
Abstract
Secondary structural and functional abnormalities of the neurovascular unit are important pathological mechanisms following traumatic brain injury (TBI). The neurovascular unit maintains blood–brain barrier and vascular integrity through interactions among glial cells, pericytes and endothelial cells. Trauma-induced neuroinflammation and oxidative stress may act as initiating factors for pathological damage after TBI, which in turn impairs cerebral microcirculatory function. Studies have shown that the tumor necrosis factor α (TNF-α)/nuclear factor-κB (NF-κB) pathway regulates inflammation and oxidative damage, but its role in pericyte-mediated cerebral microcirculation are currently unknown. Herein, we assessed TNF-α/NF-κB signaling and inducible nitric oxide synthase (iNOS), and the effects of the TNF-α inhibitor infliximab after TBI. Whether pericyte damage is dependent on the TNF-α/NF-κB/iNOS axis was also evaluated to explore the mechanisms underlying disturbances in the microcirculation after TBI. Microglia are activated after TBI to promote inflammatory factors and free radical release, and upregulate NF-κB and iNOS expression. After lipopolysaccharide treatment, the activity of TNF-α/NF-κB/iNOS in BV2 cells was also upregulated. Inhibition of TNF-α using infliximab reduced NF-κB phosphorylation and nuclear translocation and downregulated iNOS expression, which attenuated the inflammation and oxidative damage. Meanwhile, inhibition of TNF-α reversed pericyte marker loss, and improved pericyte function and microcirculation perfusion after TBI. In conclusion, our study suggests that microglia released TNF-α after TBI, which promoted neuroinflammation and oxidative stress by activating downstream NF-κB/iNOS signals, and this led to pericyte-mediated disturbance of the cerebral microcirculation.
Introduction
Secondary injury induced by traumatic brain injury (TBI) is considered to be a reversible pathological process, which involves vascular, cellular, metabolic, molecular, and other factors, of which neuroinflammation and oxidative stress are the key links. 1 -3 In the past, TBI research focused on neurons, ignoring the role of glia, and the cerebrovascular system. 4 After the concept of the neurovascular unit (NVU) was proposed, people appreciated that neurons, blood vessels, and glia as a whole maintain homeostasis and the function of the central nervous system (CNS) through crosstalk. 5 -7 The conceptual framework of the NVU may contribute to understanding the pathophysiological changes of TBI. Recent studies have demonstrated that the NVU is a key participant in secondary injury following TBI. 8,9
The NVU is composed of neurons, microglia, astrocytes, pericytes, smooth muscle cells, and endothelial cells. It is the minimum acting unit necessary to maintain the proper function of the CNS. 10 The NVU maintains blood–brain barrier (BBB) and vascular integrity through the interaction between its constituent cells. 11 After TBI, microvascular dysfunction is mainly manifested by NVU dysfunction, which is regulated by pericytes. 12 In the NVU, pericytes interact with neighboring cells and process signals to execute multiple functional responses, particularly to the regulation of the microvessels. 13,14 Recent studies have shown that pericyte degeneration after TBI led to regional microcirculatory hypoperfusion and an increase of BBB permeability, which mediated microcirculation disturbance. 15 -17 A deeper exploration of pericyte-mediated microcirculation disorders is essential to investigate the pathological mechanisms following TBI.
An increasing body of evidence shows that the pericyte-mediated microcirculation function is influenced by neuroinflammation. 18,19 Microglia are the major immunocompetent cells in the NVU, and their activation is a hallmark of neuroinflammation. 6,20 After the initial injury, the altered microenvironment and intracellular components released from damaged cells trigger local glial activation and recruitment. 7 Activated microglia induce neuroinflammation by promoting the release of inflammatory factors, inflammatory cell infiltration, as well as free radical production. 21,22 The generated free radicals not only induce oxidative stress but also act as an inflammatory mediator to amplify inflammation. 2 In the brain, a large pool of microglia are located at the proximal region surrounding the cerebrovasculature. Moreover, confocal laser scanning microscopy revealed the highest density of microglial endfeet contacting the glial basement membrane around capillaries. These structural features allow a close interaction between microglia and pericytes. Indeed, many studies have demonstrated a very tight spatiotemporal correlation between vascular activation, cerebral blood flow (CBF) restriction, BBB breakdown, and activation of microglia. 23 -25
Under pathological conditions, the inflammation-related tumor necrosis factor α (TNF-α)/nuclear factor-κB (NF-κB) pathway is activated, which is closely related to the prognosis of TBI. 26,27 TNF-α activates IKK-α/β by binding to its receptor, causing IκB phosphorylation and promoting NF-κB nuclear translocation, which in turn regulates NF-κB-related signaling. 26 As a pro-inflammatory signal, the TNF-α/NF-κB pathway plays a central role in initiating and regulating the cascade of inflammatory factors. 28,29 In addition, TNF-α/NF-κB can also participate in the inflammatory process of diseases by inducing oxidative stress. 30 -32 The overactivated inflammatory response releases abundant inflammatory factors, induces oxidative stress, and produces excessive oxidative free radicals, including reactive oxygen species (ROS) and reactive nitrogen species (RNS). Inducible nitric oxide synthase (iNOS), a subtype of nitric oxide synthase that is only induced under pathological conditions, is regulated by NF-κB. When iNOS is activated, it can release a large amount of NO, which leads to the excessive production of ROS/RNS. 33 –36 In the brain, the unchecked ROS/RNS release leads to lipid peroxidation of cell membranes. This in turn disrupts phospholipid-dependent enzymes and ionic gradients resulting in other sequelae, including disturbances of the CBF, BBB permeability changes, and the development of edema. 34 Many studies assessing the roles of ROS/RNS have demonstrated their major role in the signal transduction associated with vascular contraction and relaxation. 34,37 Oxidative stress triggered by free radicals is closely associated with the functional regulation of vascular cells. Free radicals, particularly RNS can impair pericyte function, stimulating pericyte contraction. Subsequently, the contracted pericytes die upon further activity of damaging factors, leading to sustained constriction of the microvasculature. 34,38,39
Considering that the TNF-α/NF-κB/iNOS pathway is an important regulatory factor of the neuroinflammatory response, it may be closely related to pericyte-mediated microcirculation. Therefore, we speculated that the activation of the TNF-α/NF-κB/iNOS pathway after TBI promotes neuroinflammation and oxidative stress, which may impair cerebral microcirculation by affecting the function of pericytes.
