
Abstract
Traumatic brain injury (TBI) is a global public health concern, and few effective treatments for its delayed damages are available. Oridonin (Ori) recently has been reported to show a promising neuroprotective efficacy, but its potential therapeutic effect on TBI has not been thoroughly elucidated. The TBI mouse models were established and treated with Ori or vehicle 30 min post-operation and every 24 h since then. Impairments in cognitive and motor function and neuropathological changes were evaluated and compared. The therapeutic efficacy and mechanisms of action of Ori were further investigated using animal tissues and cell cultures. Ori restored motor function and cognition after TBI-induced impairment and exerted neuroprotective effects by reducing cerebral edema and cortical lesion volume. Ori increased neuronal survival, ameliorating gliosis and the accumulation of macrophages after injury. It suppressed the increased production of reactive oxygen species, lipid peroxide, and malondialdehyde and reversed the decrease of mitochondrial membrane potential and adenosine triphosphate content, which was also identified in oxidatively stressed neuronal cultures. Further, Ori inhibited the expression of nucleotide-binding domain leucine-rich repeats family protein 3 (NLRP3) inflammasome proteins and NLRP3-dependent cytokine interleukin-1β that can be induced by oxidative stress after TBI. Regarding underlying mechanisms, Ori significantly enhanced expression of key proteins of the nuclear factor erythroid 2-related factor 2/heme oxygenase-1 (Nrf2/HO-1) pathway. Our results demonstrated that Ori effectively improved functional impairments and neuropathological changes in animals with TBI. By activating the Nrf2 pathway, it improved mitochondrial function and antioxidant capacity and suppressed the neuroinflammation induced by oxidative stress. The results therefore suggest Ori as a potent candidate for managing neurological damage after TBI.
Introduction
Traumatic brain injury (TBI), a leading cause of death and long-term disability, is a global public health concern, affecting approximately 53–69 million individuals worldwide annually. 1 A TBI can cause direct mechanical tissue damage (primary injury) and biochemical changes with delayed or progressive cell loss (secondary injury). This largely determines the outcome and prognosis of patients. 2
Currently, there are still no suitably established treatments against TBI progression, especially against secondary injuries, mainly because of the complex nature of the mechanisms associated with this injury. 3 Therefore, novel therapeutic approaches need to be developed.
Oxidative stress is a confirmed post-traumatic secondary response pathway observed in TBI, which arises when the balance between reactive oxygen species (ROS) levels and the antioxidant environment is disrupted. 4 The brain is highly sensitive to free radical-mediated injury; thus, oxidative damage can halt its cellular energy generation and cause neuronal loss. 5
Neuroinflammation also plays a key role in TBI secondary damage, 6 and the nucleotide-binding domain leucine-rich repeats family protein 3 (NLRP3) inflammasome is an essential component in neuroinflammation pathogenesis. Multiple stimuli can induce NLRP3 activation, including ROS and mitochondrial damage after tissue injury. The NLRP3 inflammasome regulates interleukin (IL)-1β and IL-18 maturation and production, 7 while both IL-1β and IL-18 can promote the accumulation of ROS, creating a vicious cycle between oxidative stress and NLRP3 activation. 8 Therefore, it is considered a promising therapeutic strategy for TBI to control oxidative stress and thereby mitigate NLRP3 inflammasome-mediated neuroinflammation.
Nuclear factor erythroid 2-related factor 2 (Nrf2), a vital nuclear transcriptional factor, binds to specific deoxyribonucleic acid antioxidant response elements (AREs) of antioxidant genes and induces the expression of antioxidant and detoxification enzymes and downstream proteins, such as heme oxygenase-1 (HO-1) and nicotinamide adenine dinucleotide phosphate (NAD(P)H): quinone oxidoreductase-1 (NQO-1). 9
Increased levels of Nrf2 ameliorate oxidative stress and present neuroprotective effects on neuroinflammatory and neurodegenerative diseases. 10 Moreover, the activation of NLRP3 inflammasome could be inhibited by the Nrf2 signaling as well, through reduction of ROS production. 11 Hence, Nrf2 has been suggested as a potential target to attenuate TBI-induced neurological damage.
Rabdosia rubescens is a commonly available herbal plant medicine used in traditional Chinese medicine, and it has successfully supplemented the treatment of those with acute and chronic inflammatory disorders, including pharyngitis, bronchitis, and tonsillitis. 12,13 Oridonin (Ori) is its major active constituent, and several studies have shown that it inhibits the neuroinflammation, prevents synaptic loss, and presents neuroprotective activities in neurodegenerative diseases. 14 As a potent activator of Nrf2, Ori can decrease the production of ROS, and it presents strong antioxidant properties. 15
Therefore, we designed the present study to fully evaluate the neuroprotective effects of Ori on TBI and investigate its potential mechanisms of action, recognizing the potential therapeutic value of Ori in TBI management.
Methods
Chemicals
Ori (≥99.5% purity) was purchased from Target Molecule (Shanghai, China). Lipopolysaccharide (LPS), adenosine triphosphate (ATP), and H2O2 were obtained from Sigma-Aldrich (St. Louis, MO.).
Experimental animals, ethics statement and TBI animal model
In total, 153 female mice, aged 6–8 weeks, SPF grade, were randomly divided into three groups: sham, TBI, and TBI+Ori groups. Simple randomization was used to assign animals to each group. Briefly, all animals were numbered from 1 to 153 by ear marks, and the animals with numbers divisible by three were assigned into the sham group, the animals not divisible by three but with remainder one into the TBI group, and the remaining animals into the TBI+Ori group. They were intraperitoneally injected with Ori at a dose of 20 mg/kg, or with the same dose of dimethyl sulfoxide+phosphate buffered saline 30 min post-injury, and every 24 h until the time of sacrifice.
All animal procedures were approved by the Experimental Animal Ethics Committee of Nanjing Medical University (ethical approval number: #IACUC-2011011). The TBI surgery used a controlled cortical impact model, as reported previously. 16 For the behavioral and neuropathological experiments and analysis, the raw data were originally evaluated by observers who were blinded to the treatments of the animals. Animals were identified by the ear marks with the assigned numbers.
Beam walking test, fear conditioning test (FCT), and blood–brain barrier (BBB) evaluation
As described previously, 17 the beam walking test was used to detect the motor coordination of mice, and the Evans blue staining was applied to determine the permeability of BBB. A FCT was used to analyze memory behaviors of the mice, as previously described, 18 with the FCT machine LE116 (Panlab, Barcelona, Spain) (n = 6–7 for behavioral tests; n = 4 for BBB evaluation).
