
Abstract
Cellular homeostasis requires critical communications between the endoplasmic reticulum (ER) and mitochondria to maintain the viability of cells. This communication is mediated and maintained by the mitochondria-associated membranes and may be disrupted during acute traumatic brain injury (TBI), leading to structural and functional damage of neurons and supporting cells. To test this hypothesis, we subjected male C57BL/6 mice to severe TBI (sTBI) using a controlled cortical impact device. We analyzed the physical ER-mitochondrion contacts in the perilesional cortex using transmission electron microscopy, Western blot, and immunofluorescence. We specifically measured changes in the production of reactive oxygen species (ROS) in mitochondria, the unfolded protein response (UPR), the neuroinflammatory response, and ER stress-mediated apoptosis in the traumatic injured cerebral tissue. A modified neurological severity score was used to evaluate neurological function in the sTBI mice. We found that sTBI induced significant reorganizations of mitochondria-associated ER membranes (MAMs) in the cerebral cortex within the first 24 h post-injury. This ER-mitochondrion coupling was enhanced, reaching its peak level at 6 h post-sTBI. This enhanced coupling correlated closely with increases in the expression of the Ca2+ regulatory proteins (inositol 1,4,5-trisphosphate receptor type 1 [IP3R1], voltage-dependent anion channel 1 [VDAC1], glucose-regulated protein 75 [GRP75], Sigma 1 receptor [Sigma-1R]), production of ROS, degree of ER stress, levels of UPR, and release of proinflammatory cytokines. Further, the neurological function of sTBI mice was significantly improved by silencing the gene for the ER-mitochondrion tethering factor PACS2, restoring the IP3R1-GRP75-VDAC1 axis of Ca2+ regulation, alleviating mitochondria-derived oxidative stress, suppressing inflammatory response through the PERK/eIF2α/ATF4/CHOP pathway, and inhibiting ER stress and associated apoptosis. These results indicate that dysfunctional ER-mitochondrion coupling might be primarily involved in the neuronal apoptosis and neurological deficits, and modulating the ER-mitochondrion crosstalk might be a novel therapeutic strategy for sTBI.
Introduction
Traumatic brain injury (TBI) remains a leading cause of death and disability. 1,2 Despite extensive basic and clinical research on TBI in the past 150 years, promising pre-clinical data have not been translated into successful development of new therapeutic to improve the outcomes of TBI and no effective pharmacological interventions are available to date. 3 TBI consists of primary mechanical insults and secondary injuries. While the primary insults are often transient, the secondary injuries evolve over hours to days, leading to neuronal death in areas that are beyond the site of a traumatic impact. 4 These secondary injuries provide a window of opportunity for therapeutic intervention. 5
Dysfunctional inter-organelle communications of neurons and supporting cells have emerged as critical events that lead secondary brain injuries during acute TBI. 6 -8 The communication between the endoplasmic reticulum (ER) and mitochondria is essential for cellular homeostasis and its disruption play a key role in the cellular injures developed in the traumatically injured brain. 9 –12 It has been demonstrated that ER interacts with mitochondria via mitochondria-associated ER membranes (MAMs), which serve as scaffold for rapid exchange of biological molecules between the two organelles to maintain cellular viability. 13 MAMs are dynamic structures that are highly conserved across eukaryotic phyla and are characterized by a unique lipid profile and the expression of a specific group of proteins involved in Ca2+ signaling, phospholipid biosynthesis, protein folding, membrane tethering, and the transfer of stress signals. 11,14,15 MAMs are therefore crucial for processing ER stress (accumulation of unfolded proteins in the ER lumen), initiating the unfolded protein response (UPR) for cellular survival. 16 MAMs have recently been shown to contribute substantially to the pathologies of obesity, insulin resistance, aging, tumorigenesis, axon regeneration, and neurodegenerative disorders. 17 –21 Dysfunctions of ER and mitochondria have been investigated individually in TBI, 22 -24 but their communications remain poorly understood for the development of cerebral injuries secondary to sTBI.
Here, we report the results from a study designed to investigate changes in MAMs of neurons in the cerebral cortex induced by sTBI in mouse models. Our study identified multiple changes related to the ER-mitochondrial coupling during acute TBI, including the expression of MAM-resident Ca2+ regulatory proteins, mitochondria-derived oxidative stress, ER stress, UPR signaling, and neuroinflammatory response. These results indicate that dysfunctional ER-mitochondrial coupling during acute injury contributes to neuronal apoptosis and neurological deficits in mice subjected to sTBI, and identified key targets for regulating these changes as new therapeutics to improve outcomes of sTBI.
Methods
The mouse experiments performed for this study were approved by the Small Animal Protection Board of Tianjin Medical University.
Animals
Adult male C57BL/6 mice weighing 22-25 g (8-10 weeks old) at the time of surgery were purchased from the Experimental Animal Laboratories of the Academy of Military Medical Sciences (Beijing, China). They were housed individually in a temperature- (20 ± 2°C) and humidity-controlled (55 ± 5%) vivarium and maintained on a standard 12-h light/dark cycle (7:00 a.m. to 7:00 p.m) with access to food and water ad libitum. Efforts were made to minimize the number of mice used and their suffering. In all experiments, data were obtained by investigators blinded to the experimental design.
Experimental design
Experiment 1
We investigated the dynamic changes in the physical contact between ER and mitochondria in neurons from the perilesional cortex after sTBI using transmission electron microscopy (TEM). Thirty-six mice were randomly assigned to sTBI or sham surgery. The TBI mice were examined at 1, 3, 6, 12, and 24 h post-injury.
Experiment 2
We investigated changes in the expression of MAM-resident proteins in purified mitochondria and the production of reactive oxygen species in lysates from the tissue collected from the perilesional cortex at 1, 3, 6, 12, and 24 h post-injury. Mice were randomly allocated to experimental groups, as discussed in experiment 1. Brain tissue samples from sTBI and sham mice were examined for the expressions of the ER-mitochondrion tethering proteins phosphofurin acidic cluster sorting protein 2 (PACS2) and mitofusin-2 (MFN2) using immunofluorescence and immunoblots. We also examined the expression of inositol 1,4,5-trisphosphate receptor type 1 (IP3R1), glucose-regulated protein 75 (GRP75), voltage-dependent anion channel 1 (VDAC1), and Sigma 1 receptor (Sigma-1R) because they are MAMs-resident proteins that have been demonstrated to regulate Ca2+ flux from ER to mitochondria. 25 reactive oxygen species (ROS) levels in mitochondria were detected by MitoSOX staining.
Experiment 3
MAMs are crucial not only for the efficient transfer of Ca2+ from the ER to mitochondria, but also for regulating ER stress, the UPR, and neuroinflammatory responses16, 26-28. We studied ER stress, UPR signaling, and neuroinflammatory response in whole–tissue lysates from the perilesional cortex collected from TBI mice at 1, 3, 6, 12, and 24 h post-injury. We used immunoblots to detect the expression of GRP78, phosphorylated protein kinase (PKR)-like ER kinase (p-PERK), PERK, p-eIF2α, eIF2α, activating transcription factor 4 (ATF4), cleaved interleukin-1β (IL-1β), and tumor necrosis factor alpha (TNFα) and quantified the expression levels using optical densitometry.
Experiment 4
We evaluated the levels of ER stress-mediated apoptosis in mice subjected to sTBI or sham surgery in tissue lysates from the perilesional cortex collected at 6, 12, 24, and 72 h post-injury. We detected the expressions of the surrogate markers for the ER stress-mediated apoptosis using immunoblots, including the cleavage of caspase-12, caspase-3, Poly(ADP-Ribose) Polymerase 1 (PARP1), C/EBP homologous protein (CHOP), B-cell lymphoma 2 (Bcl-2), Bcl-2-associated X (BAX), and cytochrome C (Cytc).
Experiment 5
We studied the effects of silencing the PACS2 gene on the expression of MAM-resident proteins, production of ROS, levels of ER stress and UPR activation, and neuroinflammatory response in lysates of cerebral tissue collected from the perilesional cortex of TBI mice at 6 h post-injury and the same region from sham mice. The experimental mice received PACS2 small interfering RNA (siRNA) 48 h before TBI. The control mice underwent sham surgery or subjected to TBI, but receiving control NC siRNA. These mice were evaluated for the expression of PACS2, MFN2, IP3R1, GRP75, VDAC1, Sigma-1R in purified mitochondria. They also were examined for the expression of GRP78, p-PERK, PERK, p-eIF2α, eIF2α, ATF4, cleaved IL-1β, and TNFα in whole–tissue lysates from the perilesional cortex using immunoblots. Levels of ROS was detected by MitoSOX staining in cells isolated from brain tissue.
