 88 and can induce oxidative damage in various components of intracellular processes. 89 The toxicity of ONOO- has been proposed to be one of the major contributors to reperfusion injury in stroke. 90 Also, nitrite (NO2 -, byproduct of NO) in the presence of H2O2 can cause oxidative burden on cells, having an opposite effect of NOs protection from oxidative stress. 91
The effect of nitric oxide is complex, as it also can also have therapeutic effects. Nitric oxide has a direct scavenging function on hydroxyl radicals. 89 Additionally, it indirectly reduces oxidative stress by binding to metals such as iron, copper, and nickel. Formation of this metal-nitrosyl complex inhibits the effect of peroxide on the metal and thus prevents Fenton-type reactions. Moreover, the various cytotoxic level of reactive nitrogen species has been summarized by the following: H2O2 > HNO/NO∙ >> ONOO∙-, which shows that the oxidative stress by reactive nitrogen species may be small. 92 The damaging effect of ONOO- may also not be significant given the short lifetime of this species. 89
In neuronal cultures, membrane damage induced by H2O2 was prevented by administration of NO donor complex. 91,93,94 In these in vitro experiments, donor complexes named NONOates (chemical formula R 1 R 2 N−(NO−)−N = O) were used to release NO without the need for enzyme activation. There was a significant increase in cell survival when cells were treated with NO. In a setting of high oxidative stress such as reperfusion injury in a rat model of cerebral ischemia, intravenous delivery of NO donor lead to 10-fold decrease in free radical production and dramatic decrease in infarct size. 94 Also, another report demonstrated that various different NO donor complexes were all shown to be protective against ROS induced toxicity. 93
In prior observations of cat vasculature by both intravital and postmortem microscopy, inhibition of NO production by agents such as NG-monomethyl-L-arginine (L-NMMA) or NG-nitro-L-arginine methyl ester (L-NAME) increased leukocyte adhesion to endothelium 95,96 and extravasation. 97 These studies also demonstrate that P-selectin and ICAM-1 mediate leukocyte adhesion and extravasation when NO is inhibited. Additionally, it can activate NADPH oxidase to inhibit O2∙- formation in neutrophils. 98 Thus, NO serves an important role in preventing pro-inflammatory changes in leukocytes which can potentially lead to cell damage.
At 4 h after moderate fluid percussion injury in rats, pericontusional tissue measurements of NO showed significant NO decrease. 99 However, another study using fluid percussion injury showed that there is an upregulation of NO levels in cerebral cortex, hippocampus and cerebellum after severe injury. 100 This increase in NO also resulted in pericontusional tyrosine nitration and was associated with decreased mitochondrial respiration, specifically complex I dysfunction. As prior studies showed that high levels of NO or ONOO∙- can directly inactivate complex I by tyrosine nitration, acute NO increase was suggested as the driver of mitochondrial dysfunction. 101,102
Studies detailing the activities of NO synthesis enzyme: nitric oxide synthase (NOS) showed that the actual time course of NOS activity may be more complicated. 103 -105 There are multiple isoforms of NOS: constitutive NOS, neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS), and each of them may have a different role and time course of activity after TBI. Constitutive NOS activity is temporarily increased from 5 min to 30 min after TBI, but there was a subsequent decrease from 1 day up to 7 days. 103 Individual isoforms of NOS may also have a different role in TBI pathophysiology. Specific inhibition of nNOS by administration of 3-bromo-7-nitroindazole (7-NI) after TBI decreased contusion volume. 103 However, eNOS deficient mice had greater perfusion deficits in pericontusional tissue 104 as well as reduced arteriolar diameter. 105 Thus, while eNOS activity may be neuroprotective, nNOS activity may exacerbate injury.
In iNOS deficient mice, TBI led to decreased CBF in multiple regions including the hippocampus, thalamus, and amygdala as detected by arterial spin-labeling MRI. 106 This demonstrated the important role that iNOS may play in post-TBI perfusion in brain tissue. Experiments using iNOS deficient mice subjected to TBI also showed increased markers of oxidative stress. 107 Similarly, when iNOS inhibitors aminoguanidine and L-N-iminoethyl-lysine were administered immediately following TBI in rats, there was worse functional outcome and neuronal loss. 108 However, another study contrasted this finding by showing that administration of iNOS inhibitor 1400 W starting at 18 h or 24 h after TBI resulted in improved histological outcomes. 109 The exact role of iNOS in injury may be more complicated, as it seems to show time dependent effect if no major secondary effect by the iNOS inhibitor drugs can be assumed in both studies. Overall, whether NO serves a neuroprotective or damaging role in the setting of TBI depends on the source and thereby location of its effect, as well as the timing of its effect.