Cell culture and treatments
The immortalized mouse microglial cell line (BV2), mouse macrophage cell line (RAW264.7), and mouse neuroblastoma cell line (N2a) (American Type Culture Collection, Rockville, MD) were cultured as described previously. 17
Hematoxylin and eosin (H&E), immunofluorescence, and terminal deoxynucleotidyl transferase dUTP nick end labeling TUNEL staining
The N2a cells and brain tissue were stained using H&E solution, TUNEL staining (Vazyme, Nanjing, China), or using immunofluorescence with primary antibodies against: glial fibrillary acidic protein (GFAP, 1:300), ionized calcium binding adaptor molecule 1 (Iba1, 1:300), neuronal nuclear protein (NeuN, 1:300), NLRP3 (1:200), or Nrf2 (1:100). Immunofluorescence intensities were detected using ImageJ (n = 3–4).
Western blot analysis and quantitative polymerase chain reaction (QPCR)
Total proteins were extracted from cells and mouse tissues (pericontusional cortical area, n = 3–4 for Western blot; n = 4–5 for QPCR), and the concentrations of proteins were determined using a bicinchoninic acid (BCA) protein assay kit (Beyotime, Shanghai, China). Standard blotting procedures were conducted as described previously. 19,20 The QPCR was performed on the Light Cycler® 96 thermocycler (Roche, Rotkreuz, Switzerland) using ChamQ SYBR qPCR Master Mix (Vazyme).
ATP measurement
The ATP concentration was measured using ATP detection kits (Beyotime). The corresponding luminescence of the samples (n = 5) was measured using a chemiluminometer (Promega, Madison, WI).
Mitochondrial membrane potential (mtΔΨ/MMP) detection
The precipitated mitochondria were isolated using the Mitochondrial Isolation Kit (Beyotime). The mtΔΨ was determined using the Mitochondrial Membrane Potential Detection Kit (Beyotime) and detected by chemiluminometer (n = 5). As for cell culture, mtΔΨ of N2a cells was analyzed by FACS Calibur flow cytometry (BD, San Jose, CA).
Malondialdehyde (MDA), ROS, and lipid peroxide (LPO) assay
The MDA activity in the pericontusional cortical area was detected with the MDA detection kit (Beyotime). Cortical tissue supernatants were collected, and the production of LPO and ROS was detected using the LPO and ROS enzyme-linked immunosorbent assay (ELISA) kit (Yifeixue Bio Tech, Nanjing, China). The concentrations were calculated by standard curves (n = 5).
Cell viability and apoptosis assay
Cell activity was evaluated with the CCK-8 kit (Vazyme). The percentage of cells undergoing apoptosis was determined using the Annexin V-fluorescein isothiocyanate/propidium iodide (Annexin V-FITC/PI) Apoptosis Kit (Yeasen, Wuhan, China) according to the manufacturer's protocol. The apoptotic cells were immediately detected by flow cytometry.
Mitochondrial morphology and mitochondria-derived ROS (mitoROS) detection
Mitochondrial morphology was stained by Mito-Tracker Red (AdipoGen, San Diego, CA) and detected using confocal microscopy (Zeiss, Oberkochen, Germany). MitoROS was labeled by MitoSox Red Reagent (Invitrogen, Carlsbad, CA) and detected using a confocal laser microscope. The data were analyzed by Image J.
Statistical analysis
All results are presented as the mean ± standard error of the mean (SEM) and analyzed with the GraphPad Prism version 5.0. Statistical comparisons between two groups were performed using the Student t test. p < 0.05 was considered statistically significant.
Results
Ori provides neurological protection by alleviating motor and cognitive impairments and reducing cerebral lesions after TBI
A timeline of the experimental treatment regimen used in this study is shown in Fig. 1A. The brain tissue schematic map in Fig. 1B exhibits the location of the injured cerebral cortex (red) of the TBI model.
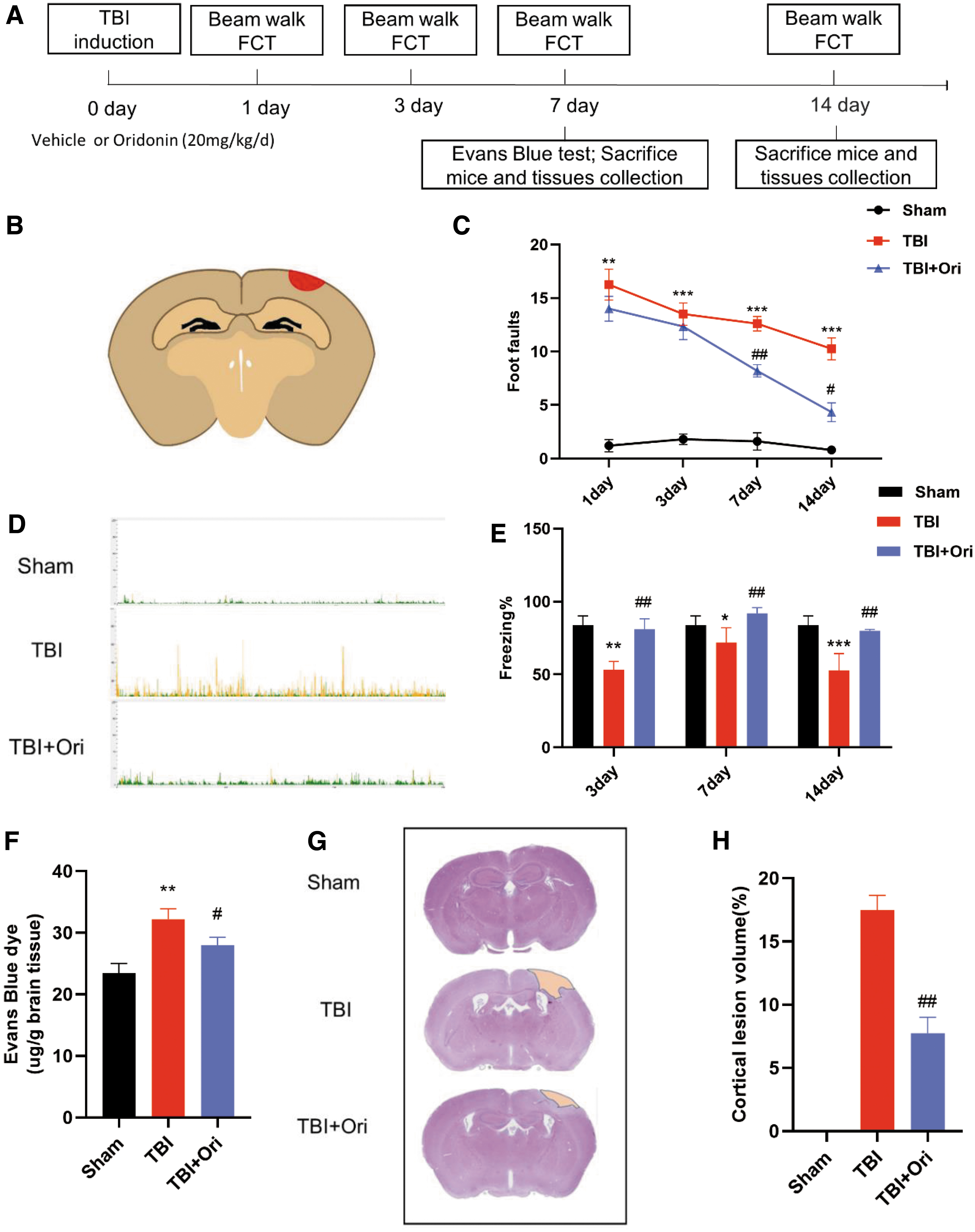
Oridonin (Ori) provides neurological protection by alleviating motor and cognitive impairments and reducing cerebral lesions after traumatic brain injury (TBI). (
First, the beam walking test was applied to evaluate motor coordination function, which was affected by TBI, with a significantly higher number of foot-slips. Ori treatment markedly improved performance in the beam walking test from day 7 after injury (Fig. 1C). Then, hippocampus-dependent cognitive deficits after TBI were evaluated by the FCT. Contextual fear conditioning was significantly impaired by TBI, and treatment with Ori restored the deficits (Fig. 1D, E). Based on the above-mentioned observations, our following evaluations mainly focus on Day 7 post-injury, the earliest time point when significant improvements were observed.