The mice were evaluated for neurological functions at the baseline and at Days 1, 3, 5, 7, and 14 post-injury. At 72 h post-injury, brain tissues collected from the perilesional cortex of these mice were examined for the cleavage of caspase-12 and the expression of CHOP, Bcl-2, BAX, and Cytc using immunoblots. The terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL) assay and immunofluorescence staining were also performed in these mice at 72 h after injury.
Severe traumatic brain injury model
Mice were subjected to injury induced by the controlled cortical impact (CCI). This model has been extensively characterized and broadly used as a pre-clinical model of head injury. 29 -31 In brief, a mouse was anesthetized with 10% chloral hydrate and morphine was used as pre-surgery analgesic, placed in a stereotaxic apparatus to drill a 4.0-mm hole through the right parietal bone midway between bregma and lambda in the parietal bone centered at 2 mm lateral from the sagittal suture with the dura matter intact, and exposed to CCI using a digital electromagnetic CCI device (eCCI Model 6.3; Custom Design, Richmond, VA) at a depth of 2.5 mm and a velocity of 5 m/sec over a period of 200 msec. The incision was closed immediately following injury, and the mice were placed in heated cages to allow recovery from anesthesia at room temperature (RT). Figure 1 shows the hematoxylin and eosin staining of the cerebral cortex at the CCI impact site 24 h after injury, showing severe cerebral injury.

Hematoxylin and eosin–stained coronal sections in the cerebral cortex 24 h after controlled cortical impact. This photomicrograph shows the severe injury. Color image is available online.
Transmission electron microscopy and MAM quantification
Tissue (1 mm × 1 mm) samples were obtained from the perilesional cortex of TBI and sham mice and fixed in a mixture of 2% paraformaldehyde and 2% glutaraldehyde in 0.1 M phosphate-buffered saline (PBS; pH 7.4) overnight at 4°C for 1 h. The tissue blocks were washed in 0.1 M phosphate buffer (pH 7.4) and post-fixed in 1% osmium tetroxide in the same buffer for 30 min at RT. The specimens were dehydrated through a graded series of ethanol and embedded in Epon resin to make serial sections on ultratome. The tissue sections were stained with uranyl acetate and lead citrate and examined under TEM set at 80 kV.
Physical contacts between ER and mitochondria and mitochondrial morphologies were observed and quantified using TEM, as previously described. 32 Briefly, TEM images were acquired from mice (60 images/mouse, n = 6/group) at × 6800 magnification and analyzed using ImageJ (National Institutes of Health, Bethesda, MD). We delineated the mitochondria and ER membranes using the free-hand tool. Two independent investigators blinded to the experimental design calculated the ratio of ER adjacent to mitochondria to mitochondrial perimeter, and the total number and area of mitochondria.
Immunofluorescence
At designated time-points, mice were euthanized under anesthesia and analgesic as discussed in the previous section and immediately perfused with PBS through cardiac puncture, followed by 4% paraformaldehyde. The brain was dissected and embedded in OCT medium (Sakura, Oakland, CA). Coronal sections (8-μm thickness) were cut using cryostat at -20°C and imprinted on poly-L-lysine-coated slides. The sections were stained for the neuronal marker NeuN, astrocyte marker glial fibrillary acidic protein (GFAP), and the ER-mitochondrion contact tethering molecules PACS2 and MFN2.
Specifically, brain sections were fixed with 2% paraformaldehyde lysine periodate, rinsed three times with PBS (pH 7.4), and blocked with 1% normal donkey serum in PBS containing 0.1% Triton X-100 PBS with Tween (PBST) at RT for 1 h. They were then incubated with a rabbit anti-PACS-2 antibody diluted at 1:1000 (Abcam, Cambridge, MA), a rabbit anti- MFN2 antibody diluted at 1:1000 (Abcam), a mouse anti-NeuN antibody diluted 1:100 (Cell Signaling Technology, Danvers, MA), or a mouse anti-GFAP antibody diluted at 1:1000 (Abcam, Cambridge, MA) in PBST containing 1% normal donkey serum at 4°C overnight, followed by extensive washing with PBS. Finally, the sections were incubated with Alexa Fluor-conjugated anti-mouse or anti-rabbit immunoglobulin G (IgG; 1:1000, Invitrogen, Grand Island, NY) for 3 h at RT. The nuclei were counterstained with Hoechst for 5 min and isotype IgGs were used as negative controls. Fluorescent images were captured under a fluorescence microscope (Olympus IX81, Tokyo, Japan), and the data were analyzed from 15 randomly selected microscopic fields (five fields per section × three sections per mouse) using the ImageJ program (National Institutes of Health).
Protein extraction and immunoblots
Mice were sacrificed as described in the section above after transcardiac perfusion of cold PBS to remove blood at designated time-points. The perilesional cortex from the ipsilateral hemisphere to injury were homogenized in ice-cold RIPA buffer (Beyotime) containing phenylmethylsulfonyl fluoride (1 mM final) for 30 min and then centrifuged for 10 min at 12,000 rpm (4℃). The supernatants were boiled in 4 x SDS sample buffer at 95℃ for 10 min. The total protein content was determined by the BCA protein assay kit (Thermo). The solubilized proteins (8 μg per lane) were separated by SDS/PAGE and transferred to PVDF membranes (Roche, Canada). After blocking with 5% nonfat dry milk in Tris-buffered saline (TBS) for 2 h at RT, the membrane wase incubated overnight at 4℃ with primary antibodies listed in Table 1, rinsed with TBS, incubated with the appropriate horseradish peroxidase–conjugated secondary IgG for 1 h at RT and then developed with the ECL system (Millipore, Billerica, MA). Protein expression was quantified by ImageJ (National Institutes of Health) according to the mean pixel density of each protein band. β-actin was used as the loading control in all immunoblots. The routine quality control and validation measures include: 1) all antibodies were tested with serial dilutions and 2) when it was possible, second antibodies from different species or clones were tested to validate the results and to ensure specificity (Supplementary Figs. S1–S3).
Antibodies for Western Blot
GRP, glucose-regulated protein; CHOP, C/EBP homologous protein; PACS2, phosphofurin acidic cluster sorting protein 2; MFN2, mitofusin-2; IP3R1, inositol 1,4,5-trisphosphate receptor type 1; IL, interleukin; TNF, tumor necrosis factor; IgG, immunoglobulin G.
Mitochondria isolation
The MAM regulatory proteins PACS2, MFN2, IP3R1, GRP75, VDAC1, Sigma-1R were assessed in purified mitochondria, as previously described. 32,33 Tissues from the perilesional cortex of sTBI and control mice were dissected, weighed immediately, and washed three times in sodium chloride solution, cut into small blocks, and homogenized in Lysis Buffer supplemented with Protease Inhibitor Solution. They were transferred to 1.5 mL pipet and incubate on an end-over-end shaker for 10 min at 4°C. The brain homogenates were centrifuged at 1000 × g for 10 min at 4℃ to collect the supernatant, which was centrifuged for 10 min at 6000 × g at 4°C to collect mitochondrial pellets. The supernatant also was collected and fractionated for ER granules by centrifuging the supernatant at 100,000 g for 60 min. Protein concentration was determined by BCA, and 25 μg of protein was separated by SDS- PAGE and immunoblotted (Supplementary Fig. S4).
Reactive oxygen species
A commercial MitoSOX-based assay was used to ROS production according to the manufacturer's instructions (Thermo Fisher, Waltham, MA) with minor modifications as described in previous studies. 34 -36 Briefly, tissue collected from the perilesional cortex were dispersed into a single-cell suspension and washed with 10 mM PBS twice. A 5 mM Mito SOXTX reagent stock solution in HBSS/Ca/Mg buffer was diluted to a working solution containing 5 μM MitoSOX reagent, in which cells were incubated for 10 min at 37°C. Intracellular fluorescence was measured. All experimental procedures were performed in the dark.
siRNA Transfection
siRNA Transfection was performed in vivo according to the method of Zhao and colleagues. 37 To knockdown the PACS2 gene, 1.32 μg/5 μL of PACS-2 siRNA (Sangon Biotech, Shanghai, China) or the control NC siRNA (Sangon Biotech) were diluted in an equal volume of EntransterTM-In Vivo Transfection Reagent (Engreen, Beijing, China). The solution was injected intracerebroventricularly into mice through a 1-mm cranial burr hole 48 h prior to TBI. 38 A 30-gauge needle on a Hamilton syringe was implanted into the lateral ventricle the following stereotactic coordinates: 1.5 mm posterior to bregma, 1.0 mm right lateral to the midline, 2 mm in depth, with an injection speed of 1 μL/min (total volume = 5 μL). The transfection rate is approximately 85 ± 5%.