Methods
Randomization and blinding
All animals were randomized for group allocation and surgical procedures and included in the analysis. The operators responsible for the experimental procedures and data analysis were blinded and unaware of group allocation throughout.
Experimental model and drug administration
All animal experiments were approved by the 900th Hospital Ethics Committee (Fuzhou, China). Adult male C57BL/6 mice (20-25 g) were purchased from the experimental animal facility of Fujian Medical University. All mice were randomly divided into three groups: a control group, a TBI group, and a TBI+infliximab (IFX) group (n = 24 per group). Each group was randomly divided equally into four subgroups. One subgroup (n = 6) was used for CBF monitoring, and paraffin sections of the brain were produced after monitoring. One subgroup (n = 6) was used for Western blot analysis, enzyme-linked immunosorbent assay (ELISA), and brain water content assessment. Another subgroup (n = 6) was used for the Evans blue (EB) extravasation assay. The last subgroup (n = 6) was used for neurological assessment.
The details of the damage caused by controlled cortical impact (CCI) have been described previously. 40 First, anesthesia was induced with 3% isoflurane in a container. After the animal was successfully anesthetized, anesthesia was maintained with 1.5% isoflurane delivered using a small-animal anesthetic machine (RWD Life Science Co., China). Then each mouse was placed in a stereotactic frame. The craniotomy was centered 2.5 mm to the right of the sagittal suture and 2.5 mm posterior to the coronal suture, and a bone window approximately 3 mm in diameter was abraded (Fig. 1A). The craniotomy area of the mice was subsequently impacted using a 2 mm metal flat-tip impactor (Brain and Spinal Cord Impactor, 68099H, RWD Life Science). The velocity was 5 m/sec, the depth was 3 mm, and the impact duration was 100 msec. Then the scalp was closed with a suture, and the mice were put back into their home cages. The control group animals received identical surgical procedures without CCI.

Traumatic brain injury (TBI) induces neuroinflammation and oxidative stress.
Pharmacological inhibition of TNF-α was performed using IFX, as previously described. 41 Approximately 30 min after TBI, the TBI+IFX group was intraperitoneally injected with IFX (diluted to 2.5 mg/mL in saline, 10 μg/g, cilag Ag) once per day. The subgroups used for neurological assessment were treated for 7 days, and the remaining subgroups were treated for 3 days. The remaining groups were injected with the same dose of saline as used in the TBI+IFX group.
Cell culture
The microglial cell line BV2 was obtained from the China Infrastructure of Cell Line Resources (Beijing, China) and cultured in a medium comprising 90% Dulbecco's Modified Eagle's Medium (Invitrogen, USA), 10% fetal bovine serum (Hyclone, USA), and 1% antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin) at 37°C in a humidified atmosphere of 5% CO2.
Preparation of paraffin-embedded sections
At 3 days after TBI, after deep anesthesia with sodium pentobarbital, the mice were transcardially perfused with phosphate-buffered saline (PBS) followed by 4% paraformaldehyde. Then, the brains were removed and post-fixed by immersion in fixative solution for 48 h. Subsequently, tissues were embedded in paraffin and 4-μm sections were prepared. The sections were then dewaxed in xylene, rehydrated in graded ethanol and deionized water, and then processed for immunohistochemistry, immunofluorescence, Nissl staining, and terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining.
Immunohistochemical analysis
Paraffin sections were subjected to antigen retrieval followed by incubation with an antibody against ionized calcium-binding adapter molecule (Iba)-1 (1:500, ab178847, Abcam, UK), a disintegrin and metalloproteinase 17 (ADAM17; 1:200, ab57484, Abcam), NF-κB p65 (1:200, ab239882, Abcam), or iNOS (1:250, ab178945, Abcam), washed and then incubated with secondary antibody. Five sections from each mouse were selected for quantification using ImageJ software. The signal intensity was evaluated as follows: 0, < 5% positive cells; 1, 5-20% positive cells; 2, 20-50% positive cells; 3, 50-75% positive cells; and 4, > 75% positive cells.
