
Abstract
When the blood–spinal cord barrier (BSCB) is disrupted after a spinal cord injury (SCI), several pathophysiological cascades occur, including inflammation and apoptotic cell death of neurons and oligodendrocytes, resulting in permanent neurological deficits. Transient receptor potential melastatin 7 (TRPM7) is involved in the pathological processes in many neuronal diseases, including traumatic brain injury, amyotrophic lateral sclerosis, parkinsonism dementia, and Alzheimer's disease. Further, carvacrol (CAR), a TRPM7 inhibitor, is known to protect against SCI by reducing oxidative stress and inhibiting the endothelial nitric oxide synthase pathway. However, the functions of TRPM7 in the regulation of BSCB homeostasis after SCI have not been examined. Here, we demonstrated that TRPM7, a calcium-mediated non-selective divalent cation channel, plays a critical role after SCI in rats. Rats were contused at T9 and given CAR (50 mg/kg) intraperitoneally immediately and 12 h after SCI, and then given the same dose once a day for 7 days. TRPM7 was found to be up-regulated after SCI in both in vitro and in vivo studies, and it was expressed in blood vessels alongside neurons and oligodendrocytes. Additionally, CAR treatment suppressed BSCB disruption by inhibiting the loss of tight junction (TJ) proteins and preserved TJ integrity. CAR also reduced apoptotic cell death and improved functional recovery after SCI by preventing BSCB disruption caused by blood infiltration and inflammatory responses. Based on these findings, we propose that blocking the TRPM7 channel can inhibit the destruction of the BSCB and it is a potential target in therapeutic drug development for use in SCI.
Introduction
Spinal cord injury (SCI) is a devastating condition that results in permanent disability, thereby influencing a profound effect on the quality of life. 1 Despite years of intensive research, there are no widely accepted therapies to reduce tissue injury and enhance functional recovery after SCI. Initial mechanical damage causes necrosis, vascular damage, and blood–spinal cord barrier (BSCB) disruption that increases vascular permeability. As a highly selective permeable vascular endothelial structure in the spinal cord, the BSCB primarily consists of vascular endothelial cells that are tightly associated with well-developed tight junctions (TJs), which regulate the restriction of the entry of plasma components and blood cells into the brain or the spinal cord. After SCI, BSCB disruption is a critical event that causes secondary damage such as programmed cell death of neurons and glia and production of inflammatory mediators like cytokines, which results in chronic permanent dysfunction after SCI. 1 -3 Therefore, the development of therapeutic agents inhibiting BSCB disruption would be useful for the restriction of secondary injury followed functional recovery after SCI.
Extensive ion imbalances, including calcium overload in the spinal cord after SCI, cause irreversible damage to various cellular functions such as protein synthesis, mitochondria, cytoskeleton, and cell membrane functions, which is one of the leading causes of cell death. 4 Transient receptor potential (TRP) ion channels, which are non-selective cation channels, are expressed in many cells and tissues, including neurons and endothelial cells of the nervous system and have been shown to play important roles in diverse cellular processes. 5,6 The TRP melastatin 7 (TRPM7) channel in particular is a metal ion-permeable, non-selective cation channel that is expressed in almost all tissues and regulates divalent cation (Mg2+, Ca2+, and Zn2+) homeostasis, cell survival, proliferation, and Ca2+-mediated neurotransmitter release. 7 –10
TRPM7 has been shown to play an important role under pathological conditions, 11 although its function has been mainly researched in neurons. Previous studies indicated that TRPM7 mediates the neuronal death by regulating Ca2+ influx during cerebral ischemia and prolonged oxygen-glucose deprivation (OGD). 12 -14 The suppression of TRPM7 after brain ischemia also facilitated neuron survival and preserved the morphology and function of neurons in hippocampal CA1. 15 Additionally, it was reported that TRPM7 is involved in the pathologic processes of some neurodegenerative diseases including western Pacific amyotrophic lateral sclerosis, parkinsonism dementia, and Alzheimer's disease. 16,17 Further, carvacrol (CAR), a TRPM7 inhibitor, is known to protect against SCI by suppressing oxidative damage and the endothelial nitric oxide synthase pathway. 18 However, the functions of TRPM7 in the regulation of BSCB homeostasis after SCI have not been examined. Here, we examined the role of TRPM7 by investigating the effect of CAR on BSCB disruption after SCI in rats.
Methods
Animal model of SCI
Adult male Sprague-Dawley rats (250-300 g, Samtako, Osan, Korea) were used in this study. Before surgery, rats were weighed and anesthetized with 500 mg/kg of chloral hydrate intraperitoneally (i.p.). Laminectomy was performed at the thoracic 9-10 (T9-T10) level and exposed spinal cord was subjected to moderate injury (10 g × 25 mm) using a New York University (NYU) impactor as previously described. 19 Rats also underwent laminectomy at T9-T10 level without injury for sham control. All animal experiments were performed in accordance with the Guidelines of Animal Care Committee of the Kyung Hee University (permission number: KHUASP(SE)-17-059).
Drug administration
CAR (5-isopropyl-2-methylphenol; Sigma-Aldrich, St. Louis, MO) was dissolved in 0.9% saline, and 50 mg/kg was administrated i.p. into rats immediately and 8 h after SCI. Then the same dose of CAR was injected once a day for 7 days. The vehicle-control group received equivolumetric intraperitoneal injection of 0.9% saline at the corresponding time-points. For sham-operated controls, rats underwent a T9-T10 laminectomy without contusion injury, and received no pharmacological treatment.
Endothelial cell culture and OGD/reperfusion
American Type Culture Collection (Manassas, VA) provided a mouse brain endothelial cell line (bEnd.3), which was cultured as previously described. 20,21 In brief, bEnd.3 cells were cultured with Dulbecco modified Eagle medium (DMEM; Sigma) containing 10% fetal bovine serum to confluence; the culture medium was then replaced with glucose-free DMEM (Sigma) and then placed in a humidified anaerobic chamber (0.1% O2, 5% CO2, and 94.9% N2; APM-30D, Astec, Fukuoka, Japan) as previously described. 22 After 6 h of OGD, cells were further cultured in DMEM with 25 mM glucose under normoxia (reoxygenation). Control cells were cultured in DMEM with 25 mM glucose under normoxia. CAR (500 μM) was dissolved in 0.9% saline and treated 30 min before OGD. For vehicle control, 0.9% saline was treated.