Nitric oxide has also been explored as a therapeutic agent focused on improving CBF given its vasodilatory effects. 110 -112 When tested at a range of 3-300 parts per million (ppm), inhaled NO does not cause major decrease in systemic blood pressure over a wide range until high concentrations such as 300 ppm. 113 In a mouse model of stroke, inhaled NO at 50 ppm was carried to the brain in the form of nitrite and S-nitrosohemoglobin and specifically increased the penumbral perfusion without increasing perfusion in other areas of the brain. 114 Similarly in animals that had TBI, perfusion in pericontusional site was increased without any significant increase in ICP when L-arginine, NO precursor was administered. 115,116 Moreover, it reduced inflammatory markers and lead to functional improvement when administered to mice immediately after TBI. 117
Large animal experiments also show improved CBF: pigs that were subjected to contusion TBI and inhaled NO (at 30 ppm for male animals and 20 ppm for female animals) showed increase in CBF measured via radioactive microspheres injected via arterial catheter. 118 Using the pig model of TBI, the effect of inhaled NO on cerebrovascular regulation was also studied. 119 Potassium channel agonists (cromakalin, NS1619, prostaglandin E2) induce vasodilation of cerebral blood vessels, but following TBI this effect is blunted. Exposure of injured pigs to inhaled NO reversed this loss of vasodilatory effect. 119 Moreover, inhaled NO reduced cerebrospinal fluid levels of ET-1, ERK, and IL-6, the major agents in cerebral autoregulation and inflammation. 120
In a randomized, blinded study of mice that underwent controlled cortical impact, inhaled NO was administered following injury. 110 There was a significant increase in CBF immediately, and ICP increase seen in control group was attenuated in the inhaled NO group over the course of 90 min of monitoring. Additionally, in a detailed study of cerebral perfusion of sheep and mice using 18 F-fluorodeoxyglucose micropositron emission tomography and intravital microscopy, a specific vasodilatory effect of inhaled NO to hypoperfused brain tissue was found. 114 Inhaled NO selectively increased CBF in areas of low perfusion without affecting normally perfused brain regions. This inverse steal phenomenon has been termed “Robin Hood Effect” by the authors. These reports showing selective targeting of hypoperfused areas by inhaled NO argue against global cerebral vasodilation leading to potential ICP crisis. As several early studies using nitroprusside, a NO donor, in patients with intracranial pathologies raised concerns about the possibility of risk of increased ICP, 121 -123 this is an important point to be noted in these more recent studies using inhaled NO.
A classic study from 1980s that raised a concern for the use of nitroprusside is a case report from of a 14-year-old girl who suffered a respiratory tract infection, who then became comatose. 121 She was found to have elevated transaminases and ammonia level, and ICP was noted to be at 30 mm Hg when intracranial pressure monitor was placed. The combination of pentobarbital, mannitol, and hyperventilation reduced the ICP. However, on Day 5 of hospitalization, arterial blood pressure rose to 200/100 mm Hg, and sodium nitroprusside drip was initiated. Immediate elevation of ICP from 20-22 to 38 mm Hg was reported with subsequent blood pressure falling to 170/90. This case report is concerning for nitroprusside causing ICP elevation, but it is also possible that ICP had been up-trending just prior to this event. If the cerebral edema had begun already and nitroprusside reduced arterial blood pressure, cerebral hypoperfusion would be exacerbated leading to worsening hypoperfusion injury. Unfortunately, the detailed timeline of events is not described in this study.