ELISA
Concentrations of TNF-α, interleukin (IL)-1β, IL-6, interferon (IFN)-γ, ROS, RNS, and cyclic guanosine monophosphate (cGMP) in brain tissue samples were measured using ELISA kits (Jingmei Biotechnology, China) according to the manufacturer's instructions.
Immunofluorescence staining
After antigen retrieval, sections were incubated with antibodies against Iba-1 (1:500, ab178847, Abcam), TNF-α (1:200, ab183218, Abcam), CD31 (1:200, ab281583, Abcam), NeuN (1:500, ab279296, Abcam), NF-κB p65 (1:200, ab239882, Abcam), iNOS (1:200, ab178945, Abcam), platelet-derived growth factor receptor (PDGFR) β (1:200, 3169T, CST), ZO-1 (1:200, ab276131, Abcam), or occludin (1:200, ab216327, Abcam). After washing, the sections were incubated with secondary antibodies for 1 h at room temperature. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Images were captured with a fluorescence microscope (Leica, Germany).
For the lipopolysaccharide (LPS) group, BV2 cells were stimulated with LPS for 24 h, while the control group was not treated with LPS. After washing with PBS, the cover-slips were fixed with paraformaldehyde. Then the cover-slips were permeabilized with 0.1% Triton X-100 and blocked with 5% bovine serum albumin, and incubated overnight with primary antibodies against Iba-1 (1:500, ab178847, Abcam) and TNF-α (1:200, ab183218, Abcam). After incubation with the secondary antibody and DAPI, images were captured with a fluorescence microscope.
TNF-α assay in BV2 cells
The supernatants of BV2 cells were collected, and the concentrations of TNF-α were measured using an ELISA kit (Jingmei, China) according to the manufacturer's instructions.
Western blot analysis
Cultured cells were lysed with radioimmunoprecipitation assay (RIPA) lysis buffer, then supplemented with protease and phosphatase inhibitors, scraped off the flasks, and collected for protein extraction. Mice were sacrificed 3 days after modeling and tissue samples were collected from the cortex around the injury and were extracted with RIPA lysis buffer. Lysates were incubated on ice and supernatants were collected after centrifugation. The protein concentration was determined using a BCA protein assay kit (Abcam). Then, 30 μg of protein was loaded on a gel and separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Subsequently proteins were transferred to polyvinylidene difluoride membranes and probed with primary antibodies against NFκB p65 (1:1000, ab239882, Abcam), p-NFκB p65 (1:1000, ab86299, Abcam), iNOS (1:1000, ab178945, Abcam), 3-nitrotyrosine (3-NT; 1:1000, ab61392, Abcam), PDGFRβ (1:500, 3169T, CST), α-smooth muscle actin (α-SMA; 1:500, 19245T, CST), cleaved caspase3 (1:1000, ab214430, Abcam), Bax (1:1000, ab32503, Abcam), Bcl2 (1:1000, ab182858, Abcam), neuron-glial (NG) 2 (1:1000, ab275024, Abcam), ZO-1 (1:500, ab276131, Abcam), occludin (1:1000, ab216327, Abcam), claudin-5 (1:1000, ab131259, Abcam), or aquaporin (AQP) 4 (1:1000, ab259318, Abcam) followed by incubation with secondary antibodies. Immunoblots were visualized using the ECL Western Blotting Detection System (Millipore). Expression levels were normalized against β-actin (1:2000, ab8226, Abcam) or Lamin B1 (1:2,000, ab16048, Abcam).
Nissl staining
After paraffin sections were stained with Nissl solution (Boster Biotech), five regions of interest (ROIs) around the injured area were randomly selected using a high magnification microscope, and the damaged neurons were counted using ImageJ software. The proportion (%) of damaged neurons was used for the statistical analysis.
TUNEL staining
A TUNEL assay was performed using an apoptosis kit according to the manufacturer's instructions (Roche, USA). Slices were incubated with NeuN (1:500, ab279296, Abcam) overnight, and after washing in PBS, the samples were incubated with TUNEL reaction mixture for 1 h. TUNEL-positive neurons around the injured area were observed and counted with a microscope at high magnification. Five ROIs were selected for quantification and averaged for statistical analysis.
Foot-fault test
To evaluate the motor function of the mice, a foot-fault test was performed before TBI and at 1, 2, 3, and 7 days after TBI. Mice were placed on a grid and their movement was observed. With each weight-bearing step, the paw may fall or slip between the wire. This was recorded as a foot fault. A total of 50 steps was recorded for the left forelimb.
Neurological score
Nerve injury was assessed by modified neurological severity score (mNSS) and Garcia test. 21, 42 The mNSS included motor, sensory, and reflex tests in mice. The mNSS test was graded on a scale of 0-18, in which a score of 0 indicated normal performance, 1-6 indicated mild injury, 7-12 indicated mean-moderate injury, and 13-18 indicated severe injury. The Garcia test consisted of seven evaluations: spontaneous activity, axial sensation, vibrissae proprioception, and limb symmetry, as well as the ability to perform lateral turning, forelimb outstretching, and climbing. Each test received a score between 0 (worst performance) and 3 (best performance). The evaluation was performed pre-injury and post-injury (1, 2, 3, and 7 days) by two investigators who were blinded to the experiments.