The BBB disruption is among the principally identified mechanisms underlying secondary injury in TBI, 21 and BBB leakage was determined by an Evans blue test. As expected, Evans blue extravasation markedly increased in the ipsilateral hemisphere after TBI. Treatment with Ori, however, significantly decreased the extravasation (Fig. 1F). Further H&E staining showed marked cortical damages after TBI (Fig. 1G), and Ori treatment significantly reduced the cortical lesion volumes (Fig. 1H). Taken together, Ori exerted its neuroprotective effect by reducing cerebral edema and cortical lesion volume after TBI.
Ori improves neuronal survival and ameliorates the accumulation of reactivated glial cells and macrophages after TBI
To assess neuronal survival in the pericontusional cortex areas and ipsilateral hippocampal subfields (CA1, CA3) after trauma, staining of NeuN, a widely used neuronal marker, was conducted (Fig. 2A). Significantly more NeuN-positive cells were detected in the Ori treatment group than in the TBI group, in the cortical region (Fig. 2B) and hippocampus (Fig. 2C, D). The TUNEL assay detected cellular apoptosis in the cortical region, and more apoptotic neurons were identified after TBI. Ori treatment significantly decreased the number of apoptotic neurons (Fig. 2E, F).

Oridonin (Ori) improves neuronal survival and ameliorates the accumulation of reactivated glial cells and macrophages after traumatic brain injury (TBI). (
The proliferation and activation of glial cells can be identified in both the early and late stages of injury. Accordingly, we used GFAP to identify astrocytes and Iba1 to identify both reactivated microglial cells and macrophages, which reside in or infiltrate into the cerebral parenchyma through the disrupted BBB.
As shown in Fig. 2G and 2I, the number of GFAP-positive cells was increased in the TBI group compared with the sham group, which revealed significant astrocytosis in the cortex, but Ori treatment reversed this change. Moreover, a significantly increased number of Iba1-positive cells was detected after trauma, which was reversed by Ori treatment (Fig. 2G, H). These results demonstrated that Ori could ameliorate gliosis and the accumulation of macrophages after TBI.
Ori inhibits oxidative stress by promoting mitochondrial function and antioxidant potential through Nrf2 activation after TBI
We evaluated ROS production in the injured cortex (Fig. 3A). The TBI markedly increased ROS production, but treatment with Ori significantly reversed this increase. Moreover, the levels of LPO and MDA were significantly increased after TBI, and treatment with Ori suppressed the increased levels (Fig. 3B,C). These data confirm the protective effect of Ori against oxidative stress.
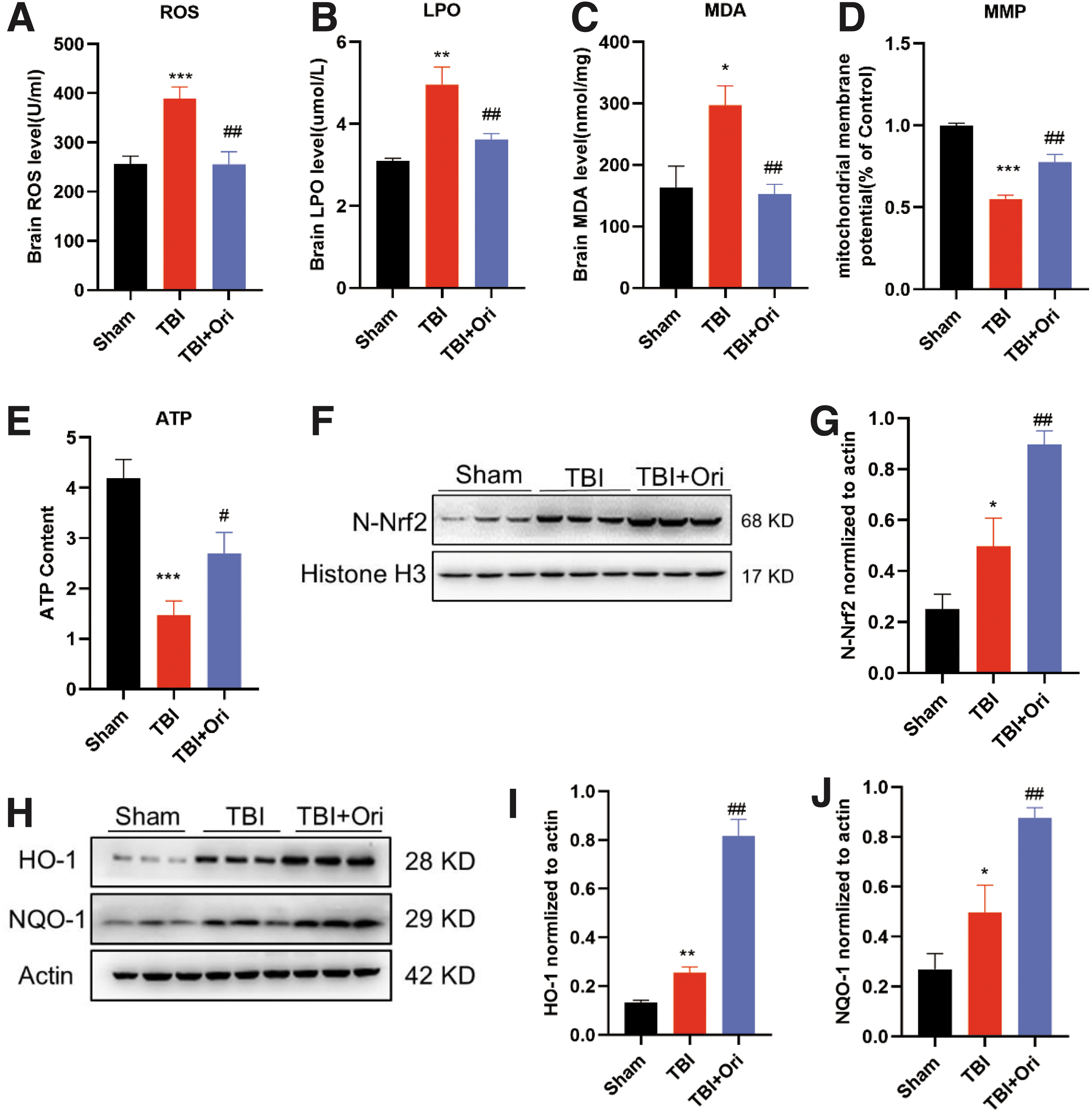
Oridonin (Ori) inhibits oxidative stress by promoting mitochondrial function and antioxidant potential through nuclear factor erythroid 2-related factor 2 (Nrf2) activation after traumatic brain injury (TBI). The production levels of reactive oxygen species (ROS) (
Mitochondrial dysfunction is reported to cause continuous production of ROS, so mtΔΨ in the pericontusional cortex was evaluated. The mtΔΨ was reduced after TBI, indicating an increased number of dysfunctional mitochondria, and this mtΔΨ reduction was significantly reversed by Ori treatment (Fig. 3D). The ATP content was measured in the same cortical areas, and a significant decrease in ATP content after TBI was detected; it markedly increased after Ori administration (Fig. 3E). These results indicate that Ori could alleviate mitochondrial dysfunction caused by TBI.