Other reports from the 1970s also showed significant decrease in blood pressure when nitroprusside was used. 122,123 Administration of nitroprusside in ten intracranial tumor patients was associated with ICP increase from 14.5 to 27 mm Hg while mean arterial pressure was reduced by 33%. 123 Decreased systemic blood pressure during the ICP crisis may have contributed to reduced cerebral perfusion. In a descriptive study of Mongrel cats that were treated with sodium nitroprusside, ICP had an early fluctuation for one minute after initiation of treatment while mean arterial pressure was reduced from the baseline of 100 mm Hg to 65 mm Hg. 122 Soon afterwards ICP was maintained at baseline and was unchanged as nitroprusside administration continued. The lack of ICP elevation during the time nitroprusside was administered argues against the direct cause and effect relationship of nitroprusside to increase ICP. In a more recent study, which explored the use of nitroprusside in ischemic stroke patients, it was applied only at a dose that caused 10 mm Hg fall in mean arterial pressure, and improvement in CBF was seen. 124
In the last 3 decades, neurointensivists and neurosurgeons avoided using nitroprusside in patients with risk of increased ICP, but inhaled NO should be approached with a different stance. Inhaled NO, as shown in the studies reviewed in this section induced no significant hypotension. While high NO concentrations (at 300 ppm) decreased mean arterial blood pressure from 62 to 55 mm Hg in rabbits, 113 the doses of 10-30 ppm used in the recent studies resulted in no significant effect on blood pressure. Although the dose-dependent relationship of inhaled NO and parameters such as cerebral blood volume, mean arterial pressure, and ICP would need to be clarified in future studies, these prior studies indicate that inhaled NO may be a useful therapy in TBI patients. Additionally, other mechanisms of its beneficial effect should be explored as it is theoretically possible that inhaled NO could improve blood flow without increasing cerebral blood volume by altering blood rheology.
To date, there have not been any large-scale clinical studies completed on use of inhaled NO in human subjects. As approximately 20-30% of patients with severe TBI develop acute lung injury 125,126 the use of NO as a selective pulmonary vasodilator has been explored in several case reports. In a 10-year-old child who suffered from a motor vehicle accident found with subarachnoid hemorrhage, diffuse axonal injury, as well as pulmonary hypertension, a trial of 10-35 ppm dose of inhaled NO resulted in decreased pulmonary artery pressure, improved cardiac index without major changes in intracranial pressure. 127 Other promising case reports have supported the potential utility of using inhaled NO in TBI patients with acute respiratory distress syndrome as shown in Table 5. 128,129
Exact length of inhaled NO administration was not reported. **These studies were focused on targeting respiratory complications and not brain injury.
TBI, traumatic brain injury; NSS, Neurological Severity Score; ICP, intracranial pressure; rCBF, relative cerebral blood flow as measured by laser doppler fluxmetry; PGE2, prostaglandin E 2; N/A, not applicable; CT, computed tomography.
In a case of a patient who fell 10 m and sustained intracranial bleeding, lumbar spine fracture, as well as development of acute respiratory distress syndrome, inhaled NO was administered at 10 ppm to treat the respiratory impairment. 129 There was successful recovery of oxygenation and no adverse effect on intracranial pressure. Thus, NO was considered as a potentially useful agent in a population of patients with TBI who also suffer from acute respiratory distress syndrome. In another case, a patient who suffered a motor vehicle accident with multiple brain parenchymal contusions and developed acute respiratory distress syndrome (ARDS) was treated with 20 ppm inhaled NO. 130 There was significant improvement of oxygenation, as well as significant decrease in intracranial pressure. Although there is relative lack of data on human subjects, these case reports give hopeful outlook for larger clinical studies using inhaled NO for TBI patients.
Nitric oxide use has different safety profiles across various ages. It has a favorable safety profiles in newborns without major side effects such as systemic hypotension. 131 Among newborns treated with inhaled NO, abrupt cessation of therapy can lead to severe hypoxemia as endogenous NO production is downregulated. 132 Thus, a gradual down titration is necessary after a course of inhaled NO therapy. Also, a minor increase in methemoglobin levels (less than 10% increase in 90% of the neonates) has been reported. 133 Aside from these, in pediatric patients randomized trials of inhaled NO in ARDS reported no major side effects. 134,135 However, more significant side effects have been demonstrated in adults. In Cochrane review of inhaled NO use for ARDS, adult patients had increased risk of renal dysfunctions. 136 Thus, in future TBI clinical studies using inhaled NO, limited dosing in patients with renal impairments and close monitoring of renal function would be necessary.