EB extravasation assay
EB (2%, 5 mL/kg, Sigma-Aldrich, USA) was injected via the common carotid artery 2 h prior to sacrifice 3 days after TBI. 17 After sacrifice, the brains were removed, weighed, and homogenized in trichloroacetic acid. The samples were then centrifuged. The absorption of the supernatant was measured using a spectrophotometer at a wavelength of 620 nm. The quantity of EB was calculated according to a standard curve and expressed as micrograms of EB/g of brain tissue.
Analysis of cerebral edema
At 3 days post-TBI, the brain tissue was removed from the injured side and the wet weight was recorded. Samples were then placed in an oven for 72 h at 90°C and reweighed to determine the dry weight. The following formula was used to determine the brain water content (%) = (total wet weight of brain - dry weight of brain)/total wet weight of brain × 100%. 17
Laser speckle contrast imaging
CBF was monitored using Laser speckle contrast imaging (LSCI). 43 The selection of the ROIs in the LSCI was performed by tools provided by the software, and the values obtained were the average blood flow values in the region. LSCI was used to observe the relative blood flow values in the capillary areas surrounding the impinging lesion in mice before and after craniotomy, and post-injury (5 min, 1 day, and 3 days). Before inducing the model, the regional CBF was recorded as the baseline.
Statistical analysis
All statistical analyses were performed using SPSS 23.0 statistical software. The results are expressed as mean ± standard deviation. The comparison between two groups was performed using an independent samples t-test, while the statistical difference between each group was evaluated by one-way analysis of variance (ANOVA) with Bonferroni correction for post hoc multiple comparisons. Differences with p < 0.05 were considered statistically significant.
Results
Trauma induces microglial activation and promotes neuroinflammation and oxidative stress
Microglia are the key driver of the inflammatory response in CNS. 6 At 3 days after TBI, immunohistochemistry showed that the microglial marker Iba-1 increased significantly, suggesting that trauma induced microglial activation and proliferation (Fig. 1B). The expression levels of inflammatory factors (TNF-α, IL-1β, IL-6, IFN-γ) were measured after TBI using ELISA, and results showed that the TBI group had significantly higher expression levels of inflammatory factors compared with the control group (Fig. 1C). The concentrations of ROS and RNS were also increased (Fig. 1D). These results suggested that microglia were activated after TBI, accompanied by inflammation and oxidative stress.
Neuroinflammation and oxidative damage secondary to TBI are positively correlated with TNF-α. 27 Here, we evaluated the changes of the downstream NF-κB/iNOS pathway by immunohistochemistry. As expected, TBI up-regulated the expression of NF-κB p65 and iNOS. ADAM17, also known as TNF-α converting enzyme, is the trigger of TNF-α pro-inflammatory activity. 29 Our results showed that the expression of ADAM17 around the injury areas in the TBI group was also increased (Fig. 1E, 1F). These results suggested that TNF-α and its signaling pathway may be closely related to the secondary injury following TBI.
Activated microglia release TNF-α and induce the activation of the NF-κB/iNOS signaling axis
From the above results, we found that TNF-α/NF-κB/iNOS axis activation may play an important role in the secondary injury of TBI. However, it is unclear whether this axis is associated with activated microglia, so we further explored the relationship between them in microglia (BV2 cells) cultured in vitro. After LPS stimulation, immunofluorescence showed a significant increase in TNF-α expression levels in BV2 cells (Fig. 2A). Meanwhile, the concentration of TNF-α in the supernatant of the culture medium also increased (Fig. 2B). Subsequently, we determined the degree of activation of the NF-κB/iNOS axis in BV2 cells. Western blot analysis revealed that LPS stimulation resulted in phosphorylation of NF-κB p65 and increased the expression of NF-κB p65 and iNOS (Fig. 2C). These results suggested that microglia activated the NF-κB/iNOS pathway by generating TNF-α after LPS stimulation.

Activated microglia upregulate the expression of tumor necrosis factor (TNF)-α, which promotes nuclear factor-κB (NF-κB)/inducible nitric oxide synthase (iNOS) signaling in vitro.
Blocking TNF-α can play a neuroprotective role in the acute phase of TBI
TNF-α is a multi-functional proinflammatory cytokine, and blocking TNF-α is neuroprotective after TBI. 26 Our results suggest that this protective effect may be associated with the inhibition of the TNF-α/NF-κB/iNOS axis. Therefore, we tested our hypothesis by inhibiting TNF-α using IFX. Compared with the TBI group, the expression of TNF-α in microglia was significantly inhibited by IFX (Fig. 3A). Meanwhile, immunofluorescence and western blot analyses showed that IFX could inhibit the phosphorylation and nuclear translocation of p65, and reduce the expression of iNOS (Fig. 3B-E). Moreover, the concentrations of inflammatory factors (TNF-α, IL-1β, IL-6, and IFN-γ) and free radicals (ROS, RNS) in the TBI+IFX group were also significantly decreased compared with the TBI group at 3 days after TBI (Fig. 3F, 3G). Excess NO produced by iNOS can quickly react with superoxide anions to form another powerful free radical, peroxynitrite. 37,44,45 Here, we determined the expression of 3-NT to laterally reflect peroxynitrite levels. 45 Western blot analysis showed that the expression of 3-NT was significantly increased after TBI, suggesting that the nitrification of protein tyrosine residues mediated by peroxynitrite increased, and IFX could down-regulate its expression (Fig. 3E).