Modified neurological severity scores
The modified neurological severity score (mNSS) was used to evaluate neurological function, as described previously. 39 Neurological assessments were performed at baseline before the injury and at Days 1, 3, 5, 7, and 14 post-injury. This score system includes motor, sensory, reflex, and balance tests. These scores were used to ensure the relative uniformity in injury severity and to compare neurological impairments among mice receiving different treatments. The tests were performed by two independent observers who were blinded to the experimental conditions and treatments.
TUNEL assay
The TUNEL assay was used to detect the DNA fragmentation of apoptotic cells in the perilesional cortex of the mouse brain at 72 h after TBI cells 40 using the In Situ Cell Death Detection Kit, POD (Roche, Germany), according to the manufacturer's instructions.
Statistical analysis
The data are presented as the mean ± standard deviation of the mean (SD) analyzed using Prism 9.3.0 (GraphPad Software, San Diego, CA). Parameters were examined by Kolmogorov-Smirnov to test the normality of data, and then compared by analysis of variance followed by Tukey's multiple comparisons test. Pearson's correlation coefficients (r) were calculated to assess the strength of relationships; p values <0.05 were considered statistically significant.
Results
ER-mitochondrion contacts underwent dynamic changes in neurons
The ER and mitochondria interact to form dynamic contact sites in cells and this interaction is responsible for integrating diverse intracellular functions such as Ca2+ homeostasis, oxidative stress, ER stress, inflammation, and survival. 19,41 Therefore, we hypothesized that ER-mitochondrion contacts could induce characteristic changes in neurons of cerebral cortex to orient intracellular signals and regulate cell survival after sTBI.
We first identified the physical interaction between ER and mitochondria on TEM sections of the perilesional cortex collected at the baseline and at 1, 3, 6, 12, and 24 h post-injury. Neuronal cell bodies were distinguished from glial cell bodies using previously described criteria. 24,42 We observed that mitochondria that had moderately dense matrixes and a regular cristae architecture at the baseline (Fig. 2a, 2g, 2m) underwent significant structural changes and reduced their numbers in neurons immediately after injury (Fig. 2b-f, 2n-r). Some exhibited disorganized morphologies with swelling and an electron-lucent matrix. At 6 h post injury, mitochondria in neurons were severely swollen and had inner membrane-associated dense granular inclusions, which is a distinct mitochondrial feature of calcium overload. These mitochondria had a markedly high degree of apposition to the ER (Fig. 2d,2 j, 2p). The quantitative analysis of cerebral sections showed that the proportion of the ER in close contact with mitochondria to the mitochondrial perimeter began to increase at 1 h, reached the peak level at 6 h, and returned to slightly above baseline 24 h post injury (Fig. 2B). Mitochondria in neurons was significantly reduced in numbers (Fig. 2C), but increased in areas measured in the brain tissue collected at 6 h post-injury (Fig. 2D), indicating swollen mitochondria, which resulted in enlarged MAMs and their close contact with the ER.
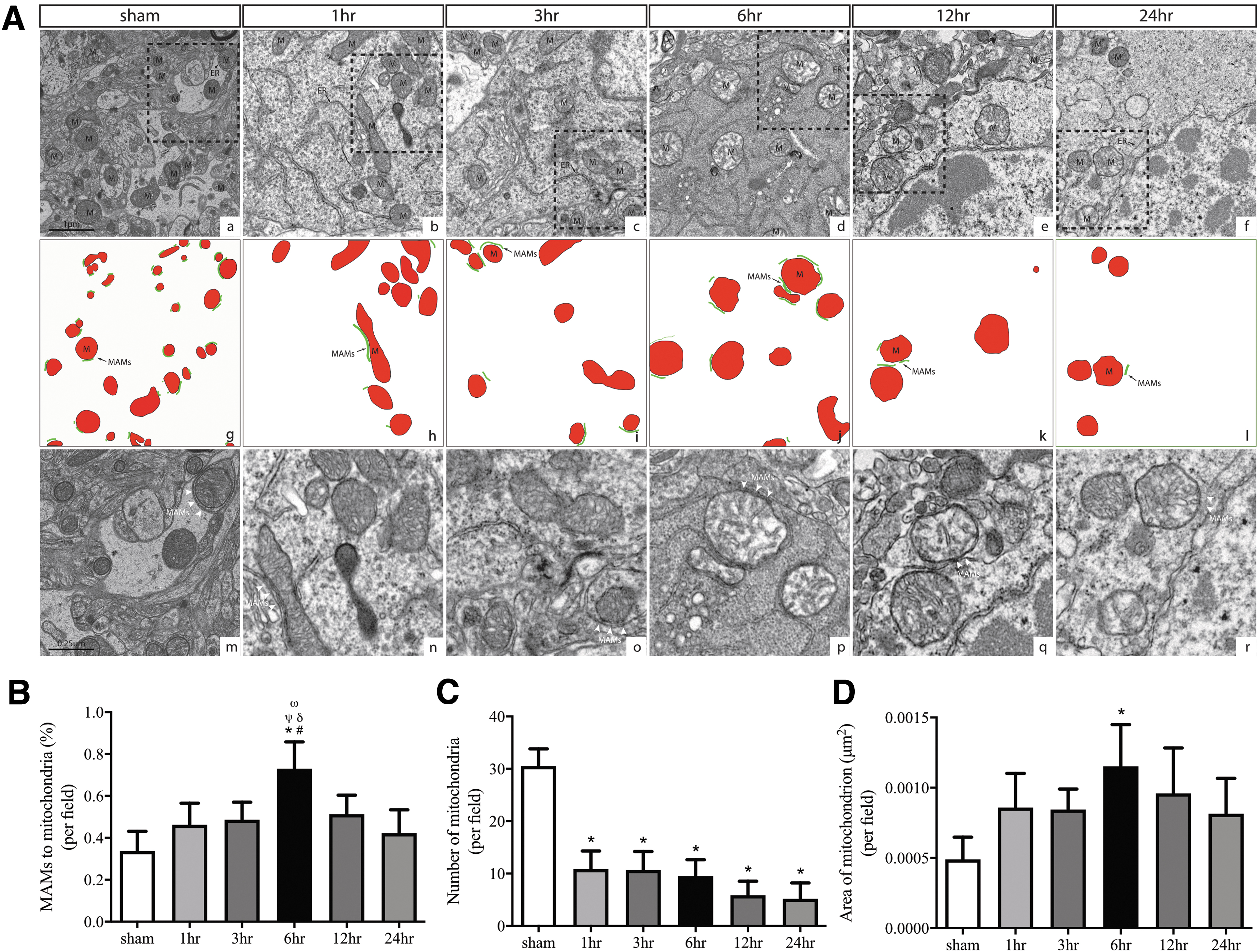
Traumatic brain injury (TBI)–induced the mitochondria-associated membrane (MAM) formation and changed mitochondrial morphologies during acute severe TBI in mice.
Severe TBI increased expression of MAM-resident proteins and mitochondrial oxidative stress
The detection of more ER-mitochondrion physical contacts suggests structural changes in mitochondria-associated ER membranes (MAMs), where the ER-mitochondrial communication occurs, 10 during acute TBI. We thus measured the expression of MEM residential proteins, which maintain the ER-mitochondrial communications through the tether proteins PACS2 and MFN2. 43,44 The expression of PACS2 and MFN2 was increased rapidly after injury, reaching the peak levels at 6 h post-injury, and declined gradually thereafter to the baseline by 24 h (Fig. 3B, 3C). The findings from immunoblots were validated by immunofluorescence staining of PACS2 in neurons identified by the neuronal marker NeuN and in astrocytes identified by a GFAP antibody in the ipsilateral cerebral cortex at the highest levels 6 h after TBI, but ER was more frequently attached to mitochondria in neurons (Fig. 3A).