Measurement of BSCB disruption
As previously described, the permeability of the BSCB was measured using Evans blue dye extravasation at 1 day after SCI. 22 In brief, at 1 day after SCI, 5 mL of 2% Evans blue dye (Sigma) solution in saline was administered intraperitoneally. Three hours later, the animals were anesthetized with chloral hydrate (500 mg/kg) and sacrificed by intra-cardiac perfusion with saline. The T9 spinal cord segment was removed and homogenized in a 50% trichloroacetic acid solution. After homogenization, samples were centrifuged at 10,000 g for 10 min, supernatants were collected, and fluorescence was quantified at an excitation wavelength of 620 nm, and an emission wavelength of 680 nm using a microplate reader (SpectraMax Gemini EM, Molecular devices, Sunnyvale, CA). Dye in samples was determined as micrograms per gram of tissue from a standard curve plotted using known amounts of dye. 23 For qualitative examination of Evans blue extravasation, the animals were perfused with phosphate-buffered saline (PBS) and subsequently with 4% formaldehyde as described above. The spinal cords were sectioned 20 μm thickness with a cryostat. The fluorescence of Evans blue in spinal tissues was observed with a fluorescence microscope and the relative fluorescence intensity was determined by MetaMorph software (Molecular devices).
Tissue preparation
Injured rats were anesthetized at indicated time points with chloral hydrate and perfused with 0.1 M PBS and then with 4% paraformaldehyde in 0.1 M PBS. The spinal cord (1.5 cm) was dissected out and post-fixed by immersing it in the same fixative for 5 h and placing it in 30% sucrose in 0.1 M PBS. After embedding the spinal segment for frozen sections, longitudinal or transverse sections were cut on a cryostat at 10 or 20 μm (CM1850; Leica, Germany). For molecular work, the spinal cord segment (1 cm) with the lesion epicenter were also isolated after perfusion with 0.1 M PBS and frozen at -80°C.
RNA isolation and reverse transcription polymerase chain reaction
TRIZOL Reagent (Invitrogen) was used to prepare total RNA, and 1 μg of total RNA was reverse-transcribed into first strand cDNA using MMLV according to the manufacturer's instructions (Invitrogen) as described. 22 The primers used for TRPM7, interleukin (IL)-6, tumor necrosis factor (TNF)-α, COX-2, inducible nitric oxide synthase (iNOS), monocyte chemoattractant protein (MCP)-1, macrophage inflammatory protein (MIP)-1α, MIP-1β, growth-regulated oncogene (GRO)-α, MIP -2α, and glyceraldehyde-3-phosphate dehydrogenase were synthesized by the Genotech (Daejeon, Korea); the primer sequences are shown in Table 1. The relative band intensity was measured by the AlphaImager software (Alpha Innotech Corporation, San Leandro, CA) and compared with sham control value.
Nucleotide Sequences of Primers and Conditions used for Reverse Transcription Polymerase Chain Reaction
TRPM7, transient receptor potential melastatin 7; IL, interleukin; TNF, tumor necrosis factor; iNOS, inducible nitric oxide synthase; MCP, monocyte chemoattractant protein; MIP, macrophage inflammatory protein; GRO, growth-regulated oncogene; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Western blot
Total protein was isolated using a lysis buffer as described previously. 22 Sodium dodecyl sulfate-polyacrylamide gel electrophoresis was used to separate protein samples (30 μg) and transfer them to nitrocellulose membrane (Millipore, Billerica, MA). The membranes were incubated for 1 h at room temperature with blocking solution (Tris-buffered saline containing 0.1 % tween-20 and 5% skim milk or bovine serum albumin), then overnight at 4°C with primary antibodies listed in Table 2. Secondary antibodies conjugated with horseradish peroxidase (Jackson ImmunoResearch, West Grove, PA) were used to detect the primary antibodies and immunoreactive bands were visualized using chemiluminescence (Thermo Scientific, Rockford, IL). Immunoblot image and the densitometric values were obtained by using AlphaImager software (Alpha Innotech Corporation). β-Tubulin was normalized to internal standard.
Antibody Information for Western Blot
TRPM7, transient receptor potential melastatin 7; iNOS, inducible nitric oxide synthase.
Immunohistochemistry
Frozen sections were processed for immunohistochemistry as previously described 24 using antibodies against TRPM7, neuronal-specific nuclear protein (NeuN), CC1, RECA1, ZO-1, myeloperoxidase (MPO), ED-1, cleaved caspase-3, 200 kDa neurofilament (NF200), and 5-hydroxytryptamine (5-HT), which are listed in Table 3. Fluorescein isothiocyanate (FITC)- or cyanin 3-conjugated secondary antibodies (Jackson Immunoresearch) was used for double labeling. The manufacturer's protocol was used to label nuclei with 4′,6-diamidino-2-phenylindole (Molecular Probes). For quantification of MPO and ED-1 intensity, serial transverse sections were collected every 200 μm section rostral and caudal 3000 μm to the lesion site (total 30 sections) as previously described. 22 Digital images of MPO- or ED-1-stained tissues were obtained, and we quantified the entire fluorescent intensity of each transverse section above threshold by using MetaMorph software (Molecular Devices) and averaged. For quantification of cleaved caspase-3-positive oligodendrocytes (cleaved caspase-3/CC1 double positive), serial transverse sections were collected every 200-μm interval rostral and caudal 4000 μm to the lesion site (total 40 sections). Cleaved caspase-3-positive oligodendrocytes tin the white matter (WM) in each section were counted and averaged.
Antibody Information for Immunohistochemistry
TRPM7, transient receptor potential melastatin 7; NeuN, neuronal-specific nuclear protein; MPO, myeloperoxidase; NF200, 200 kDa neurofilament; 5-HT, 5-hydroxytryptamine.
To quantify the spared axon, serial transverse sections (20 μm thickness) were collected every millimeter section rostral and caudal 7000 μm to the lesion site per animal and stained with NF200 antibody. The number of spared axons at preselected areas within the ventral funiculus (including vestibulospinal tract) and dorsolateral funiculus (including rubrospinal tract) was counted as described. 25 To define the location of the ventral funiculus and dorsolateral funiculus axons, 200 × bright-field images were captured using well-established methodology: the ventral funiculus region for quantification was 250 μm from medial to the ventral edge of the spinal cord and the dorsolateral funiculus region for quantification was 350 μm from medial to the lateral edge of the spinal cord. 26 Axons were manually counted in equivalently sized (diameter larger than 10 μm) and similarly located regions from each section as described. 26,27 We were counted the number of axons in each section and averaged them across all animals per group. All of digital images were obtained using MetaMorph software (Molecular device). Serial sections were also stained with Cresyl violet acetate for histological analysis.