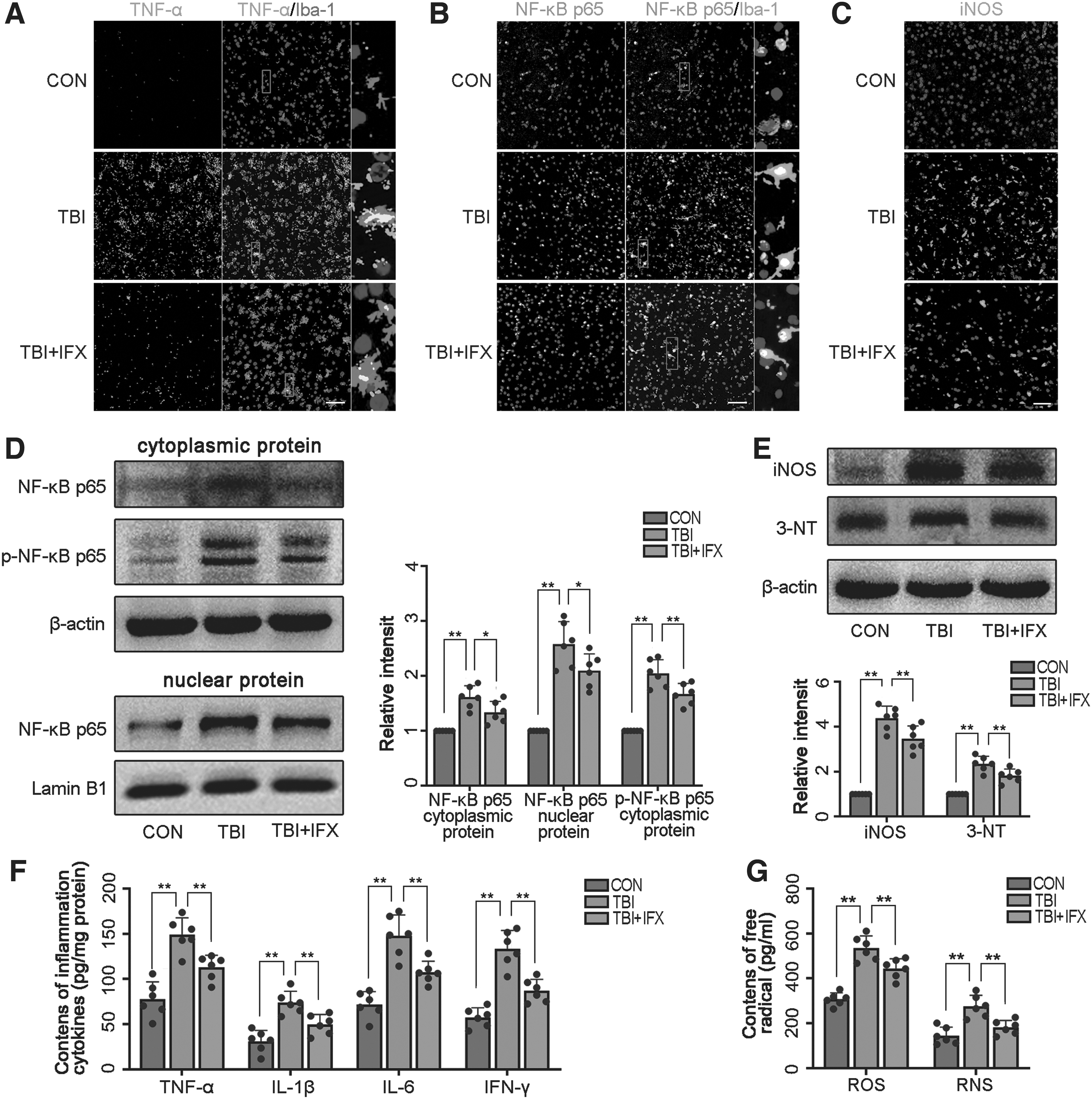
Infliximab (IFX) reduces neuroinflammation and oxidative damage by inhibiting the activation of the tumor necrosis factor (TNF)-α/nuclear factor-κB (NF-κB)/inducible nitric oxide synthase (iNOS) pathway.
Then, we used Nissl staining to assess the damage in neurons. At 3 days after injury, the percentage of injured cells in the TBI group was significantly higher than that in the control group (Fig. 4A). TUNEL staining was used to evaluate the apoptosis of neurons. The results demonstrated that TUNEL-positive neurons were increased in the TBI group compared with the control group (Fig. 4B). Western blot analysis further revealed that TBI resulted in the upregulation of apoptotic factors (cleaved caspase3, Bax) in the cortex after TBI, whereas the anti-apoptotic factor, Bcl-2, was decreased (Fig. 4C). However, these phenomena were inhibited to a certain extent by IFX, indicating that inhibition of TNF-α was beneficial for improving neuronal injury and apoptosis (Fig. 4A-C).