Meanwhile, Ori treatment significantly upregulated the expression of nuclear Nrf2 (N-Nrf2) and its downstream antioxidants HO-1 and NQO-1 in the TBI animals (Fig.3 F–J), suggesting that Ori may enhance cerebral antioxidant capacity by activating the Nrf2 pathway after TBI. That was further investigated in neuronal cultures.
Ori inhibits H2O2-induced oxidative stress in N2a cells by promoting mitochondrial function and antioxidant potential through Nrf2 activation
Oxidative stress was induced by H2O2, and H2O2-exposed N2a cells were treated with serial concentrations of Ori. Ori exhibited promising protective effects in a dose-dependent pattern (Fig. 4A). The 4-μM concentration, exerting the highest protective effect without affecting cell viability, was used in the subsequent experiments.
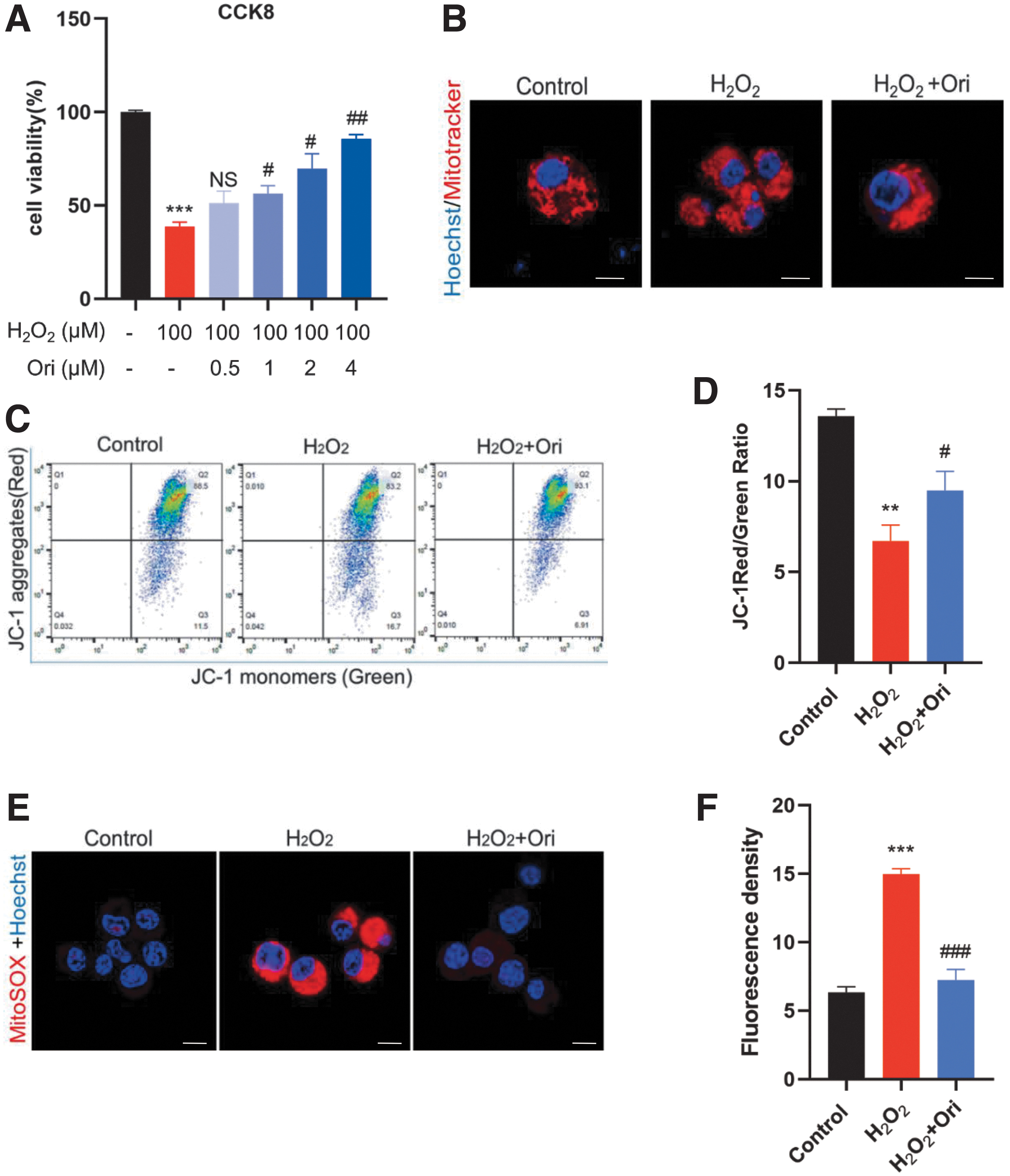
Oridonin (Ori) inhibits H2O2-induced oxidative stress in N2a cells by promoting mitochondrial function. (
Mito-Tracker Red was applied to observe mitochondrial morphology. Punctate, injured mitochondria were detected in H2O2-exposed N2a cells, and Ori treatment restored them to a relatively normal appearance (Fig. 4B). JC-1 staining was used to evaluate mtΔΨ, and ratios of J-aggregates (red) to monomers (green) in all groups were calculated (Fig. 4C, D). H2O2-exposed N2a cells showed a markedly low ratio, suggesting large numbers of damaged mitochondria, and Ori treatment significantly reversed the decreased ratio. The production of mitoROS was detected by mitoSOX staining. H2O2 significantly induced mitoROS accumulation in N2a cells that was reversed by Ori treatment (Fig. 4E, F).