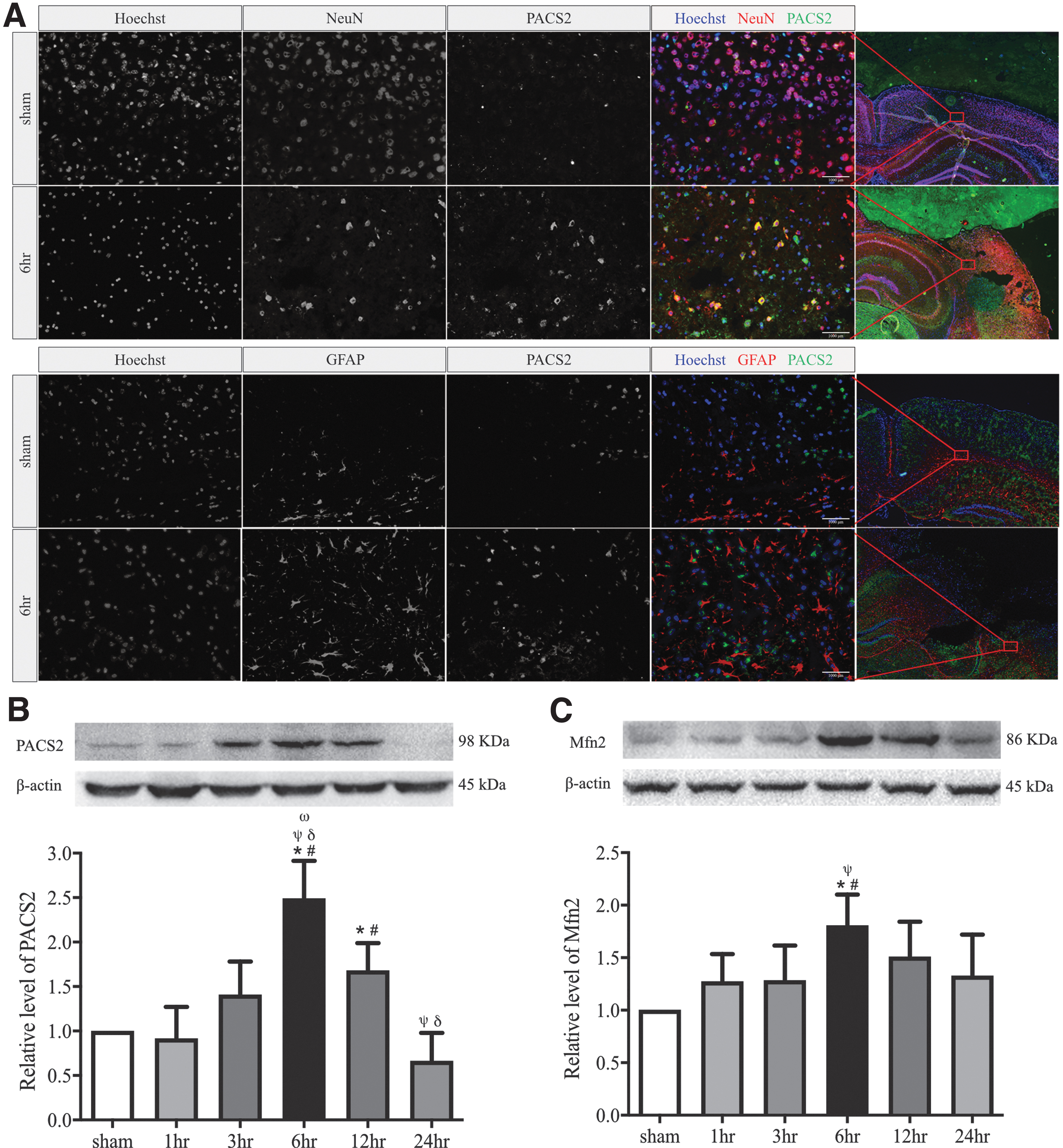
The dynamic expression of endoplasmic reticulum-mitochondrion tethering proteins in the pericontusional cortex of severe traumatic brain injury mice.
In addition to PACS2 and MFN2, we also detected enhanced expression of inositol 1,4,5-trisphosphate (IP3) receptor type 1 (IP3R1) which is a component of the ER-mitochondrion tethering complex located on the surface of ER membrane), the voltage-dependent anion channel 1 (VDAC), which is expression on the outer membrane of mitochondria but linked to IP3R1 through the chaperone GRP75, 45 and Sigma-1R, which is a Ca2+-sensitive and ligand-operated receptor chaperone that regulates ER-mitochondrion inter-organelle Ca2+ signaling. The enhanced expression was in parallel with the increase in ROS production at 6 h after injury, consistent with the increase in the MAM formation (Fig. 4A-C). These results support our TEM observation of increasing ER-mitochondrion contacts during acute sTBI and further suggest that molecules involving Ca2+ transport are changed specifically at MEMs and that the ER-mitochondrial communication contribute to oxidative stress.
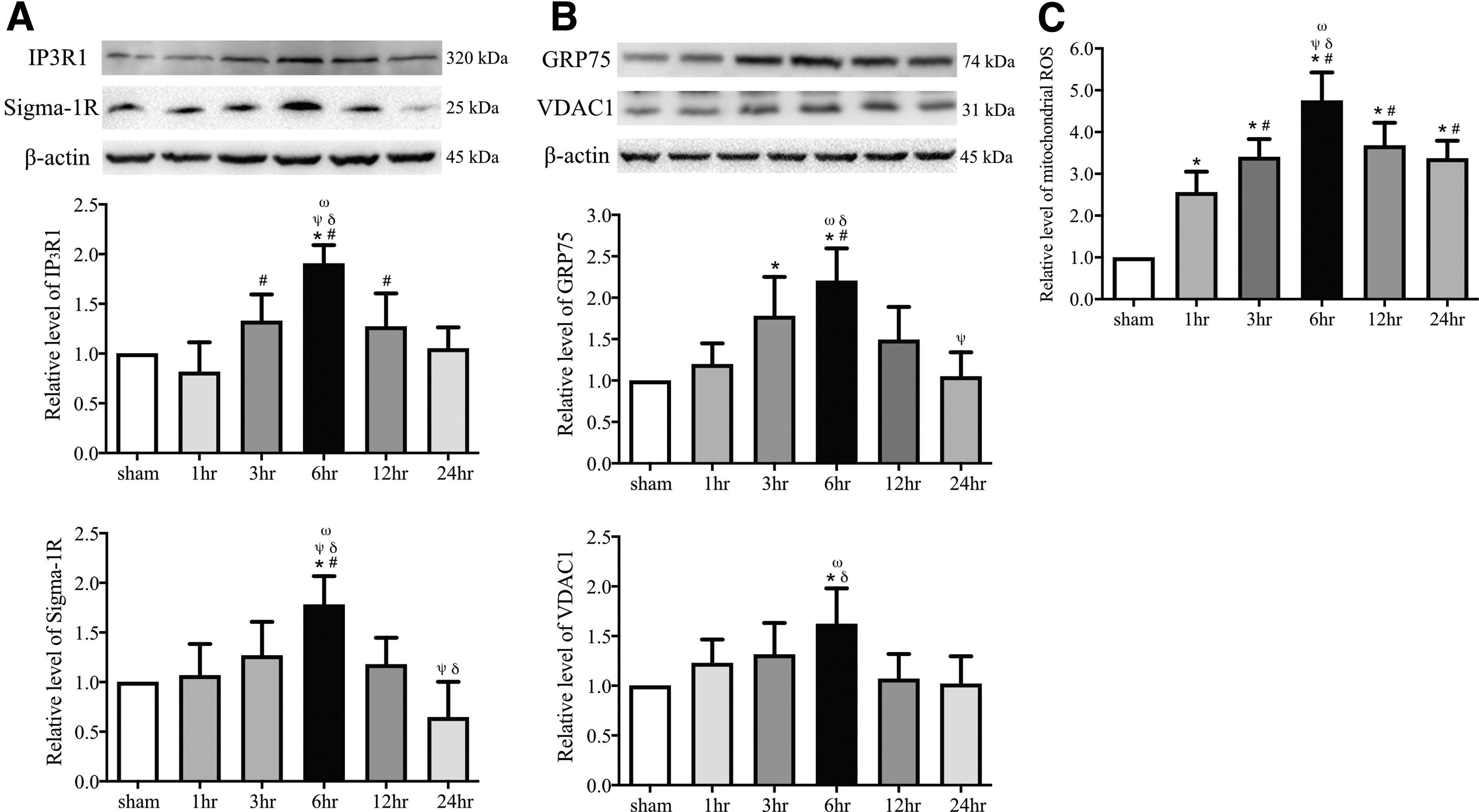
Traumatic brain injury (TBI) alters the expression of mitochondria-associated membrane–resident Ca2+ regulatory proteins and the rate of reactive oxygen species (ROS) production in the pericontusional cortex from severe TBI and sham mice.