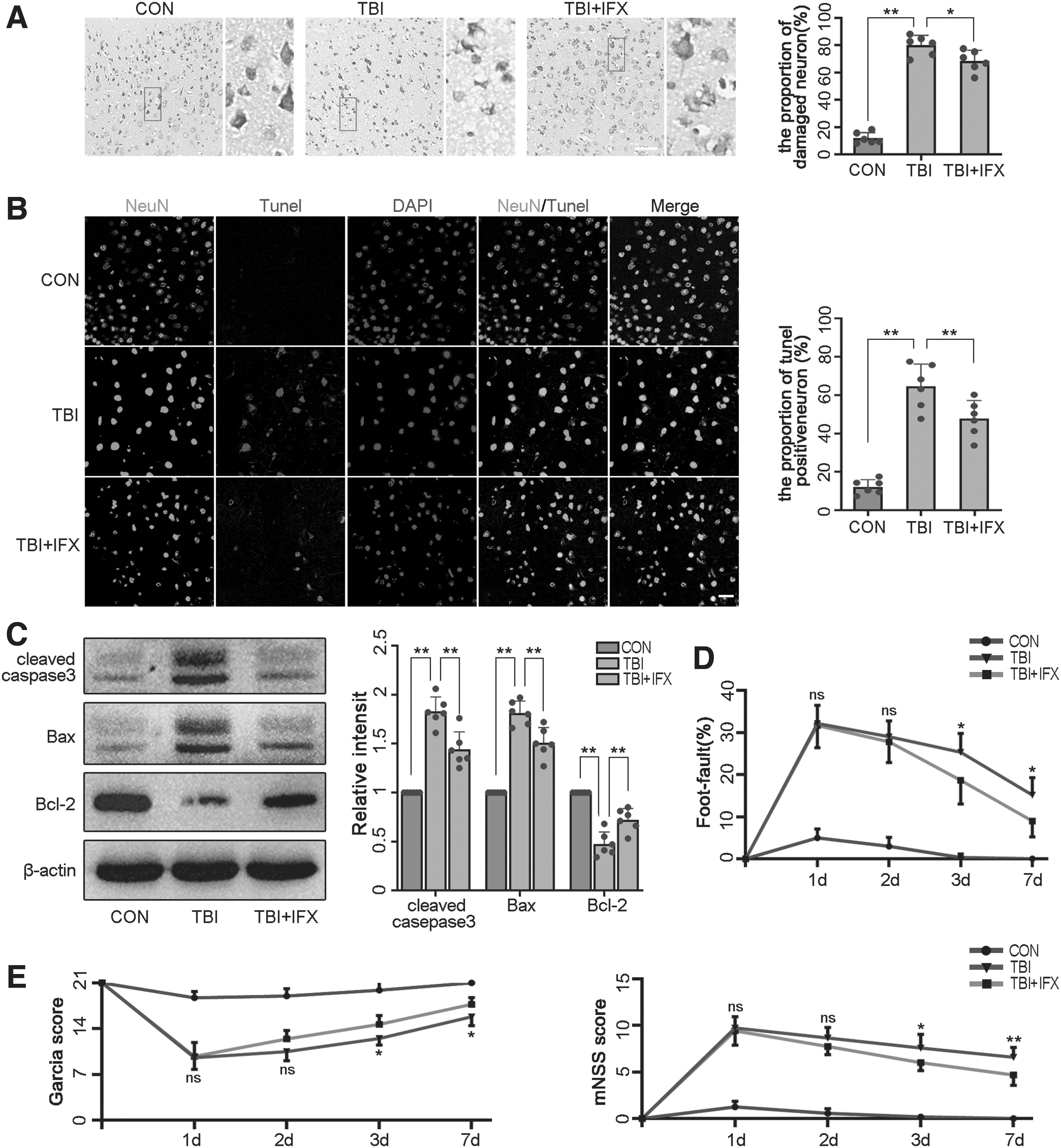
Inhibition of tumor necrosis factor (TNF)-α can play a neuroprotective role after traumatic brain injury (TBI).
Then, we used the foot-fault test, mNSS and Garcia test to assess neurological function. The neurological deficit in the TBI group and TBI+IFX was most severe at 1 day and gradually recovered with time. At 3 days, the neurological function in the TBI+IFX group was significantly improved when compared with the TBI group (Fig. 4D, 4E; Supplementary Tables S1-S3). These results supported our hypothesis that blocking TNF-α after TBI could play a neuroprotective role in the acute phase of injury by reducing neuroinflammation and oxidative stress, and this protective effect was associated with the inhibition of the NF-κB/iNOS pathway.
Neuroinflammation and oxidative stress secondary to TBI induce pericyte degeneration
We aimed to understand the effect of TBI on pericytes by detecting the changes in pericyte markers. Immunofluorescence imaging showed that the pericyte marker (PDGFRβ) and endothelial cells (CD31) were co-located in cerebral capillaries. The expression of PDGFRβ in the TBI group decreased significantly compared with the control group. Interestingly, the level of PDGFRβ in the TBI+IFX group was significantly higher than that in the TBI group (Fig. 5A). Then, we analyzed the expression of PDGFRβ and the other pericyte markers α-SMA and NG2 using western blot analysis. The data showed that the expression of PDGFRβ, α-SMA, and NG2 in the TBI+IFX group was significantly increased compared with that in the TBI group (Fig. 5B). Our results suggest that pericyte degeneration after TBI is associated with inflammation and oxidative stress mediated by the TNF-α/NF-κB/iNOS axis.