Measurement of transendothelial electrical resistance
Endothelial permeability was determined by measuring transendothelial electrical resistance (TEER) according to the manufacturer's instructions (Millipore) as described. 22,24 Briefly, bEnd.3 cells were seeded (5 × 105 cells) and seeded and cultured for 24 h on Transwell inserts (Transwell-COL, Corning) coated with rat tail collagen. Cells were than subjected to OGD/reperfusion injury and TEER was determined using with a Millicell ERS-2 Volt-Ohm Meter (Millipore). The surface area of the Transwell inserts was used to calculate the values in ohms per square centimeter (Ω·cm2).
Cell counting of viable ventral motor neuron
Serial transverse spinal cord sections (20 μm thickness) were collected every 500 μm section rostral and caudal 8 mm to the lesion site and stained with Cresyl violet acetate to count the number of ventral motor neurons (VMN). Specifically, VMN were counted by their presence in the lower ventral horn, below the deep apex where the ventromedial gray and white matter meets, about 140 μm below the central canal. To avoid double counting of adjoining sections, we defined the neurons with soma size lager than 200 μm2 in the ventral horn as VMN and counted only the neurons which having diameters ranging from larger than half the size of the sampling square (20 × 20 μm) using MetaMorph software (Molecular Device) as previously described. 28 We counted and averaged the number of VMN in three serial sections at each location, and subsequently the values of the individual animals were averaged to obtain the group mean. All counting and data analysis was performed by two trained investigators who were blind as to the experimental conditions.
TUNEL staining
Terminal deoxynucleotidyl transferase (TdT) dUTP nick-end labeling (TUNEL) staining was performed on injured spinal cord (1 and 5 days after injury) serial transverse sections (20 μm thickness) collected every 200 μm interval using an Apoptag in situ kit (Millipore) according to the manufacturer's instructions. TUNEL-positive cells with morphological features of nuclear condensation and/or compartmentalization in gray matter (GM) at 1 day and WM at 5 days after injury were counted and quantified as described in the previous report. 22,29
Behavioral tests
For behavior tests, animals were randomly divided into three experimental groups (sham, vehicle, and CAR-treated group). We used 10 rats respectively for the use of motor function evaluating of each group. All behavior tests were performed on the same day in randomized order by two investigators who were blind to the experimental conditions. Behavioral tests including Basso-Beattie-Bresnahan (BBB), inclined plane test, grid walk, and footprint were performed according to previously described. 25,30 -32
BBB locomotion scale, which is a 22-point scale (scores 0-21) was used to evaluate hindlimb locomotor function, starting with no spontaneous movement of the hindlimbs up to plantar stepping, weight support, forelimb-hindlimb coordination, and trunk stability. 31 For the inclined plane test, rats were placed on the testing apparatus in one of two positions (right side or left side up), and the maximum angle maintained without falling for 5 sec was recorded and averaged. 32 Rats were also tested on a horizontal grid to assess their ability to precisely place the hindlimb. 30 The animals had to walk on a 1 m long horizontal runway of metal grid bars with 10 mm square hole elevated 30 cm from the ground. The animals were allowed to freely explored and performances were recorded with a video camera and hindpaw placings are evaluated from video. When the hindpaw falls down, it is considered a foot slip. The total number of steps and foot slips for each hind paw were counted within a 1 m horizontal grid and the mean percentage of foot slips was calculated. For the footprint analysis, both forepaws and hindpaws were dipped in nontoxic red and blue dye before walking across a narrow box (1 m long and 7 cm wide). 25,33 The footprints were then scanned, and resulting digitized images were examined.
Luxol fast blue staining
To determine myelin loss, selected slides were stained with 0.1 % Luxol fast blue (Solvent Blue 38; Sigma) as previously described. 34 MetaMorph software (Molecular Device) was used to create digital images of Luxol fast blue-stained tissues.
Measurement of lesion volume
Serial longitudinal sections (10 μm) through the dorsoventral axis of the spinal cord were used to determine lesion volume as previously described. 35 Every 50 μm section (total 20 sections) was stained with Cresyl violet acetate and the lesion volume was calculated by measuring the area of cavitation at the injury epicenter with a low-power (1.25 × ) objective and then using a MetaMorph software (Molecular Device). Areas at each longitudinal level were determined, and the total lesion volume was derived by means of numerical integration of sequential areas.
Statistical analysis
The data were presented as the mean, standard deviation (SD), or standard error of the mean (SEM). To determine the statistical significance of the difference between the vehicle- and CAR-treated groups, the unpaired Student's t test was used. For immunohistochemical, and molecular analyses comparisons were based on a one-way analysis of variance (ANOVA). BBB locomotor scale and inclined plane test were analyzed using repeated measurement ANOVA (time vs. treatment) with post hoc Tukey's multiple comparison. SPSS 15.0 (SPSS Science, Chicago, IL) was used for statistical analysis. A p value <0.05 was considered to be statistically significance.
Results
Expression of TRPM7 is up-regulated after SCI
To test the hypothesis that TRPM7 channels may contribute to biological events following SCI, we first examined the profile of TRPM7 expression in injured rat spinal cords. TRPM7 expression was markedly up-regulated after SCI (Fig. 1A, 1B). Reverse transcription polymerase chain reaction (RT-PCR) and Western blot analysis revealed that TPRM7 expression was increased and peaked at 7 days after SCI. TRPM7 was mainly localized in neurons in the GM and oligodendrocytes in the WM, in the uninjured spinal cord (Fig. 1C; Sham). Interestingly, after SCI, TRPM7 was not only expressed in neurons and oligodendrocytes, but also in blood vessels near the injury site (Fig. 1C; SCI 7 days). Double immunostaining revealed that TRPM7 immunoreactivity was observed in neuron (NeuN positive) and oligodendrocytes (CC1 positive) in both sham and injures spinal cord (Fig. 1D). In addition, TRPM7 was also colocalized with blood vessel endothelial cells (RECA1 positive) 7 days after SCI (Fig. 1E; 7 days). By contrast, TRPM7 immunoreactivity was not detected in blood vessel of uninjured rats (Fig. 1E; Sham). These results indicate that TRPM7 is up-regulated in the blood vessels after SCI.