Possible mechanism of the action was investigated further. Ori treatment significantly elevated p-Akt/Akt and p-GSK3β/GSK3β ratios and the expression of N-Nrf2, HO-1, and NQO-1 in oxidatively stressed cells, which is in accord with our results obtained from the in vivo animal experiments (Fig. 5A–E and 5G–I). Immunofluorescence staining was used to confirm the location of Nrf2 (Fig. 5F). In the control group, Nrf2 was mostly detected in the cytoplasm, but was mainly detected in the nuclei after H2O2 exposure. After Ori treatment, its expression in the nuclei was significantly increased further.
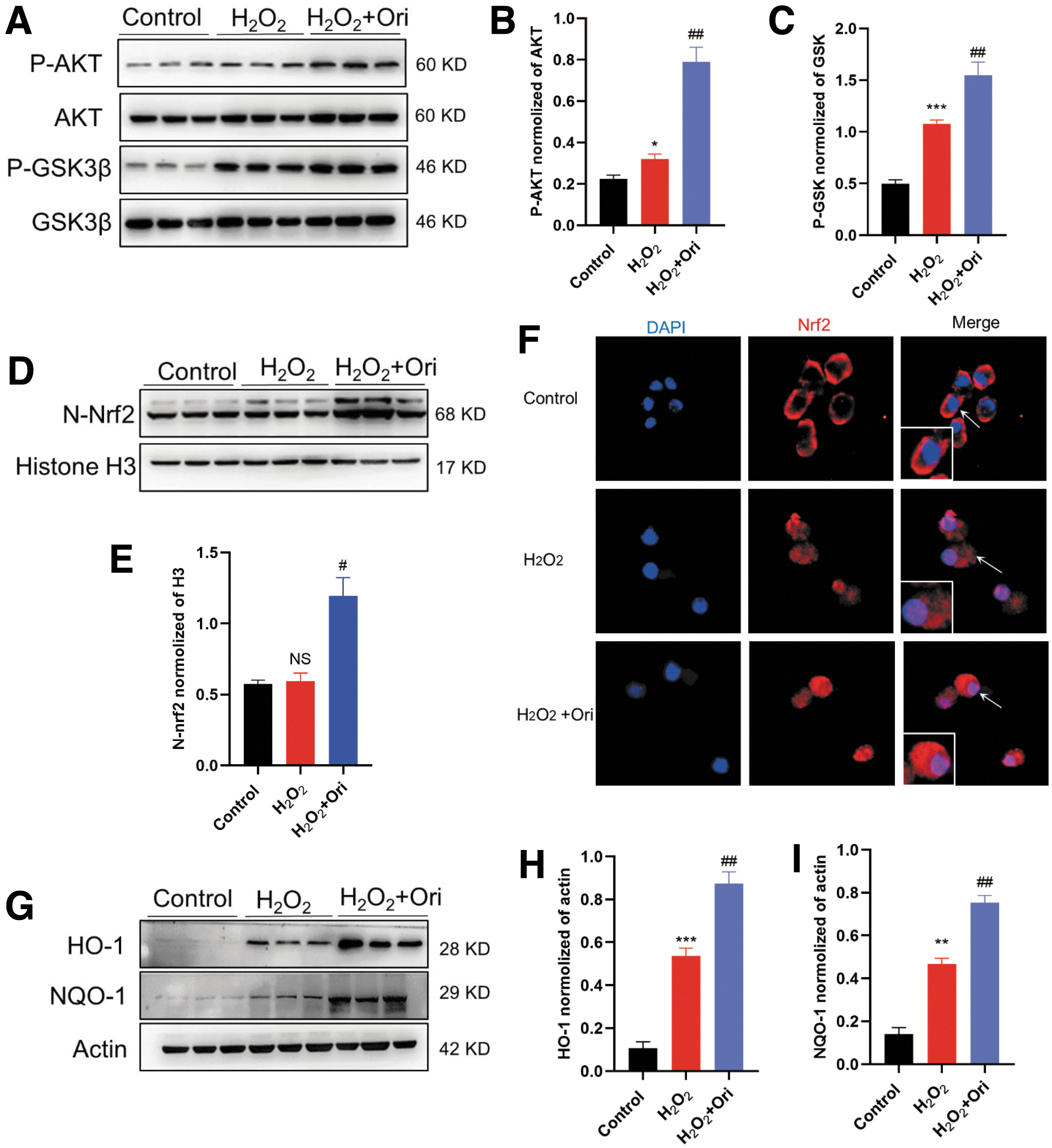
Oridonin (Ori) inhibits H2O2-induced oxidative stress in N2a cells by enhancing nuclear factor erythroid 2-related factor 2 (Nrf2)-mediated antioxidant potential. 4 μM Ori was applied in the following experiments. (
Collectively, our data suggest that Ori protected N2a cells from H2O2-induced oxidative damage by protecting the mitochondria and enhancing the antioxidant capacity through Nrf2 pathway activation.
Ori inhibits TBI-induced activation of the NLRP3 inflammasome
The QPCR and immunoblotting were applied to measure the expression of the NLRP3 complex in the pericontusional cortex. The expression of NLRP3, caspase-1, apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain (ASC), and IL-1β at the protein (Fig. 6A-E) and messenger ribonucleic acid (mRNA) (Fig. 6F) levels markedly increased after TBI, but significantly decreased after Ori treatments.