ER stress, UPR signaling, and neuroinflammatory responses were initiated
It has been extensively shown that MAMs are crucial for regulating not only the mitochondrial and ER calcium homeostasis, efficient transfer of Ca2+, but also for ER stress, the UPR, and neuroinflammatory responses. 16,26 -28 The latter suggests that MAMs transfer stress signals from the ER to mitochondria by engaging the UPR, 12 most notably under the condition of loss of ER proteostasis. Since the RNA-dependent PERK, a key ER stress sensor, is located in the ER membrane as a component of the ER-mitochondrion tethering complex, 46,47 changes in crosstalk between ER and mitochondria are likely a part of ER stress that contributes to neuroinflammation. We analyzed the expression profiles of ER stress-related molecules and proinflammatory cytokines in the pericontusional cortex of TBI mice at different time-points up to 24 h post-injury. The expression of the 78 kDa glucose-regulated protein (GRP78, also referred to as BiP), which is a common marker for ER stress, was significantly elevated at 3 h and peaked at 6 h following TBI (Fig. 5D; p < 0.05), while the levels of p-PERK/PERK, p-eIF2α/eIF2α, and ATF4 exhibited comparable dynamic changes with peak expression at 6 h (Fig. 5A-C; p < 0.05). The production of the procytokines cleaved IL-1β and TNFα also peaked at 6 h (Fig. 5E, 5F; p < 0.05). Pearson's correlation analysis showed that the PACS2 expression was significantly corrected with the expression of MFN2, IP3R1, VDAC1, Sigma-1R, GRP75, ROS, GRP78, p-PERK/PERK, p-eIF2α/eIF2α, ATF4, cleaved IL-1β, and TNFα within 24 h of injury (Fig. 6; p < 0.05).
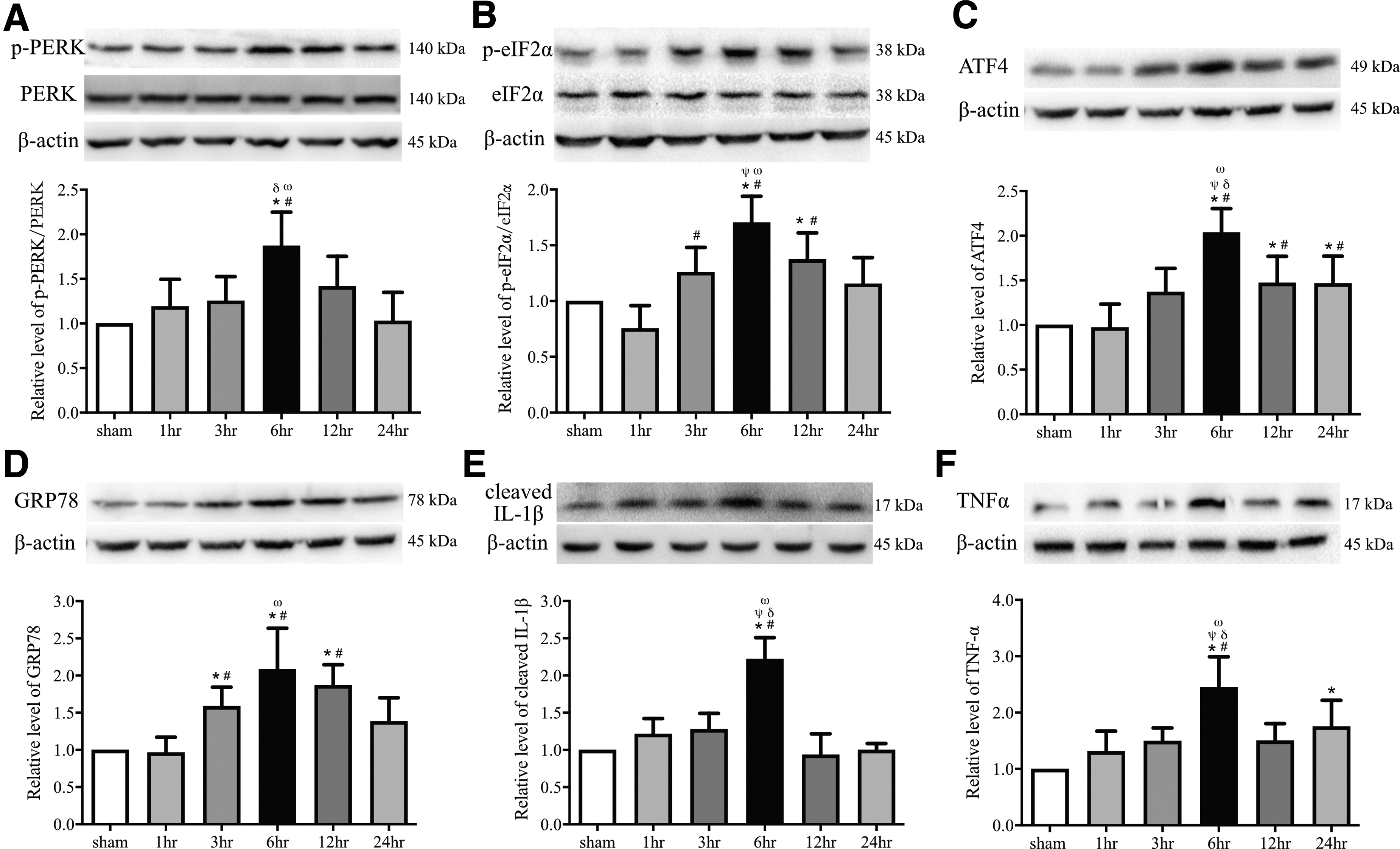
Endoplasmic reticulum stress, unfolded protein response activation, and neuroinflammation in the pericontusional cortex after traumatic brain injury.