Nitric oxide is a naturally expressed chemical with its role in blood vessel dilatation as well as antioxidant and anti-inflammatory function. The use of NO in TBI has been limited as it was only reported in several case reports, in which patients had both TBI and ARDS. In ARDS, high positive ventilation pressures for alveolar recruitment and permissive hypercapnia are tolerated. However, these strategies may increase intracranial pressure in a TBI patient. In these patient populations with conflicting therapeutic strategies for each disease entity, NO was shown to be able to increase pulmonary ventilation while not significantly affecting intracranial pressures. It may also improve perfusion in cerebral blood vessels without increasing intracranial pressure. Moreover, as mentioned above it may have additional benefits in preventing the most damaging oxidative stress by H2O2. Nitric oxide therapy is an exciting avenue of TBI research that has great future potential for clinical use.
Noble Gases
Noble gases such as helium, neon, argon, krypton, and xenon have very low chemical reactivity given their stability due to full valence electron shells. However, many in vitro and in vivo experiments have shown that they certainly have biological activity, as xenon and argon can be neuroprotective as listed in Table 6. Other noble gases such as helium, neon, and krypton have been trialed in in-vitro hippocampal model of TBI, without notable neuroprotective effect. 137
TBI, traumatic brain injury; Xe, xenon; Ar, argon.
Of particular interest in the recent years has been application of xenon in several neuropathological conditions such as stroke, perinatal asphyxia, and TBI. 137 -139 In an intrauterine perinatal asphyxia model of rats, 35% xenon provided neuroprotection, as demonstrated by reduced apoptotic cell death in hippocampal neurons and improved cognitive function on Morris water maze at 50 days. 140 Application of 50% atmospheric xenon also prevented damage from hypoxic-ischemic injury in hippocampal brain slices. 141 This study also demonstrated that NMDA receptor glycine binding site may be the focus of xenon's activity, as application of glycine blocked the neuroprotection by xenon. The amount of xenon that can be given in inhalation is limited by supplemental oxygen that is needed for the patient. Prior trials of xenon as an anesthetic agent utilized 30% oxygen and 70% xenon combination or 20% oxygen and 80% xenon combination. 142 However, when acute lung injury such as ARDS is present in addition to TBI, higher oxygen supplementation may limit the amount of xenon that can be administered.
Xenon exists in a monoatomic form with low reactivity, stable at a gaseous state. 143 It also exhibits little to no toxicity in hepatic and renal function, platelet function, coagulation activity, and cardiac hemodynamics. 144 Additionally, it is eliminated quickly from the body given its low blood-gas partition coefficient that allows it to diffuse through the blood–brain barrier with ease. 145 The neuroprotective effect of xenon gas has been demonstrated initially in hippocampal slices experiments. In this in vitro study, hippocampal slices were subjected to focal weight drop of a stylus. 146 Xenon gas at 75% applied immediately after injury, or with a 2- to 3-h therapeutic window reduced injury as measured by propidium iodide staining. Also, similar to the previous study on neonatal asphyxia model, 141 glycine application blocked the xenon's neuroprotection in hippocampal slice TBI model. 137 Neuroprotection by xenon was also confirmed using a blast injury model of in vitro TBI, with reduced injury assessed by propidium iodide staining. 147 In a mouse model of controlled cortical impact, xenon administration at 75% 148 and 30-50% 149 were shown to reduce injury volume and improve neurological outcomes. Histological analysis showed reduced astrogliosis and microglia in hypothalamus and amygdala, respectively. Also, there was an attenuation of hippocampal neuronal loss. 147
Xenon has additionally been explored as an anesthetic agent due to its analgesic effect and due to subjects having faster emergence from anesthesia compared with other gaseous anesthetics. 150,151 However, its high costs currently at $10 per liter is prohibitive in its widespread use. 152 During a 2-h anesthetic procedure, reaching an anesthesia level at 70% using 0.5 L/min would result in an efficiency lower than 20% with 34 L of xenon gas to be wasted into the atmosphere. 153 To reach a much more cost-effective solution, closed circuit rebreathing devices are currently explored by various companies to reach 70-90% of the xenon being delivered into the system. However, even with the use of a closed-circuit system, xenon at the current prices would be 2-5 times more costly than isoflurane for a typical anesthetic procedure. 154,155 There is also limitation in annual production of xenon 153 and each liter of xenon requires 220 watt-hours of energy to produce (million times that of nitrous oxide). 155 Thus, an efficient delivery technology that can further increase the efficiency of xenon delivery is much needed.