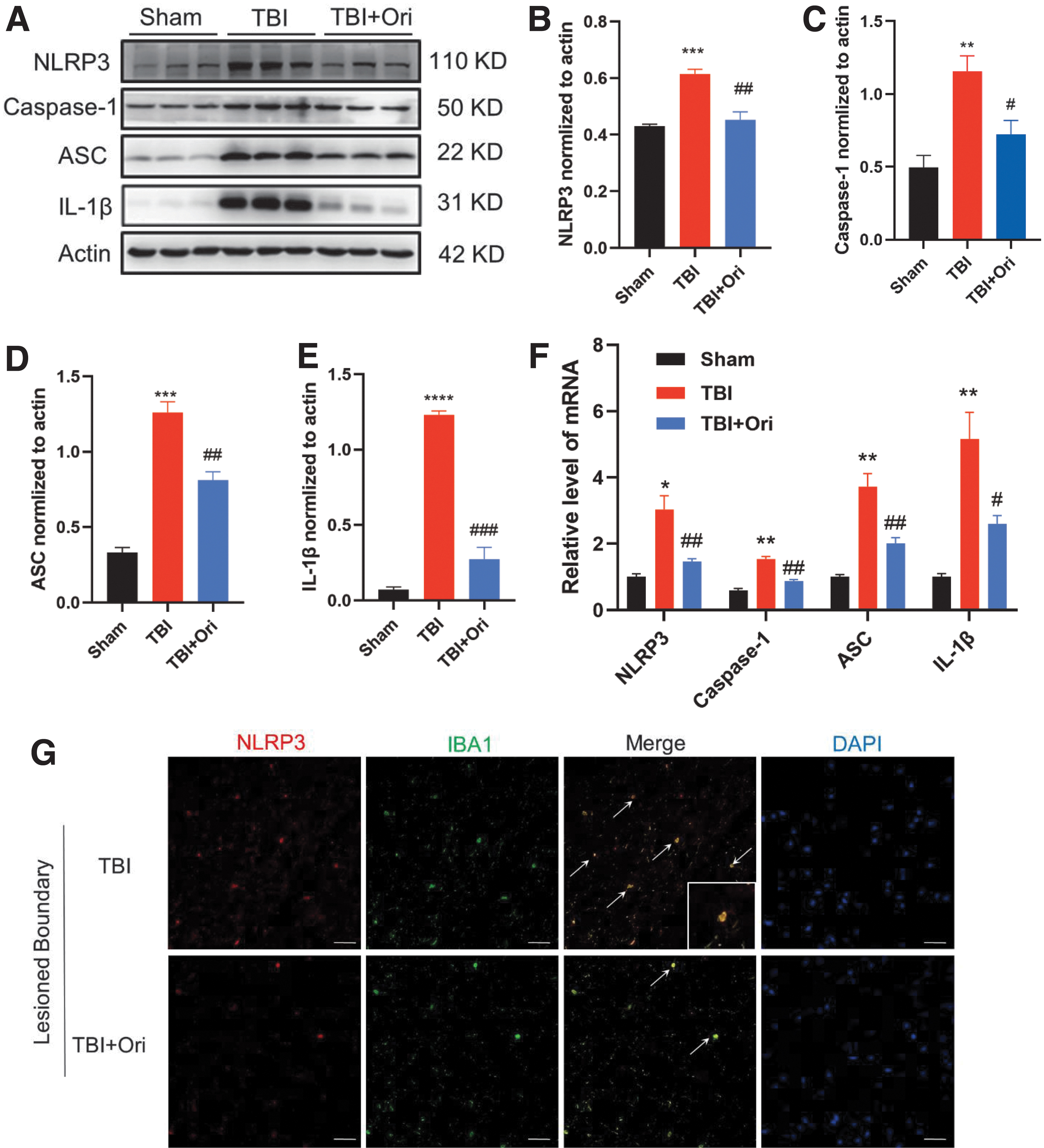
Oridonin (Ori) inhibits traumatic brain injury (TBI)-induced activation of the nucleotide-binding domain leucine-rich repeats family protein 3 (NLRP3) inflammasome. (
To identify cellular resources of NLRP3 inflammasome, double immunofluorescence staining was conducted. The NLRP3 inflammasome-positive cells (red) were hardly colabeled with GFAP (data not shown), but were mainly colabeled with Iba1 (green), and the majority of Iba1-positive cells were colabeled with NLRP3 inflammasome. Ori administration significantly suppressed the accumulation of both Iba1- and NLRP3 inflammasome-positive cells. Among the Iba1-positive cells with already reduced proportions, the number of cells colabeled with the NLRP3 inflammasome further decreased (Fig. 6G). The results suggest that Ori could inhibit the accumulation of Iba1-positive inflammatory cells and reduce the formation of the NLRP3 inflammasome in these cells.
Ori inhibits LPS-induced NLRP3 inflammasome activation in BV2 and RAW264.7 cells, consequently increasing neuronal survival
The BV2 and RAW264.7 cell cultures were employed to further prove the potential effect of Ori on NLRP3 inflammasome assembly. The cells were treated with LPS to induce NLRP3 inflammasome activation, and serial concentrations of Ori were added to the cell cultures. Ori exerted significant protective effects on both BV2 (Fig. 7A) and RAW264.7 cells (Fig. 7G), showing a dose-dependent pattern.
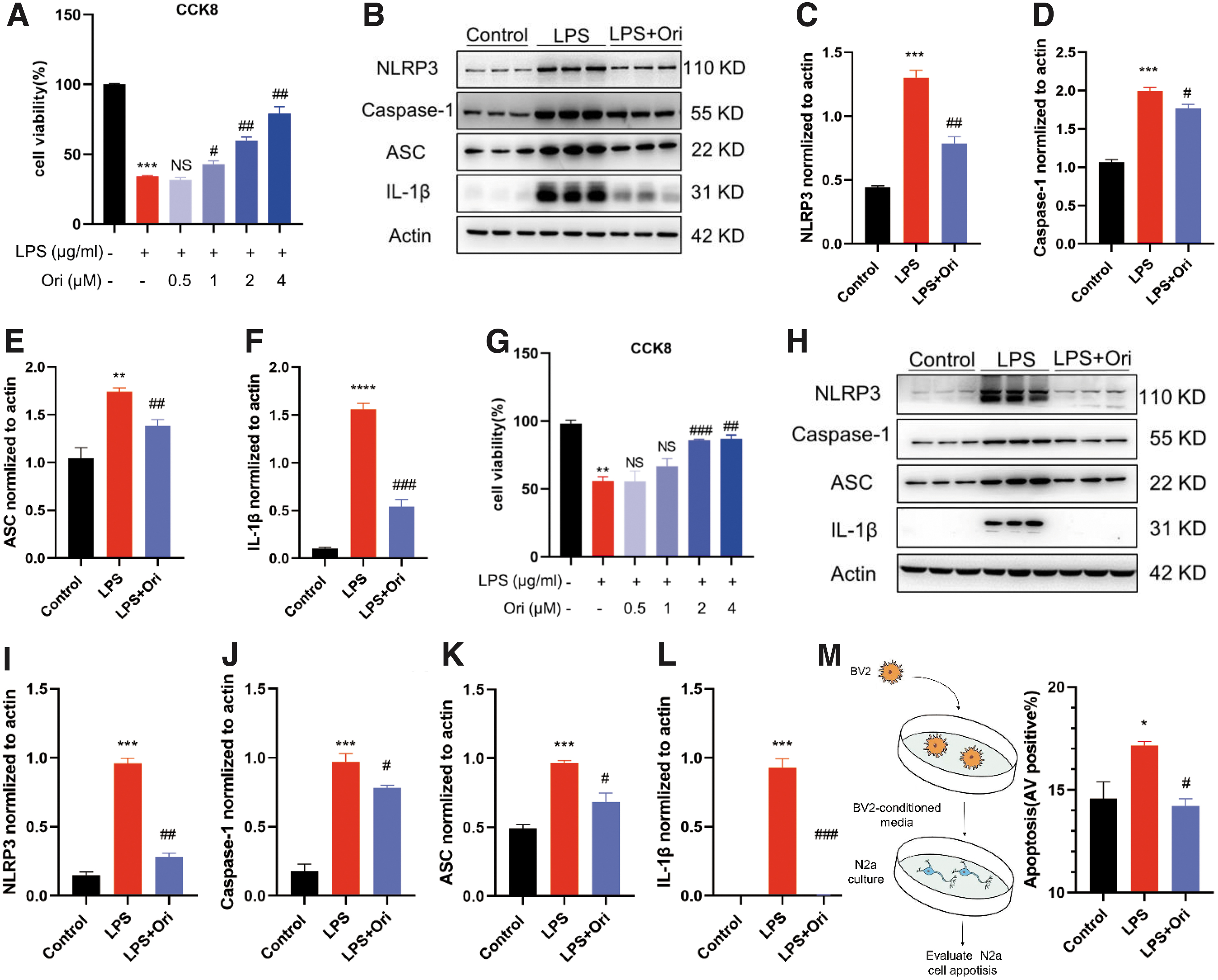
Oridonin (Ori) inhibits lipopolysaccharide (LPS)-induced nucleotide-binding domain leucine-rich repeats family protein 3 (NLRP3) inflammasome activation in BV2 and RAW264.7 cells, consequently increasing neuronal survival. The LPS-induced cells were treated with different concentrations of Ori, and cell viabilities were examined. The bar graphs exhibit its protective effect on BV2 (
Ori significantly downregulated expression of NLRP3, ASC, caspase-1, and IL-1β in the LPS-induced BV2 (Fig. 7B-F) and RAW264.7 (Fig. 7H-L) cell cultures, indicating that Ori suppressed NLRP3 inflammasome activation in both microglial cells and macrophages. The supernatant of LPS-activated BV2 cell culture was then transferred into N2a cell culture to determine the protective effects of Ori on neuronal survival by inhibiting microglial activation. The supernatant of reactivated BV2 cells significantly induced apoptosis of N2a cells, but Ori treatments efficiently protected N2a cells (Fig. 7M). Our results indicate that Ori markedly inhibited the inflammatory response and thereby enhanced neuronal survival.
Discussion
In this study, we confirmed the therapeutic effects of Ori on animal models of TBI and investigated its possible mechanisms of action. Our results indicated that Ori ameliorated behavioral and cognitive impairments in mice with TBI. Neuropathologically, Ori reduced cerebral edema and cortical lesion volume, alleviated neuronal loss in the pericontusional cortex and ipsilateral hippocampus, and ameliorated gliosis after TBI. By activating the Nrf2 pathway, Ori improved mitochondrial function and antioxidant capacity and suppressed neuroinflammation induced by oxidative stress.
Recently, ROS scavengers and ROS-degrading agents have shown promising therapeutic effects against TBI in animal models. 22 Suppressing oxidative stress by targeting ROS generation has also been suggested as a possible approach for TBI management. In this study, we found that Ori markedly attenuated TBI-induced oxidative stress by reducing ROS production and decreasing the levels of MDA and LPO, which are the products of ROS in the post-mortem tissue of patients with TBI.