Transient receptor potential melastatin 7 (TRPM7) is up-regulated and expressed in blood vessel endothelial cells, neuron and oligodendrocyte after spinal cord injury (SCI).
CAR inhibits BSCB disruption and preserves TJ integrity after injury
To determine whether ion influx through TRPM7 affects the BSCB disruption, we evaluated the effect of CAR, a TRPM7 inhibitor, on BSCB disruption at 1 day after injury by Evans blue assay. Compared with sham group, the amount of Evans blue dye extravasation was markedly increased at 1 day after SCI, implying BSCB leakage (Fig. 2A). Further, CAR administration significantly reduced the amount of Evans blue dye extravasation as compared with the vehicle (Veh) group (mean ± SEM; Veh 72.69 ± 5.05 vs. CAR 15.61 ± 2.51 μg/g tissue, p < 0.05). To confirm the perivascular accumulation of Evans blue around blood vessels of the spinal cord after SCI, we examined the standard histochemistry of Evans blue dye extravasation. Histological data showed that any signal of Evans blue dye was not detected in the sham group. However, the fluorescence intensity of Evans blue dye was significantly increased in the vehicle group in the lesion epicenter at 1 day after SCI. Further, Evans blue dye was accumulated around blood vessels in the spinal cord after SCI. Quantification analysis showed that the fluorescence intensity of Evans blue dye was lower in the CAR-treated group than in the vehicle group (Fig. 2B).
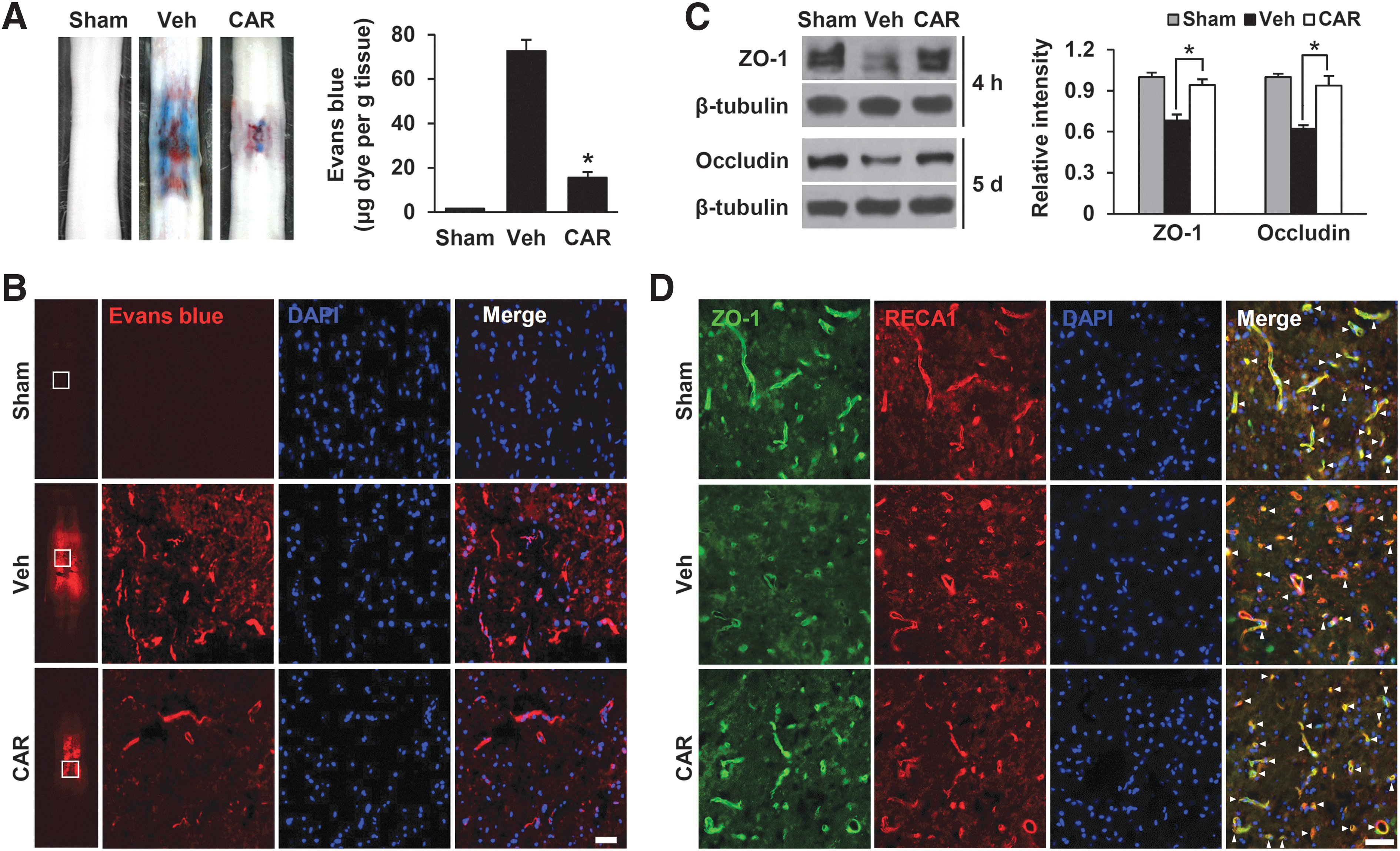
Carvacrol (CAR) inhibits blood–spinal cord barrier disruption and loss of tight junction proteins after injury.
Next, we examined the effect of CAR on the TJ protein levels of ZO-1 and occludin at 4 h and 5 days after injury. As shown in Figure 2C, CAR significantly attenuated the decrease in TJ protein levels after injury compared with the vehicle control (mean ± SD; ZO-1, Veh 0.68 ± 0.04 vs. CAR 0.94 ± 0.04; occludin, Veh 0.62 ± 0.03 vs. CAR 0.94 ± 0.07, p < 0.05). In addition, double immunofluorescence staining for ZO-1 and RECA1 showed that the fragmentation of capillary blood vessel was increased after SCI and ZO-1 immunoreactivity was decreased upon injury, compared with the sham control, whereas CAR treatment attenuated the fragmentation of the capillary blood vessels and ZO-1 loss (Fig. 2D), indicating that TRPM7 affected TJ integrity and BSCB disruption upon SCI.