In addition to closed circuit inhalational system, methods to deliver xenon in various lipid emulsions have been studied, as approximately 20-fold more xenon can deliver in lipid solutions compared with aqueous solutions. 156 The xenon gas dissolved in lipid emulsion was also shown to successfully inhibit NMDA receptors. 157 Moreover, specific lipid emulsion carriers were shown to enhance NMDA receptor antagonist activity of xenon. 156 Liposomes that encapsulate xenon and release them upon ultrasonic stimulation have also been devised as a novel delivery method to the area of injury. 158 As specific delivery to an injured tissue will help in limiting the dose needed for the therapeutic benefit, this strategy may enhance potential efficacy in TBI. With additional progress in xenon delivery methods, xenon encapsulated lipid bubbles, which also have clear ultrasound visualization have been developed. 159 This is an exciting field as further development may allow theranostic (simultaneously diagnostic and therapeutic) use of xenon encapsulated lipid bubbles in ischemic tissue as well as TBI. 160 -162
Although less extensively studied than xenon, argon gas also was shown to have narcotic as well as neuroprotective effects. 163 It is believed to operate by a different mechanism compared with xenon, likely by stimulation of γ-aminobutyric acid type-A receptors (GABAAR). 164 When applied in hippocampal brain slices for oxygen-glucose deprivation, its neuroprotective effect was not blocked by glycine application. 165 Similarly in a hippocampal slice model of TBI, application of argon reduced cell damage measured by propidium iodide. 166 The duration of argon administration is important for neuroprotection, as administration for 2 h failed to show significant neuroprotection, 167 but 72-h exposure was able to attenuate cellular injury. 166 Argon exhibits neuroprotective effect via NMDA-receptor independent mechanism in TBI: when hippocampal slice was subjected to trauma, glycine administration blocked the neuroprotective effect of xenon but not argon. 166
Use of noble gases as therapeutic intervention in TBI is a recently discovered field of interest. As they are generally inert chemicals with specific mechanisms of neuroprotection, they may be great candidates for future clinical trials in TBI. As the price of these agents are high, efficient delivery mechanisms are necessary before making them widely used in clinical settings. Novel delivery mechanisms of noble gases via microbubbles and localized release at the site of injury may also make them achieve therapeutic effects even at lower doses.
Systemic administration of neuroprotective gas agents can result in systemic side effects, reduce local therapeutic efficacy and may be expensive. To overcome these limitations, studies have examined whether targeted, ultrasound-guided delivery of gas loaded ultrasound contrast agents, shown to be safer than other imaging contrast agents in vivo, can achieve neuroprotection. 168 The concept behind therapeutic use of microbubbles is to load neuroprotective gas into ultrasound contrast agents that serve as stable carriers which can undergo acoustically triggered cavitation, a process in which high acoustic power induces oscillation and subsequent collapse of the microbubble for localized gas release. This would achieve high concentration of gas in the brain in a short amount of time, in the range of minutes, without the need for hours of gas inhalation therapy. For therapeutic applications, these microbubbles can be modified such that the inner core can encapsulate any gas agent of choice. In fact, a study recently showed that Xenon encapsulated within the liposome bilayers when released via high acoustic power at the level of the carotid can locally deliver Xenon to the injured site in models of stroke and achieve neuroprotective efficacy. 158,169,170 More studies will be needed to validate therapeutic efficacy of these gas loaded microbubbles in large animal models of TBI. The acoustic visibility of these microbubbles also raises the possibility of using these agents as potential theranostic agents.
Conclusions
Thus far, much of the pre-clinical and some clinical data on these inhalational gases hold a very promising outlook on their widespread uses as summarized in Fig. 1. Although both normobaric and hyperbaric oxygen treatments have been investigated for many years, a rigorous clinical trial is needed to clarify their efficacy in TBI. Hydrogen gas, hydrogen sulfide, and NO have shown promising effects in some pre-clinical studies and have had limited clinical investigation. They have, however, yet to be able to be translated into clinical research. Additionally, xenon and argon gases also have promising pre-clinical results but are also in need of clinical validation. The specific mechanisms of each of these agents will be further clarified in future research, but given their ease of delivery many of these will likely be seen in clinical trials in the near future. As TBI is a heterogenous injury involving multiple concurrent mechanisms, a potential avenue of future exploration is using each of these agents for different injury types as the TBI community is working on categorizing TBI subtypes via various imaging and blood biomarkers. 171 Also with continued advances in the field of targeted gas delivery, these interventions may play an important role in clinical TBI management in the near future.