Extensive data support the role of mitochondrial dysfunction and oxidative damage in the pathogenesis of different neurodegenerative and neuroinflammatory disorders, such as Alzheimer. In a transgenic APP-mutant mouse model, energy metabolism inhibitors markedly aggravated cognitive deficits and neuropathological changes, including increased brain Aβ levels, plaque deposition, and gliosis. 23 At least six nuclear genes (α-synuclein, parkin, DJ-1, PINK1, LRRK2, and HTRA2) directly or indirectly involved with mitochondria have been identified as Parkinson disease (PD) or affecting PD risk. 24 Acute brain insults, such as cerebral ischemia and TBI, 25 are also associated with mitochondrial dysfunction, wherein ROS overproduction triggers and aggravates neuronal apoptosis.
Our results demonstrated Ori could rescue mitochondrial disruption by improving the abnormally reduced mtΔΨ and ATP content in mice with TBI. Ori also significantly protected H2O2-exposed oxidatively stressed N2a cells from cytotoxicity. After treating with Ori, we observed markedly healthy mitochondria, improved mtΔΨ, and significantly decreased production of mito-ROS in H2O2-exposed N2a cells.
The Nrf2 is a critical regulator of numerous antioxidants and detoxifying enzymes, and several Nrf2 activators have progressed to human clinical trials. 26 Further, the Nrf2 system is positively associated with mitochondrial biogenesis and quality control. The downregulation of Nrf2 expression may be highly relevant to mitochondrial fragmentation in neurological diseases, despite the precise profiles of Nrf2 and mitochondrial dynamics/mitophagy differing in each disease type. 27
Our research confirmed Ori was an activator of the Nrf2 pathway and that it could contribute to Nrf2 nuclear localization and increase HO-1 and NQO-1 expression significantly in mice with TBI and H2O2-exposed oxidatively stressed N2a cells. The mechanism underlying Nrf2 activation by Ori was complicated, and we found the activated p-Akt and p-GSK3β levels were significantly higher in Ori-treated oxidatively stressed cells.
There are other upstream pathways activating Nrf2, however, including p38 MAPK and PKC delta, 28 and crosstalk pathways among Nrf2, mitochondrial biogenesis, and NLRP3. 29 Therefore, the signaling pathways involved in the neuroprotective effects of Ori against TBI, particularly against oxidative stress, could be more complicated and require further investigation.
Dysregulated or persistent neuroinflammation triggers various neuropathological changes. Several recent studies have found that pharmacological treatments targeting the NLRP3 inflammasome can reduce the neuroinflammation after moderate-to-severe TBI. 7
Our research identified Ori ameliorated the astrocytosis, microcytosis, and the accumulation of macrophages in the cortex of mice with TBI. It also suppressed the increased formation of the NLRP3 inflammasome, which was in accord with the in vitro experiments results observed in both microglial and macrophage cultures. By inhibiting the inflammatory response of reactivated microglia cells, Ori increased neuronal survival consequently. The NLRP3 inflammasome activation, however, is just one of the main processes involved in neuroinflammation propagation; other inflammatory mechanisms during TBI need further study.