TRPM7 is also up-regulated and regulates the integrity of TJ in OGD-induced bEnd.3 cells
To understand more about the molecular mechanism underlying TRPM7-mediated regulation in injured blood vessels, an in vitro OGD/reperfusion model with a bEnd.3 mouse brain microvessel endothelial cell line was used. Under the optimized condition of OGD/reperfusion injury to induce the maximal expression of the TRPM7 gene in bEnd.3 cells (data not shown), the TRPM7 messenger RNA (mRNA) and protein expression were increased in the bEnd.3 cells subjected to 6 h of OGD treatment followed by 0.5 h, 1 h, and 3 h of reperfusion (Fig. 3A, 3B). By Western blot analysis with anti-ZO-1 and occludin antibodies, the expression of the TJ proteins, ZO-1 and occludin, was decreased in the OGD/reperfusion injury-induced bEnd.3 cells (Fig. 3C, 3D; +OGD). However, CAR substantially attenuated the decrease in ZO-1 and occludin expression in the bEnd.3 cells after OGD/reperfusion injury (Fig. 3C, 3D; +OGD/CAR; mean ± SD; ZO-1, +OGD 0.6 ± 0.03 vs. +OGD/CAR 1.0 ± 0.04; occludin, +OGD 0.7 ± 0.07 vs. +OGD/CAR 0.9 ± 0.04, p < 0.05). Consistently, TEER was decreased in the OGD/reperfusion injury-induced bEnd.3 cells compared with the untreated control (Fig. 3D; +OGD). In parallel with these results, the decrease of TEER by OGD/reperfusion injury was significantly inhibited by CAR treatment (Fig. 3E; +OGD/CAR; mean ± SD; +OGD 35 ± 6 vs. +OGD/CAR 72 ± 7, p < 0.05). These results indicate that TRPM7 affects TJ integrity by increasing the degradation of TJ molecules.
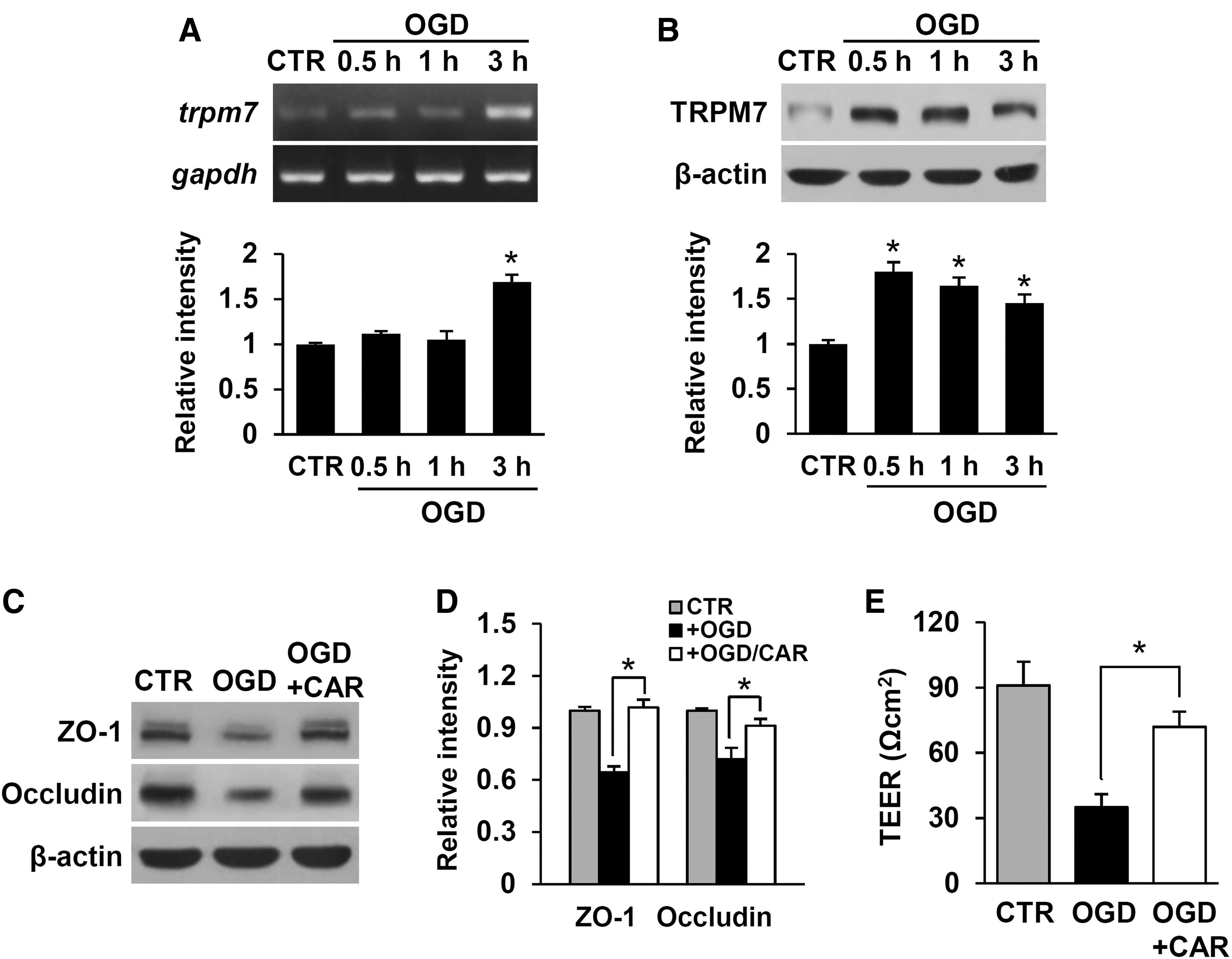
Transient receptor potential melastatin 7 (TRPM7) is up-regulated and contributes to the loss of tight junction protein in bEnd.3 cells at 0.5 h, 1 h, and 3 h reoxygenation after oxygen-glucose deprivation (OGD) for 6 h.
CAR inhibits leukocyte infiltration and the expression of inflammatory cytokines and mediators after injury
Since CAR prevented BSCB disruption following, the effect of CAR treatment on blood cell infiltration was examined by immunofluorescence staining and Western blot with neutrophil and macrophage. In general, neutrophils at 1 day and macrophages at 5 days infiltrated after SCI are known. 36 Thus, we examined by immunofluorescence staining for neutrophil (MPO) at 1 day and macrophage (ED-1) at 5 days after SCI. Immunofluorescence staining showed that numerous MPO- and ED-1-positive cells were observed in the dorsal column of the injured spinal cord. However, CAR treatment attenuated the infiltration of these cells compared with the vehicle control (Fig. 4A, 4B). Quantitative analysis of fluorescence intensity showed that CAR injection significantly reduced the MPO- and ED-1-immunoreactivity at 1 day and 5 days after injury, respectively (Fig. 4C; mean ± SD; MPO, Veh 1.0 ± 0.07 vs. CAR 0.6 ± 0.05; ED-1, Veh 1.0 ± 0.02 vs. CAR 0.5 ± 0.08, p < 0.05). Western blot and quantitative analyses also revealed that CAR treatment significantly decreased the level of ED-1 at 5 days after injury as compared with vehicle control (Fig. 4D) (mean ± SD; Veh 1.0 ± 0.07 vs. CAR 0.6 ± 0.06, p < 0.05).