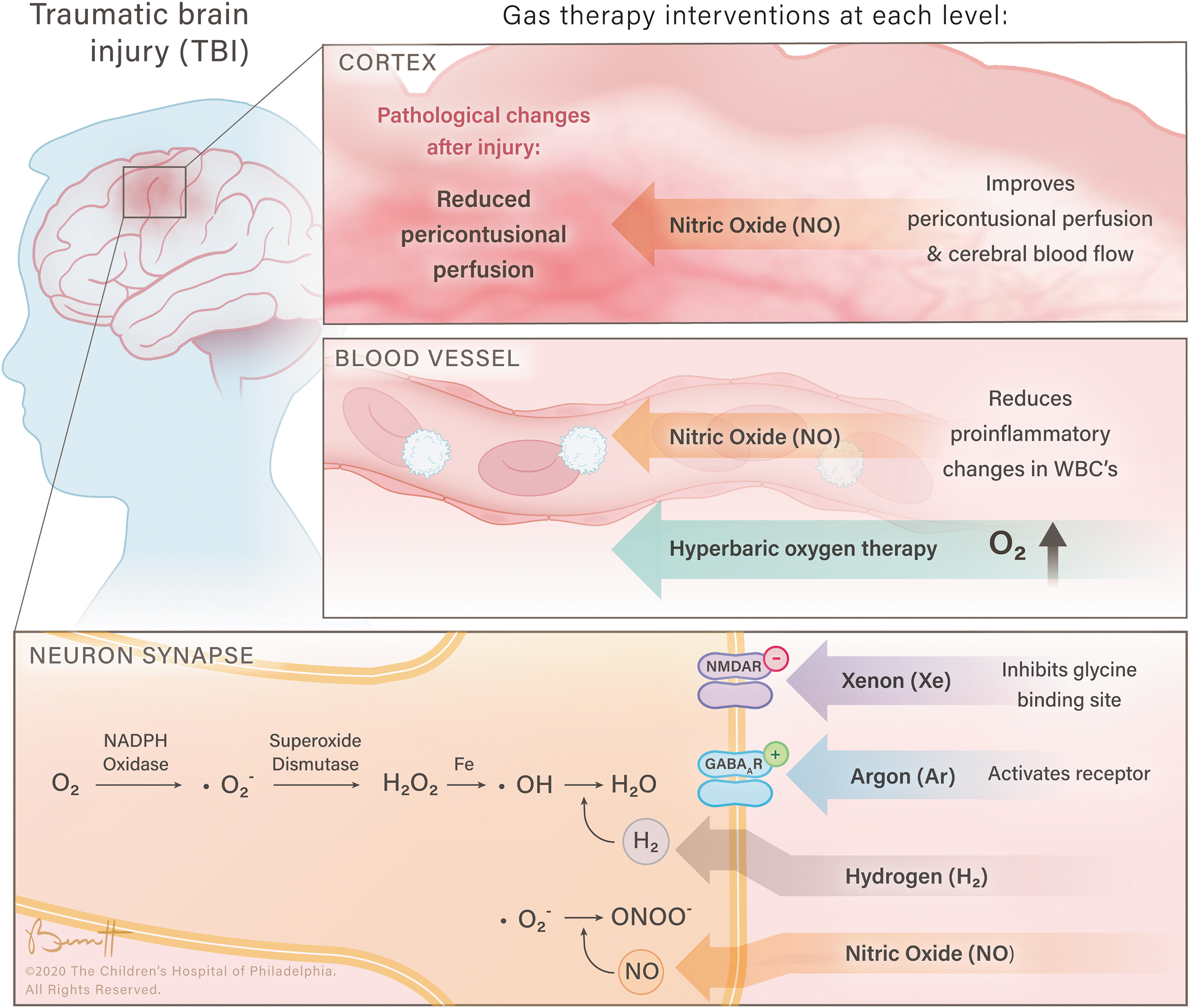
The neuroprotective effects of inhalational gases at various physiological levels.
Footnotes
Funding Information
T.K. is supported by NIH, NHLBI R01HL141386. M.H. is supported by Foerderer Grant from Children's Hospital of Philadelphia and Junior Faculty Pilot Grant.
Author Disclosure Statement
No competing financial interests exist.