There are, however, several limitations deserving attention in our study. Other possible upstream pathways activating Nrf2 and possible crosstalk pathways should be taken into consideration and could be further investigated. Ori showed protective effects on TBI at relatively early time points in the present study; its long-term effects, however, could be evaluated in the future. In addition, Ori's promising therapeutic effects in clinical situation need to be validated with preliminary clinical trials.
Conclusions
Our data indicated that Ori increased neuronal survival and improved functional impairments and neuropathological changes after TBI. By activating the Nrf2 pathway, Ori suppressed oxidative stress in neurons through enhancing mitochondrial function and antioxidant potential, and inhibited NLRP3 inflammasome-mediated inflammatory response in microglial/macrophages that can be induced by oxidative stress and further aggravate oxidative damage in turn (summarized in Fig. 8).
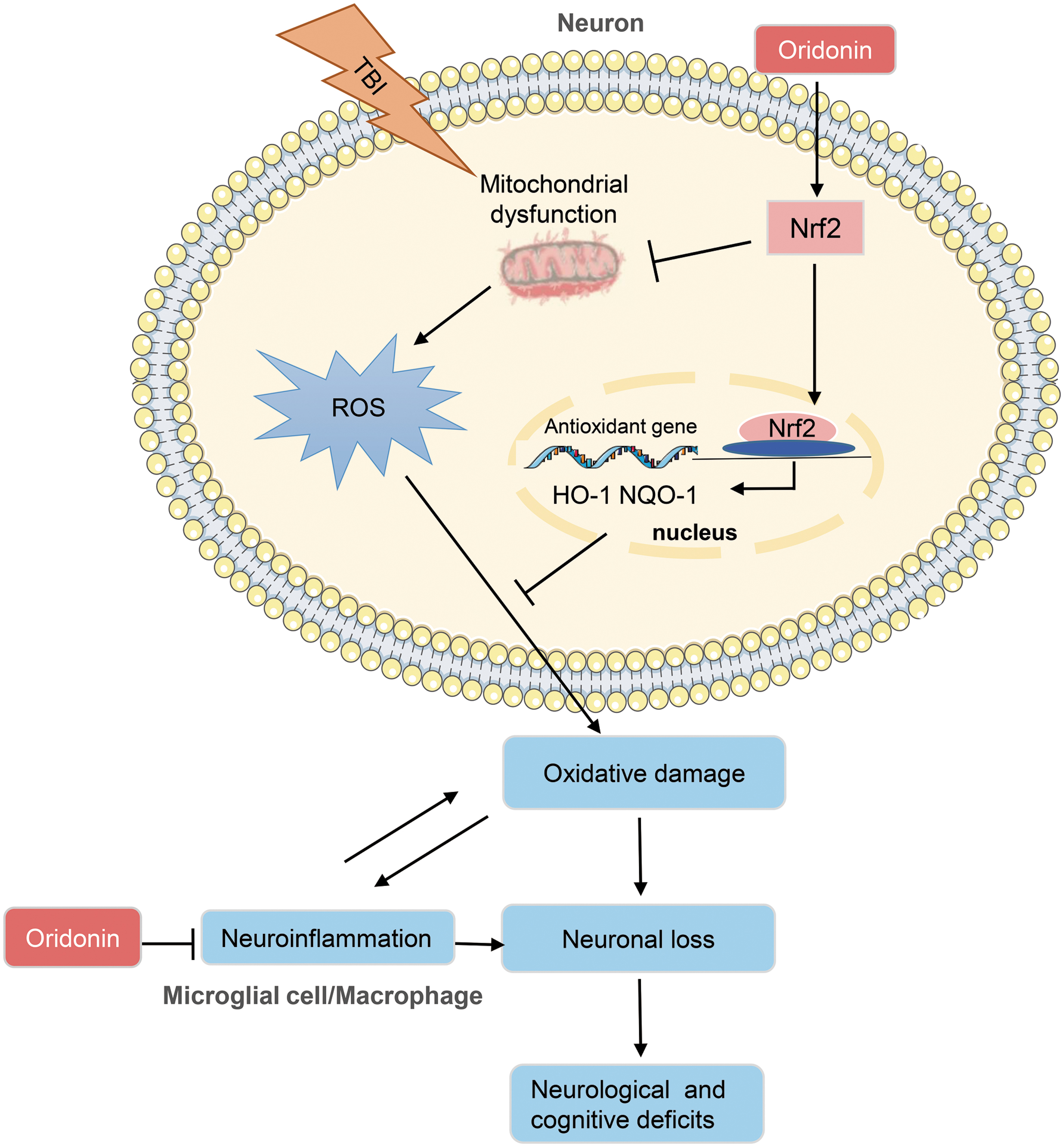
Schematic diagram depicts the mechanism underlying neuroprotective effects of Oridonin (Ori) after traumatic brain injury (TBI). The mitochondrial function of neurons is impaired after TBI, and continuous reactive oxygen species (ROS) production induces oxidative stress, further causing neuronal loss. Moreover, the neuroinflammation, which can be induced by and further aggravate oxidative damage, also plays a central role in neuronal damage after TBI. In this study, by activating the nuclear factor erythroid 2-related factor 2 (Nrf2) pathway, Ori alleviated oxidative stress in neurons through enhancing mitochondrial function and antioxidant potential, and inhibited nucleotide-binding domain leucine-rich repeats family protein 3 (NLRP3) inflammasome-mediated inflammatory response in microglial/macrophages, consequently increasing the survival of neurons and improving neurological and cognitive functions after TBI. HO-1 NQO-1, heme oxygenase-1 (HO-1) and nicotinamide adenine dinucleotide phosphate (NAD(P)H): quinone oxidoreductase-1 (NQO-1). Color image is available online.
Collectively, these results establish the beneficial effects of Ori and its possible mechanism of action, suggesting it as a promising therapeutic agent for the management of TBI in humans.
Footnotes
Authors' Contributions
Xiao-Jing Zhao and Hai-Yan Zhu performed most of the experiments and analyzed the data. Xiao-Wei Lu, Xiao-Liang Wang, Lu Xu, Cai-Long Pan, and Xue Liu participated in the experiments and contributed to the data collection. Ning Xu contributed to the design of the project and wrote the draft of the article. Zhi-Yuan Zhang designed and oversaw the whole project and wrote the final version of the article. All authors agree to be accountable for all aspects of work ensuring integrity and accuracy.
Funding Information
This work was supported by National Natural Science Foundation of China [grant numbers 81771171, 82071209, 81771286]; Nanjing Medical Science and Technology Development Foundation [grant number YKK19076]; and Science and Technology Development Foundation of Nanjing Medical University [grant number NMUB2019002].
Author Disclosure Statement
No competing financial interests exist.