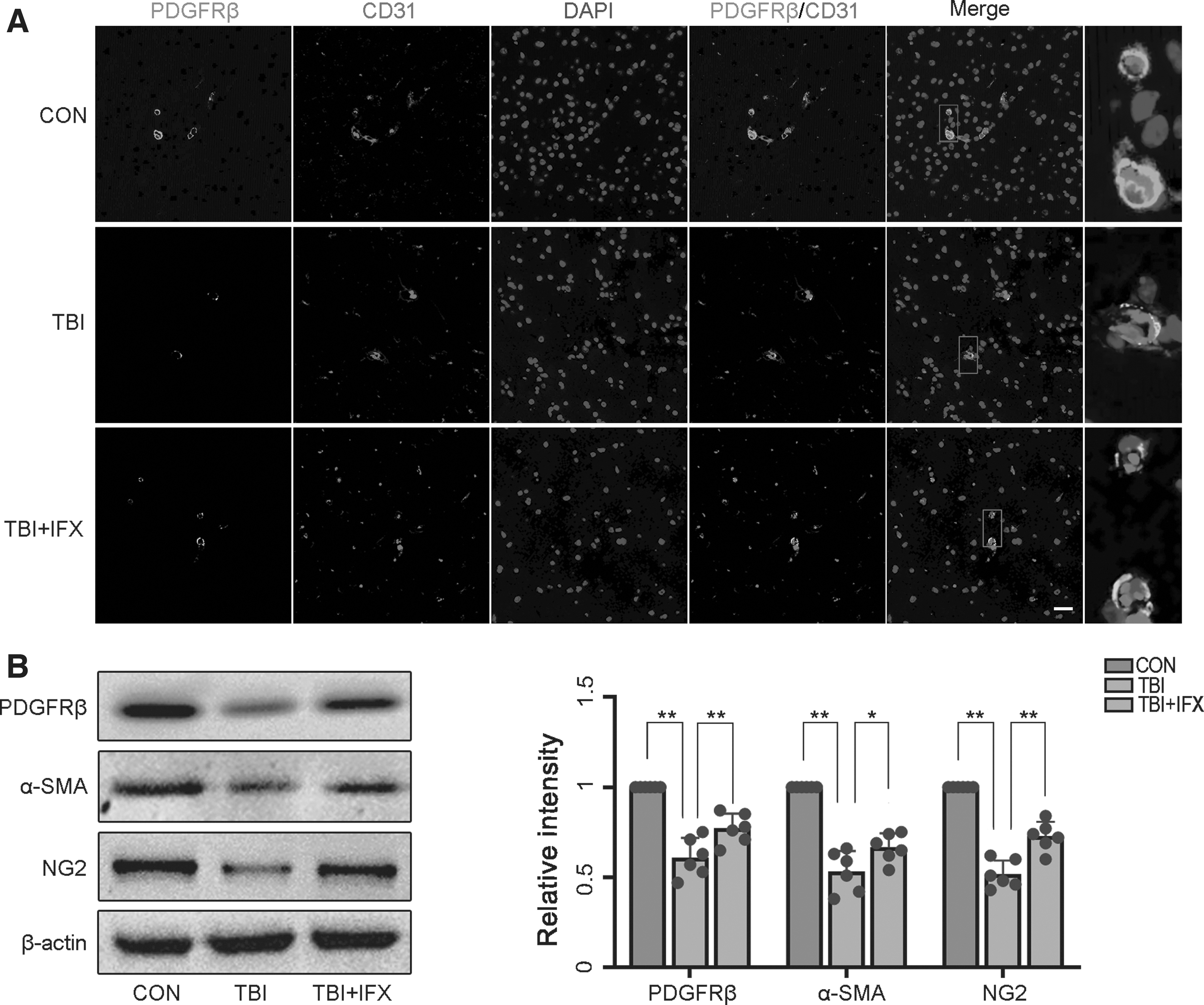
Traumatic brain injury (TBI) results in loss of pericyte coverage.
Pericyte degeneration mediates the destruction of the BBB and secondary vasogenic edema
Pericytes are involved in regulating the function and structural integrity of the BBB, including the formation of tight junction (TJ) proteins. 17 Using immunostaining, we analyzed the expression of TJ proteins (occludin and ZO-1) and assessed their co-localization with endothelial cells (CD31). Our results confirmed the expression of occludin and ZO-1 were significantly decreased after TBI, and IFX reduced the decrease in proteins caused by trauma (Fig. 6A). Subsequently, we analyzed the expression of TJ proteins including occludin, claudin-5, and ZO-1 by western blot analysis. Similar to the results of the immunofluorescence, occludin, claudin-5, and ZO-1 expression were increased in the TBI+IFX group compared with the TBI group (Fig. 6B). Next, we investigated the effect of the reduction in TJ proteins on BBB permeability by measuring the exudation of EB. The results showed that compared with the control group, the exudation of EB significantly increased in the brain tissues in the TBI group, but decreased in the TBI+IFX group (Fig. 6C).