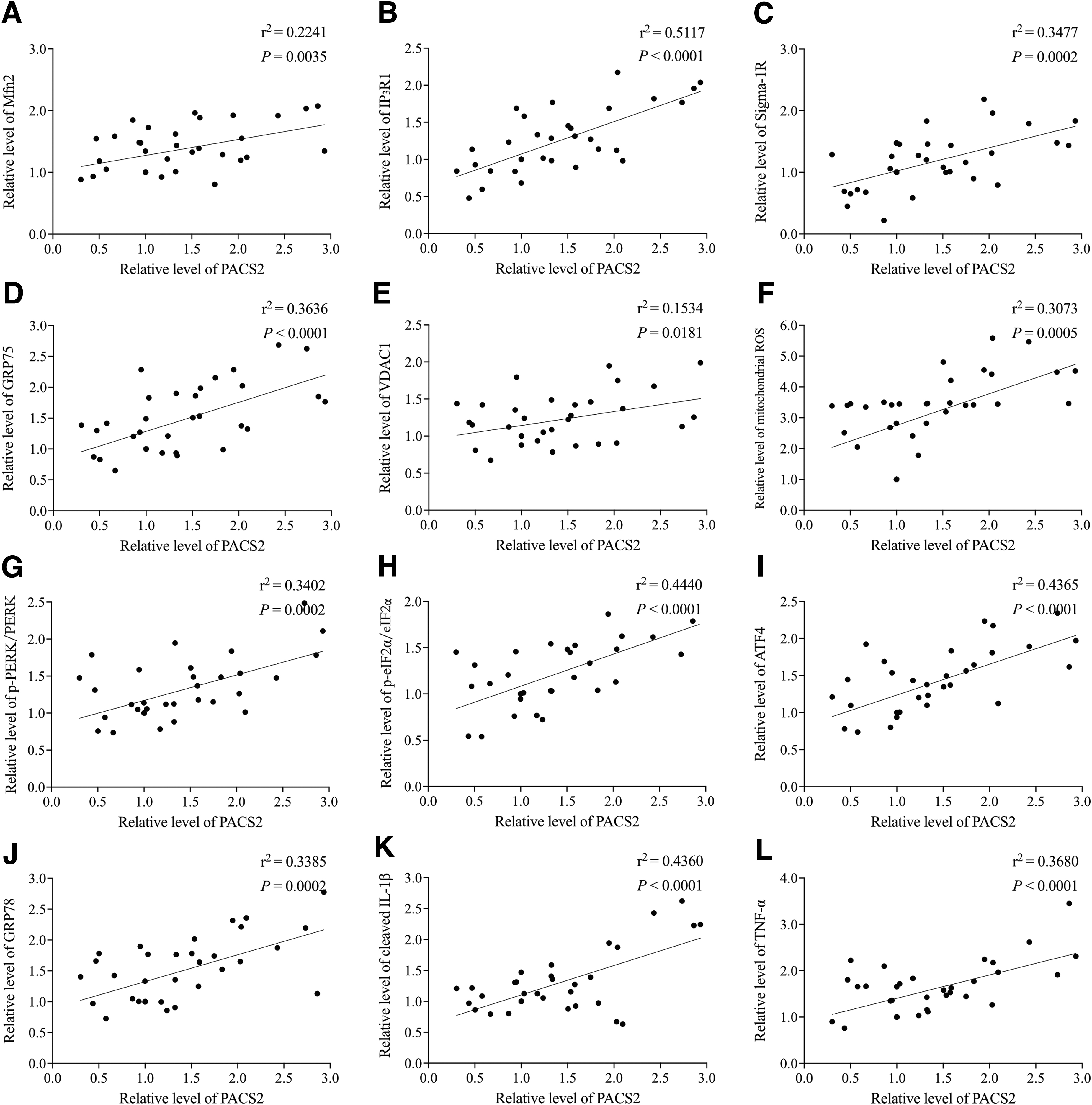
Correlation among the expression of phosphofurin acidic cluster sorting protein 2 (PACS2) with mitochondria-associated membrane formation, endoplasmic reticulum-mitochondrion Ca2+ regulation, oxidative stress, unfolded protein response signaling, and inflammatory responses in samples collected within 24 h post-injury. The correction was performed using Pearson's correlation analyses.
Together, our results suggest that the enhanced ER and mitochondria coupling in the acute stage of sTBI is associated with increased expression of MAM resident proteins, especially those involved in transporting calcium from the ER to mitochondria, ROS production, ER stress and UPR signaling, which together can enhance TBI-induced inflammatory response.
ER stress-mediated apoptosis increased following acute TBI
ER stress-induced apoptosis involves multiple processes that trigger the PERK-eIF2α-ATF4 branch of the UPR, upregulate CHOP expression, and activate caspase-12. 10,30,48 We found that the expression of cleaved caspase-12, cleaved caspase-3, cleaved PARP1, CHOP, Bcl-2, Bax, and Cytc started to increase immediately after the ER-mitochondrion crosstalk became more active at 6 h post-injury, reaching the peak level at 72 h, whereas the Bcl-2/Bax ratio decreased significantly to its lowest level (Fig. 7A-F). These data demonstrated that enhanced ER-mitochondrion communication preceded ER stress-induced apoptosis.
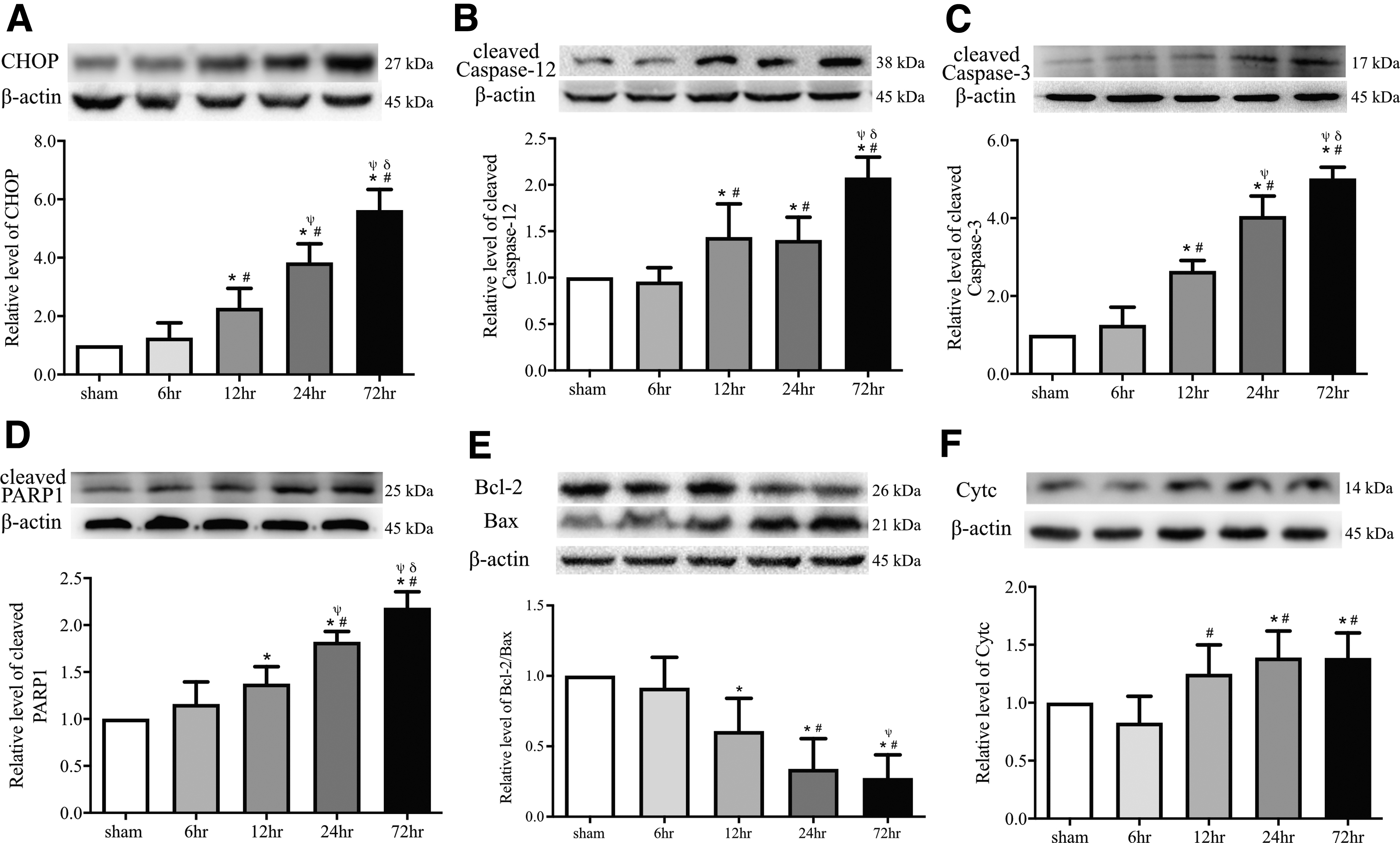
Caspase-12 cleavage and endoplasmic reticulum (ER) stress-mediated apoptosis occurred after the enhancement of ER-mitochondrion crosstalk. Top panels: representative Western blot images for staining with C/EBP homologous protein (CHOP;
Silencing PACS2 reduced sTBI-induced MAM and resultant structural changes
If increasing ER-mitochondria communication is responsible for oxidative stress, ER stress, UPR activation, and neuroinflammatory responses that occurred during acute sTBI, one would expect that blocking the communication could reverse the secondary injuries associated with TBI. To test this hypothesis, we suppressed the ER-mitochondrion physical connection by silencing the PACS2 gene in the brain using PACS2 siRNA. The siRNA reduced the ER-mitochondrion contacts (Supplementary Fig. S5) and the expression of PACS2 and MFN2 by approximately 50% (Fig. 8A, 8B) measured at 6 h post-injury. It also modestly reduced expression of IP3R1, GRP75, VDAC1, Sigma-1R and suppressed ROS production at 6 h post-injury (Fig. 8C-E). In addition, silencing the PACS2 gene reduced the expression of GRP78, p-PERK/PERK, p-eIF2α/eIF2α, ATF4, cleaved IL-1β, and TNFα that were significantly elevated in TBI mice at 6 h post-injury (Fig. 8F-K). These results suggest that blocking the ER-mitochondrion communication reduces oxidative stress, ER stress and UPR activation, and neuroinflammatory responses in the traumatically injured brain.