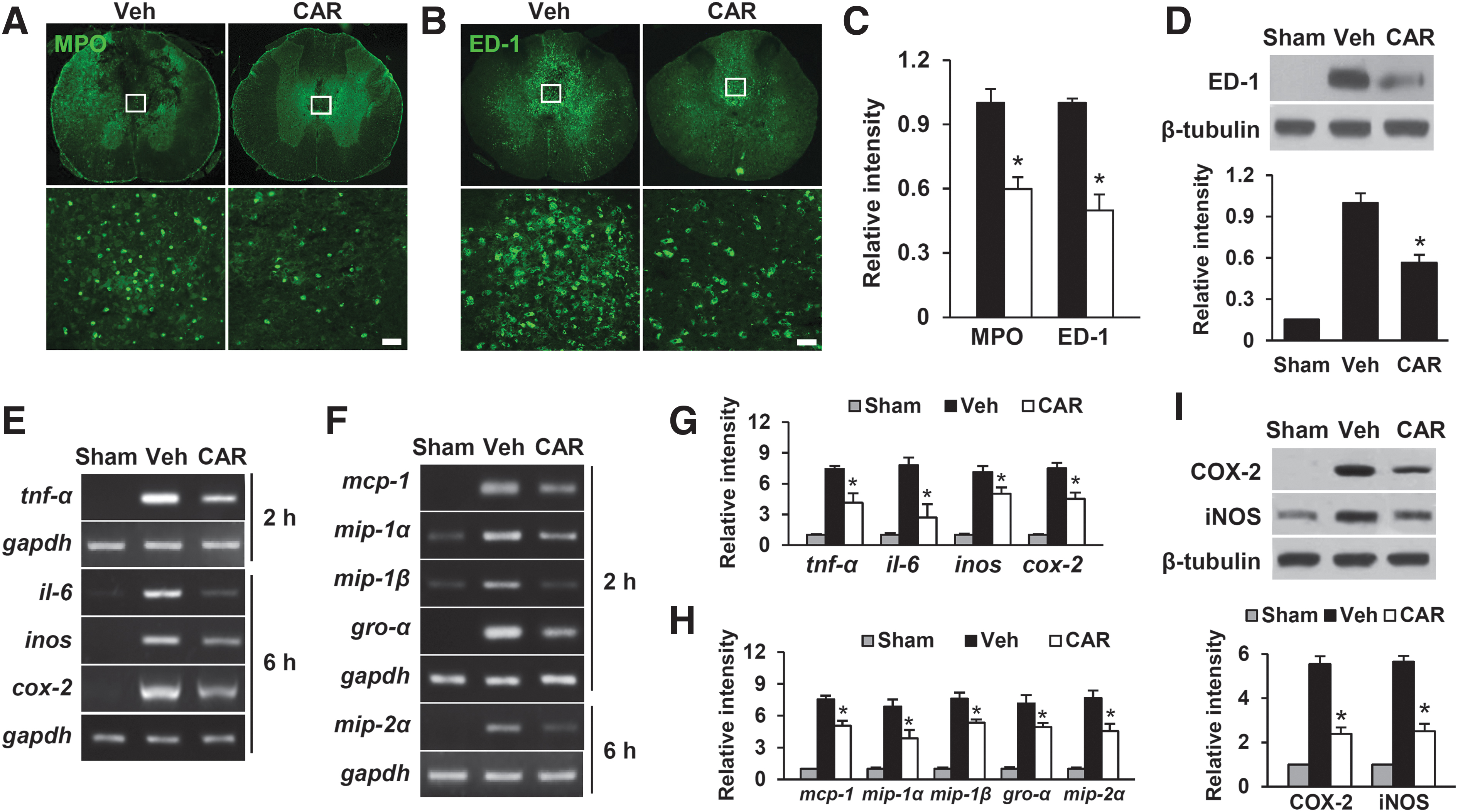
After spinal cord injury (SCI), carvacrol inhibits the infiltration of neutrophils and macrophages as well as the expression of cytokines and chemokines.
BSCB disruption and blood cell infiltration after SCI are known to mediate inflammatory responses, thereby contributing to the secondary injury cascade by releasing inflammatory mediators such as IL-6, TNF-α, COX-2, and iNOS, 20,37,38 and chemokines such as MCP-1, MIP-1β, Gro-α, and MIP-2α. 39 –42 Next, RT-PCR and Western blot tests were used to look at the effect of CAR on the expression of inflammatory mediators and chemokines after SCI. The results revealed that CAR treatment inhibited the increases in TNF-α, IL-6, COX-2, and iNOS mRNA levels after SCI (Fig. 4E and 4G) and suppressed the increases in the mRNA levels of MCP-1, MIP -1α, MIP -1β, GRO-α, and MIP-2α following injury (Fig. 4F and 4H). CAR also decreased COX-2 and iNOS protein levels at 1 day after injury as compared with vehicle control (Fig. 4I).
CAR inhibits the apoptotic cell death of neurons and oligodendrocytes
TRPM7 is known to play an important role in neuronal cell death in various neurodegenerative disease models, including SCI. 10,12,18 Further, after BSCB damage, infiltrated blood cells such as neutrophils and macrophages are known to produce inflammatory mediators such as cytokines and chemokines, which contribute to cell death. 36,43,44 Thus, based on our results showing that TRPM7 is involved in the BCSB disruption after SCI, we next examined whether CAR inhibits apoptotic cell death by attenuating inflammatory responses following BSCB disruption. As previously mentioned, 35 a massive loss of VMN was observed in the lesion area following injury and CAR treatment reduced VMN loss rostrally and caudally including the lesion epicenter as compared with the vehicle control (Fig. 5A, 5B). Next, to examine the apoptotic cell death, TUNEL staining was performed. TUNEL-positive cells were observed within the lesion site in the GM at 1 day and in the WM at 5 days. Our previous reports 29,45 demonstrate that by double-labeling, most TUNEL-positive cells in the GM are neurons at 1 day and oligodendrocytes in the WM at 5 days after injury.