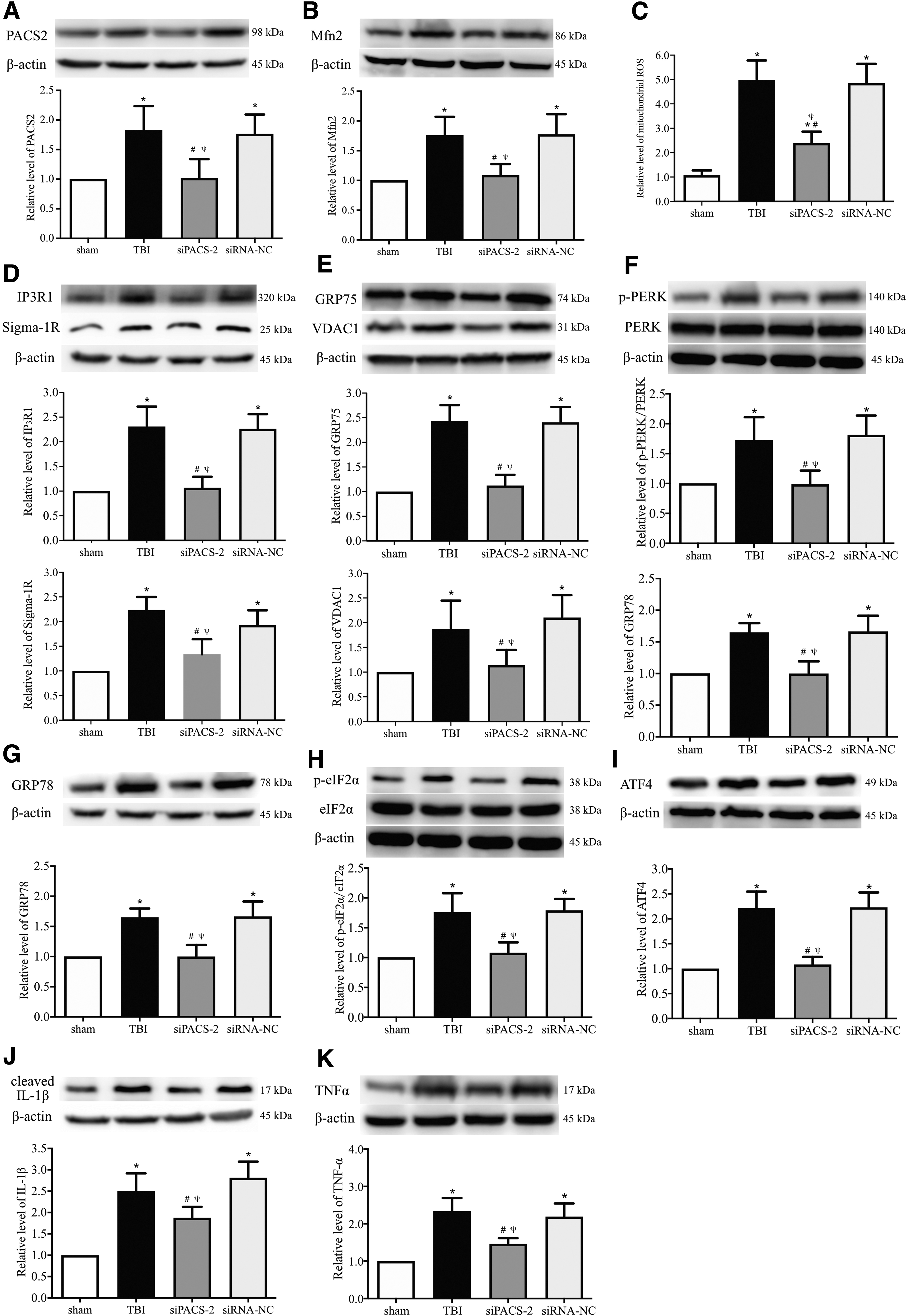
Experimental silencing the PACS2 gene suppressed the expression of mitochondria-associated membrane–resident Ca2+ regulatory proteins and mitochondrial oxidative stress, diminished endoplasmic reticulum (ER) stress and unfolded protein response (UPR) activation, and suppressed neuroinflammatory responses. Western blot and quantification analyses showing the expression levels of the ER-mitochondrion tethers phosphofurin acidic cluster sorting protein 2 (PACS2;
Knocking down PACS2 reduced ER stress-mediated apoptosis
Consistent with results reported in Figure 8, mice with the PACS2 gene silencing significantly improved neurological function during a 14-day follow-up period after sTBI (Fig. 9A). The PACS2 silencing reduced the number of TUNEL+ neurons in the perilesional cortex (Fig. 9B, 9C) and decreased the expression of the cleaved caspase-12, CHOP and Cytc, increasing the Bcl-2/Bax ratio (Fig. 9D-G). These results suggest that reducing ER-mitochondrion crosstalk at the acute stage of TBI improved cerebrovascular function, increased cellular viability, and ameliorated neurological deficits.
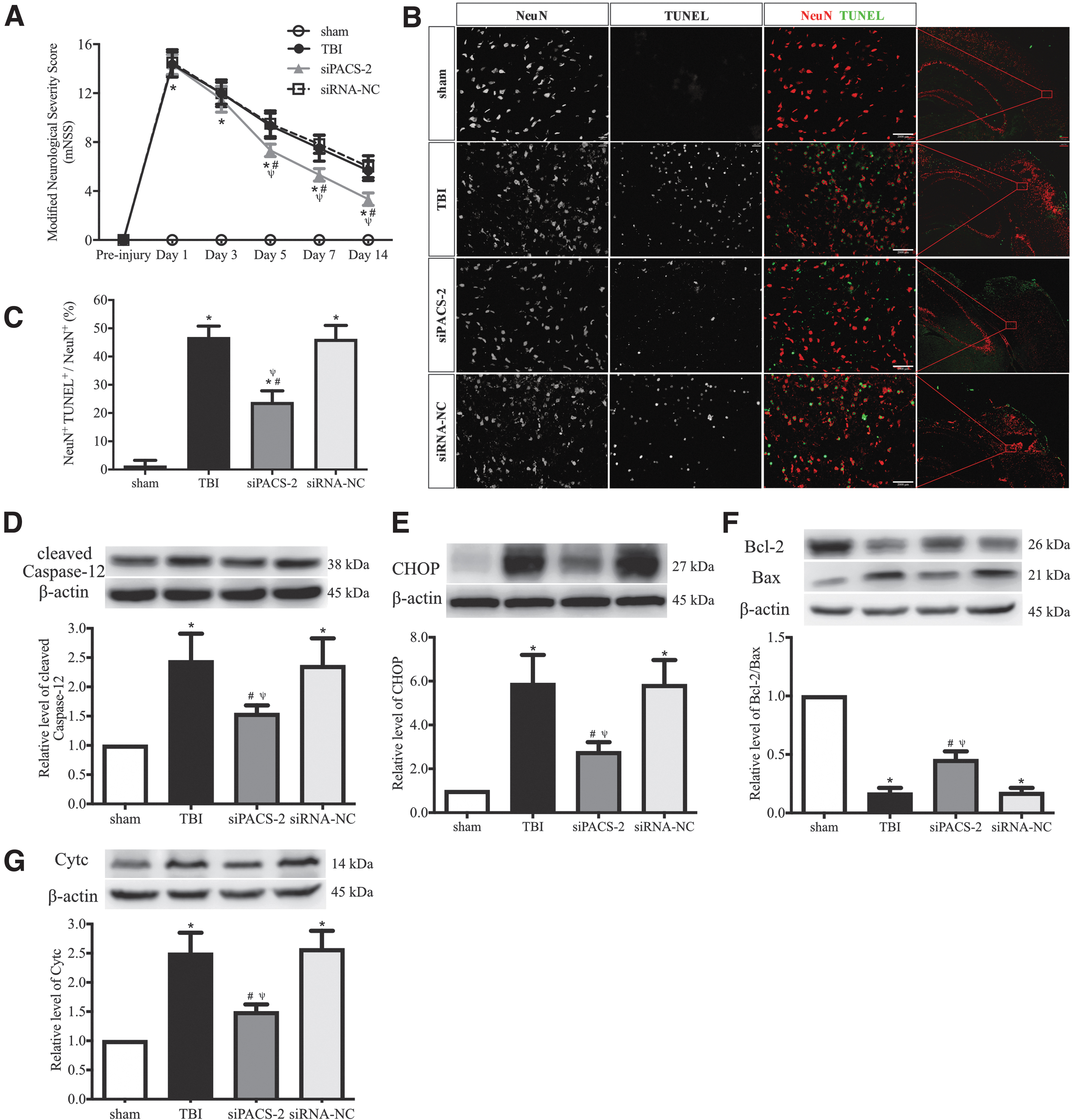
Changes induced by silencing the PACS2 gene.