Carvacrol alleviates apoptotic cell death of neurons and oligodendrocytes after spinal cord injury (SCI).
As shown in Figure 5C and 5D, CAR treatment resulted in a significant reduction in the number of TUNEL-positive cells when compared with the vehicle control in the GM at 1 day and the WM at 5 days (mean ± SD; 1 day, Veh 405 ± 36 vs. CAR 262 ± 4; 5 days, Veh 325 ± 12.2 vs. CAR 185 ± 8.3, p < 0.05). Western blot analysis also revealed that the level of cleaved caspase-3 was increased at 1 day and 5 days after injury, which was significantly reduced in CAR-treated group as compared with vehicle control (Fig. 5E, 5F; mean ± SD; 1 day, Veh 4.1 ± 0.4 vs. CAR 2.2 ± 0.4; 5 days, Veh 4.9 ± 0.4 vs. CAR 2.0 ± 0.3, p < 0.05). The cleaved caspase-3-positive cells in the WM at 5 days after SCI were CC1-positive oligodendrocytes, according to double immunofluorescence staining. CAR treatment significantly decreased the number of active caspase-3-positive cells in the WM at 5 days after SCI as compared with the vehicle control (Fig. 5G, 5H; mean ± SD; Veh 221 ± 16 vs. CAR 141 ± 11, p < 0.05). Thus, our findings show that the inhibition of TRPM7 with CAR prevents apoptotic cell death of neurons and oligodendrocytes following injury.
CAR improves functional recovery of hind limb after SCI
To assess the effect of CAR on functional recovery, rats were immediately injected with CAR (50 mg/kg, i.p.) after SCI and further injected once a day for 7 days. Functional recovery of hindlimb was assessed for 35 days after injury. As shown in Figure 6A, BBB locomotor scores were significantly increased in CAR-treated groups during 14-35 days after SCI when compared with those in vehicle-treated group (mean ± SEM; BBB scores at 35 days, CAR 12.1 ± 0.9 vs. Veh 9.1 ± 1.1, p < 0.05). In addition, the angle of incline after injury was also significantly higher in CAR-treated rats from 14 to 28 days compared with the vehicle-treated rats (Fig. 6B; mean ± SEM; at 28 days, CAR 66.3 ± 3.1 vs. Veh 55 ± 2.7, p < 0.05). At 35 days after injury, we also assessed grid walking test to determine the sensorimotor deficits and motor incoordination of injured rats. CAR treatment significantly reduced the number of footfalls on the grid walk as compared with the vehicle-treated rats (Fig. 6C; mean ± SEM; At 28 days, CAR 43 ± 1.9 vs. Veh 73.6 ± 4.2 %, p < 0.05). In footprint analysis, a little toe dragging was observed and the forelimb-hindlimb coordination was fairly constant in the CAR-treated group, whereas severe toe dragging and mismatched forelimb coordination were observed in the vehicle-treated rats (Fig. 6D).

Carvacrol improves functional recovery after spinal cord injury (SCI).
CAR reduces lesion volume and attenuates myelin and axon loss after injury
To assess the tissue loss with serial longitudinal sections at 35 days after injury, Cresyl violet staining and the measurement of the lesion volume were performed. Histological evaluation revealed that the cystic cavity by the tissue loss was extended rostrocaudally from the epicenter in injured spinal cord. Further, the tissue loss and the volume of lesion area were significantly reduced in CAR-treated group as compared with the vehicle control (Fig. 7A; mean ± SD; CAR 4.5 ± 0.3 vs. Veh 7.6 ± 0.9 mm3, p < 0.05). Next, we examined the extent of myelin loss after injury by Luxol fast blue staining. Extensive myelin loss near the lesion area was also observed in the injured spinal cord at 35 days after injury but not in the sham control (Fig. 7B; Veh); however, CAR treatment alleviated myelin loss as compared with the vehicle control (Fig. 7B; CAR).

Carvacrol decreases lesion volume and inhibits the loss of axon and myelin after spinal cord injury (SCI).
To examine the effect of CAR on axon loss after injury, immunohistochemistry with anti-NF200 and anti-5-HT antibodies was performed. As shown in Figure 7D and 7E, NF200-positive axons in dorsolateral and ventral funiculus including rubrospinal tract and vestibulospinal tract, respectively (Fig. 7C) was markedly decreased after injury when compared with sham control. However, the number of axons was significantly higher in CAR-treated group than in vehicle group (mean ± SD; rostral 2 mm, ventral funiculus, CAR 61 ± 5.3 vs. Veh 26 ± 5.4 %; dorsolateral funiculus, CAR 57 ± 5.3 vs. Veh 29 ± 6.0 %, p < 0.05). Further, 5-HT axon density in the lumbar spinal cord was significantly higher in CAR-treated group than in vehicle-treated group at 35 days after SCI (Fig. 7F). These findings imply that axon loss after SCI is alleviated by CAR treatment.
Discussion
In this study, we demonstrated that TRPM7, a calcium-mediated non-selective divalent cation channel, plays a critical role in BSCB disruption after SCI. Both in vitro and in vivo studies have shown that the expression of TRPM7 was increased in the endothelial cells consisting of BSCB after SCI. In addition, CAR suppressed BSCB disruption by preserving the TJ integrity after SCI, suggesting that blocking the TRPM7 can inhibit the disruption of the BSCB. We also found that CAR treatment significantly inhibited the infiltration of blood cells and production of proinflammatory cytokines and chemokines, thereby reduced inflammatory responses after SCI. Further, pathological consequences such as neuronal and oligodendroglial apoptosis and loss of axons and myelin after SCI were significantly alleviated by CAR, resulting in improved functional recovery. Thus, our results imply that TRPM7 can be a potential target for therapy after SCI.