Discussion
In the present study, we investigated changes in changes in the ER-mitochondrion communications in the cerebral cortex of mice subjected to sTBI and linked the changes to development of the ER stress-mediated neuronal apoptosis and neurological deficits. We demonstrated: sTBI increased MAM formation in the cerebral cortex, with the most enhanced ER-mitochondrion tethering occurring in neurons at 6 h post-injury, suggesting that this is an early event in TBI-induced secondary brain injuries. Increase in ER-mitochondrion coupling was correlated with increasing expression of IP3R1-GRP75-VDAC1 Ca2+-transfer complex, ROS production, ER stress, and UPR signaling to augment the neuroinflammatory response. This strengthened ER-mitochondrion coupling resulted in ER stress-induced apoptosis. Blocking the ER-mitochondrion connection reduced these pathological changes to prevent ER stress-induced apoptosis and improve neurological function, suggesting that modulating ER-mitochondrion crosstalk might be a novel therapeutic strategy for sTBI.
Increasing evidence suggests that the dysfunctional ER-mitochondrion crosstalk occurs in a variety of neurological diseases. Mutations in presenilins upregulate MAM function and increase ER-mitochondrion communication in patients with both the familial and sporadic forms of Alzheimer's disease (AD), indicating that AD is fundamentally a disorder of ER-mitochondrion communication. 28,49 Further, dysfunctional MAM signaling impairs neuronal calcium transport, mitochondrial dynamics, ER function, and autophagy, eventually leading to axonal degeneration in amyotrophic lateral sclerosis and hereditary motor and sensory neuropathy. 9 The loss and impairment of Sigma-1R, a MAM protein, leads to axonal and motor neuron degeneration by affecting calcium homeostasis, ER stress, mitochondrial dynamics and transport, 50 and mutation of MFN2 alters the interplay between ER and mitochondria, contributing to the development of Charcot-Marie-Tooth type 2A (CMT2A), a dominant axonal form of peripheral neuropathy. 51 Research and clinical interest have been increasingly focused on understanding the critical role of ER-mitochondrion crosstalk in the pathological course of sTBI. 52 -54 To the best of our knowledge, no data on the spectrum of ER-mitochondrion interactions in TBI are available to date. Because MAM dysfunction might be the common denominator underlying disease development, we propose that dysfunctional ER-mitochondrion coupling lie at the heart of TBI pathogenesis.
Mitochondria are in close proximity to the ER to modulate communications between the two organelles, especially with Ca2+ signal transfer from the ER to mitochondria. 9,55 This transfer is mediated the physical association between the ER-resident IP3Rs and VDAC1 on the outer mitochondrial membrane with the mitochondrial chaperone GRP75 serving as the coupling actor. 56 We have shown the overexpression of these calcium regulatory proteins in cells from mice subjected to sTBI (Fig.4A, 4B), suggesting that this calcium-mediated signal between the ER and mitochondria is activated or enhanced from the baseline level. This observation is important also because the enhanced expression occurs at MEMs through increased ER-mitochondrial contacts observed under TEM (Fig. 2A). It is also interesting to note that the increased contact is not caused by more ERs or mitochondria because the number of mitochondria was reduced in injured neurons, but likely by swollen mitochondria as shown in Figure 2C and 2D. Further, these swollen mitochondria remain metabolically competent because they produce more ROS that are responsible for the local oxidative stress environment. This notion is supported by the finding that blocking the MAM formation by silencing the PACS2 gene decreases ROS production.
While being the product of mitochondrial ATP production, the overproduction of ROS is known to cause oxidative damage to neurons, astrocytes, and vascular cells and is a key causal factor for the secondary brain injuries developed after TBI. 23,57 Excessive ROS is believed to be a major stimulus that triggers ROS-dependent ER stress, which occurs when the capacity of the ER to fold proteins becomes saturated. 11,58 -60 The ER stress activates the UPR signal to restore ER homeostasis and promote survival and adaptation. 61 We demonstrated that the suppression of MAM formation alleviated ROS-associated ER stress. Preventing or reversing ER stress through a similar mechanism has also been reported for decreasing NO production and downregulating the expression of ICAM-1, NOS, NF-κB, and VEGF 62 and improving endothelium-dependent vasorelaxation. 63,64 The MAM formation can therefore be considered an early event for the development of secondary brain injuries following TBI.
The UPR not only regulates ER stress but also play a major role in apoptosis induced by ROS-based ER stress. 46 Under ER stress, PERK dissociates from GRP78 and undergoes oligomerization and autophosphorylation, which leads to phosphorylation of the eukaryotic initiation factor eIF2α. The downstream phosphorylation of eIF2 leads to increased expression of ATF4 and translocation into the nucleus, where it binds to the unfolded protein response element (UPRE), resulting in transcriptional modification of CHOP, a proapoptotic gene transcription factor that initiates inflammation as well as programmed cell death. 13,47,65,66 This ER stress-driven apoptosis is demonstrated by the dysregulation of caspase-12, which is localized to the ER and activated by ER stress. 67 –70 Here, we demonstrated that TBI-induced MAM formation led to the activation of the PERK/eIF2α/ATF4/CHOP signaling pathway, procytokine release, caspase-12 cleavage, and ER stress-mediated neuronal apoptosis. Interfering with ER and mitochondria coupling successfully suppressed this PERK-dependent branch of the UPR to reduce caspase-12 cleavage, increase the Bcl-2/Bax ratio, promote neuronal survival and improve neurological function (Fig. 9).
While not directly examined, neuron-glial communications likely attribute to pathophysiological changes we observe. One potential pathway is that microglia and astrocytes sense the signals derived from lesioned neurons and express and release a pro-inflammatory cytokine (TNF-α, IL-1, IL6) by activating Toll-like receptor, as previously suggested. 71 The microglial cells activated by neuronal damage signal could induce more neuronal injuries. 72,73 We have shown that MAM dysfunction results in neuronal apoptosis and exaggerated neuroinflammation (Fig.5E, 5F; Fig. 7). In the current study we did not investigate the effect of blocking caspse-12 directly and differentiate the distinct function of ER-mitochondria crosstalk in neurons and glial cells. Cell-type specific changes in MEMs may further define the nature and sequence of neuronal death and inflammation during acute TBI.
Conclusion
In conclusion, dysfunctional ER-mitochondrion coupling at the acute stage of injury is involved in neuronal apoptosis and neurological deficits induced by sTBI. Modulation of ER-mitochondrion crosstalk might be a promising therapeutic strategy for patients with TBI.
Footnotes
Acknowledgments
The authors are grateful to Li Liu, Weiyun Cui, and Lei Zhou from the Tianjin Neurological Institute, and Zhijuan Chen, Baobin Liu, and Shuyuan Yue from the Department of Neurosurgery, Tianjin Medical University General Hospital, for their excellent technical support.
Authors' Contributions
X.C. conceived and designed the study. L.M., G.G., X.G., M.S., Y.C., and F.C. developed the methodology and performed the experiments. X.C., W.Y., and J.Z. interpreted the results. G.G., X.G., and M.S. performed the data analysis and prepared the figures. X.C., L.M., G.G., and X.G. wrote the manuscript. X.C., W.Y., and J.Z. reviewed and revised the manuscript and supervised the study. All authors read and approved the manuscript.
Funding Information
This work was supported by grants from the National Natural Science Foundation of China (no. 81671902), the Project of Tianjin Applied Basic and Cutting-edge Technological Research (no. 17JCYBJC25200), Scientific Research Program of Tianjin Education Commission (Natural Science) of China (No. 2019ZD034), the Tianjin Health Care Elite Prominent Young Doctor Development Program, and the Young and Middle-aged Backbone Innovative Talent Program.
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.