In this study, we used CAR as an inhibitor of TRPM7 channel function. CAR, a food additive, is naturally occurring in various plants belonging to the family Lamiaceae and abundant in the essential oil fraction of oregano and thyme. 46 CAR has also been recognized as a neuroprotective agent through its anti-oxidative, anti-inflammatory, and anti-apoptotic effects in previous studies. 47 Recent studies also reported the neuroprotective effect of CAR in CNS disease animal models such as global ischemia model and neonatal hypoxic-ischemic brain injury by using 30-50 mg/kg of CAR. 48 -50 In this study, CAR was administrated via intraperitoneal injection at a dose of 50 mg/kg immediately after SCI and then same dose of CAR at 8 days after injury, and then further treated once a day for 7 days. When we preliminary tested the efficacy of various concentration of CAR (25, 50, and 100 mg/kg), 50 mg/kg of CAR significantly decreased Evans blue extravasation compared with vehicle control (data not shown). Therefore, we determined that 50 mg/kg was the optimal concentration of CAR in this study and any significant change in body weight was not observed during our experiment. In addition, neither significant side effect nor increasing of mortality by CAR treatment was also observed.
The breakdown of the BSCB after SCI induces changes in the spinal microenvironment by facilitating immune cells infiltration into the spinal cord and triggers the post-traumatic inflammatory response, which results in additional spinal cord damage. Matrix metallopeptidase 9 (MMP-9) up-regulation is linked to BSCB disruption because it degrades the basal components of BSCB, such as TJ proteins. Further, blood–brain barrier disruption was reduced in MMP-9 knockout mice after cerebral ischemia by reducing the protein degradation of ZO-1 relative to wild type mice. 52 Further, our recent studies have shown that inhibiting MMP-9 expression and activation significantly reduces BSCB disruption. 34,38,53 Specially, we recently found that JMJD3, histone H3K27 demethylase, plays important role as an epigenetic regulator in MMP-9 expression in vascular endothelial cells after SCI. 24 CAR treatment significantly prevented BSCB disruption, according to our findings. TJ proteins were also decreased after SCI, but CAR treatment retained these molecules considerably. However, it is not yet known how the inhibition of TRPM7 prevents BSCB disruption after SCI. The precise mechanism underlying TRPM7-mediated inhibition of BSCB disruption after SCI will be investigated in a future study.
Reactive oxygen species (ROS) plays critical roles in the apoptotic cell death after SCI. 35,54 After injury, The accumulation of free radicals such as superoxide anion (O2∙-), hydroxyl radical, and peroxynitrite triggered apoptotic cell death. 55 Further, it was reported that nitric oxide and peroxynitrite play important roles in the blood–brain barrier disruption and endothelial cell permeability. 56,57 Via the RhoA, phosphatidylinositol 3 kinase, and protein kinase B signaling pathways, superoxide anion has also been shown to control endothelial TJ proteins and improve blood–brain barrier permeability. 57 The article by Jiang and colleagues showed that carvacrol exhibits a neuroprotective effect after SCI by inhibiting oxidative stress and the endothelial nitric oxide synthase signaling pathway. 18 It was also reported that the application of 2-aminoethoxy-diphenyl borate, an inhibitor of TRPM7, or knockdown of TPRM7, protects the neurons from H2O2- mediated injury. 58 Thus, we cannot exclude the possibility that carvacrol has a direct neuroprotective effect or the possibility that it prevents BSCB breakdown via its antioxidant effects, although we focused on the role of TRPM7 in the disruption of BSCB after SCI in this study. Future studies will be performed to elucidate the precise mechanism underlying TRPM7-mediated cell death and BSCB disruption by ROS after SCI.
Neuroinflammation is known as one of major factors in the secondary injury after SCI. After SCI, BSCB disruption results in leukocyte infiltration into the parenchyma of spinal cord and It has been known that these cells release inflammatory cytokines and chemokines, causing cell death, tissue injury, and functional deficit after SCI. 59 -61 Several studies have shown that the inhibition of leukocyte infiltration, such as neutrophils and macrophages, reduces apoptotic cell death and enhances functional recovery after SCI. 62,63 In this study, we found that CAR mitigates neuronal and oligodendroglial apoptosis and enhances functional recovery in part by preventing BSCB disruption and immune cell infiltration after SCI. However, according to previous studies, TRPM7 is expressed in neurons or oligodendrocytes. 10 Further, our data showed that TRPM7 is expressed in neurons and oligodendrocytes in the uninjured and injured spinal cord. Thus, it is possible that the neuroprotective effect of CAR can be mediated by directly affecting the ion influx into neurons and oligodendrocytes, and this possibility will be examined in a future study.
In this study, we showed that CAR treatment effectively improves functional recovery after SCI. As known, the hindlimbs were paralyzed immediately after SCI. However, the differences in behavior tests between the vehicle control and CAR treated groups were evident at 14 to 35 days after SCI (Fig. 6). It is known that sparing of axons within the ventral funiculi (which contain the vestibulospinal tract) is related to control of posture, balance, and coarse by regulating axial and proximal muscles. The dorsolateral funiculi (which contain the rubrospinal tract) is also related to voluntary movements of arms and legs by controlling proximal and distal muscles. 35,64,65 Our data showed that the density of spared axons within the ventral and dorsolateral funiculi was higher in the CAR treated group than in the vehicle control (Fig. 7D, 7E). These observations agree with the rubrospinal tracts and vestibulospinal tracts were highly correlated with behavior tests such as BBB score (Fig. 6A) and inclined plane test (Fig. 6B) in SCI rats. In addition, the number of footfalls (mistakes) was lower than the vehicle control, suggesting fine motor control was improved by CAR treatment (Fig. 6C). Further, CAR treatment reduced degeneration of 5-HT positive serotonergic fivers in the ventral horn, which are correlated with locomotor function as 5-HT is one of the key neurotransmitters responsible for initiating locomotion. 35,66
Conclusions
TRPM7 is a non-selective cation ion channel and is known to play an important role in pathological conditions of central nervous system diseases, but its function has been mainly researched in neurons. Our findings suggest an important role for TRPM7 and its potential mechanisms in BSCB disruption, which might aid in gaining further understanding of BSCB disruption and cell death after SCI.
Footnotes
Authors' Contributions
C.S.P., J.Y.L., and T.Y.Y. designed the research. C.S.P, H.Y.C., and J.Y.L. performed the experiments and analyzed the data. C.S.P., J.Y.L., and T.Y.Y. wrote the manuscript.
Funding Information
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT, No. NRF-2019R1A2C2003750), by the Brain Research Program through the National Research Foundation of Korea funded by the Korean Government (MIST; 2017M37A1025369), and by a grant from Kyung Hee University in 2021 (KHU-20210142) to T.Y.Y. J.Y.L is a recipient of funding from the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT, No. NRF-2019R1A2C1005791).
Author Disclosure Statement
No competing financial interests exist.