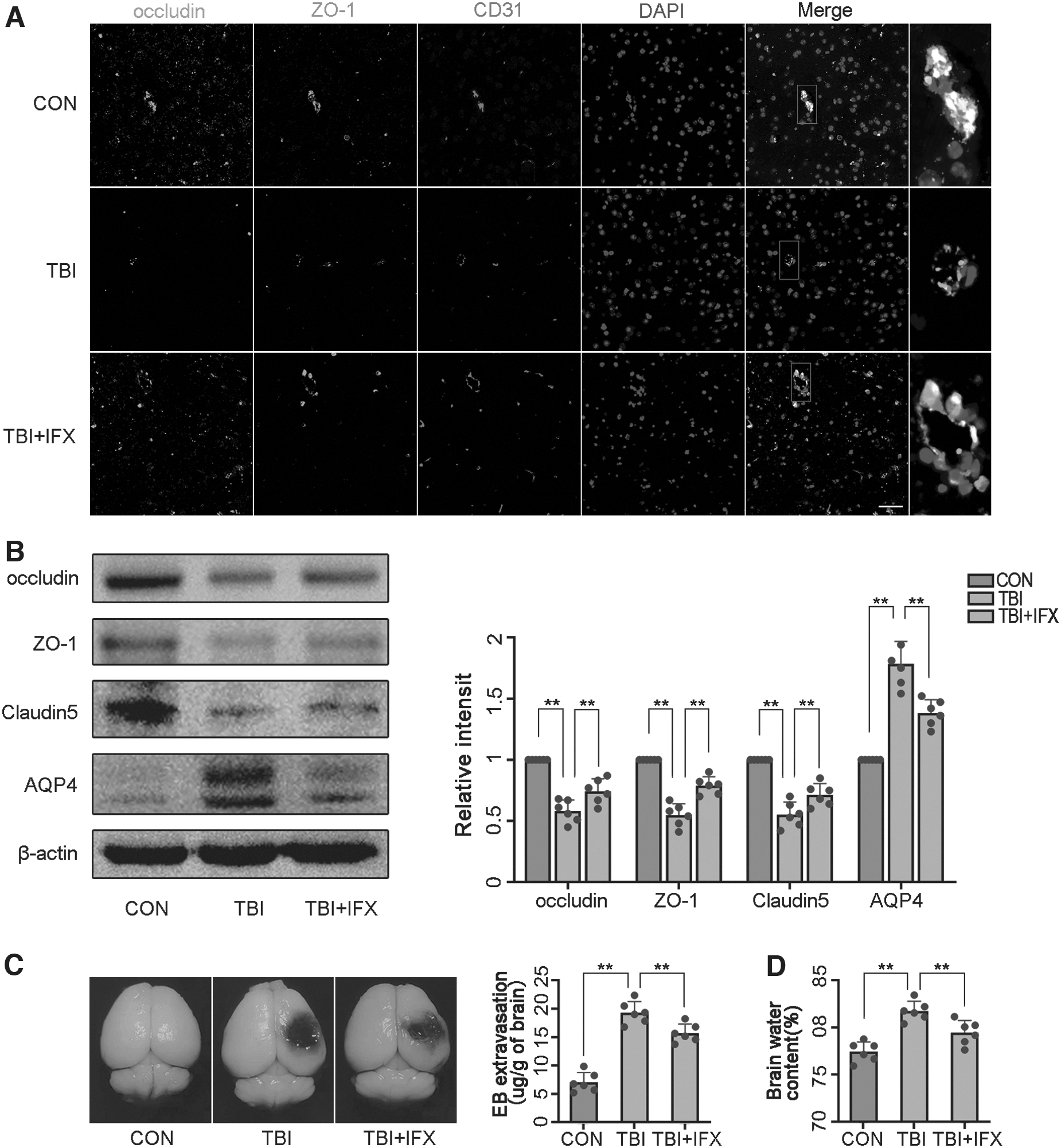
Traumatic brain injury (TBI) destroys the blood brain barrier and aggravates brain edema by mediating pericyte degeneration.
After TBI, BBB disruption leads to increased cerebrovascular leakage, resulting in vasogenic edema. AQP4, as the most important water channel protein in the mammalian brain, is closely related to vasogenic brain edema. 40 The results of the Western blot analysis showed that AQP4 expression was enhanced after TBI, which was inhibited by IFX (Fig. 6B). To further verify the consequences of BBB destruction and high AQP4 expression, we analyzed the water content of damaged brain tissue to assess brain edema. The results showed that brain water content in the TBI+IFX group was reduced compared with that in the TBI group (Fig. 6D).
Pericyte degeneration mediates limited perfusion into the cerebral microcirculation
Recent studies suggest that microvessels covered by pericytes play a key role in local cerebral blood flow regulation. 13,46 LSCI was used to monitor the changes in microcirculation blood flow at different time points. We found that craniotomy caused minor damage to the cortical vessels, which slightly reduced the blood flow of the local microcirculation. In the TBI group, the cerebral blood flow decreased significantly after impact, especially in the microcirculation around the injury areas. With the extension of time, the microcirculation perfusion gradually recovered in each group. In the TBI+IFX group, the microcirculation blood flow around the lesioned areas was improved at 3 days compared with that in the TBI group, which was close to the preinjury level. However, the blood flow values at other time points were not significantly different from those in the TBI group (Fig. 7A-C). This phenomenon is consistent with the determination of cGMP in the affected side of the brain, which is an important mediator of NO-mediated vasodilation. 33,47 The cGMP levels in the TBI+IFX group were increased at 3 days compared with the TBI group but remained lower than the control group (Fig. 7D).
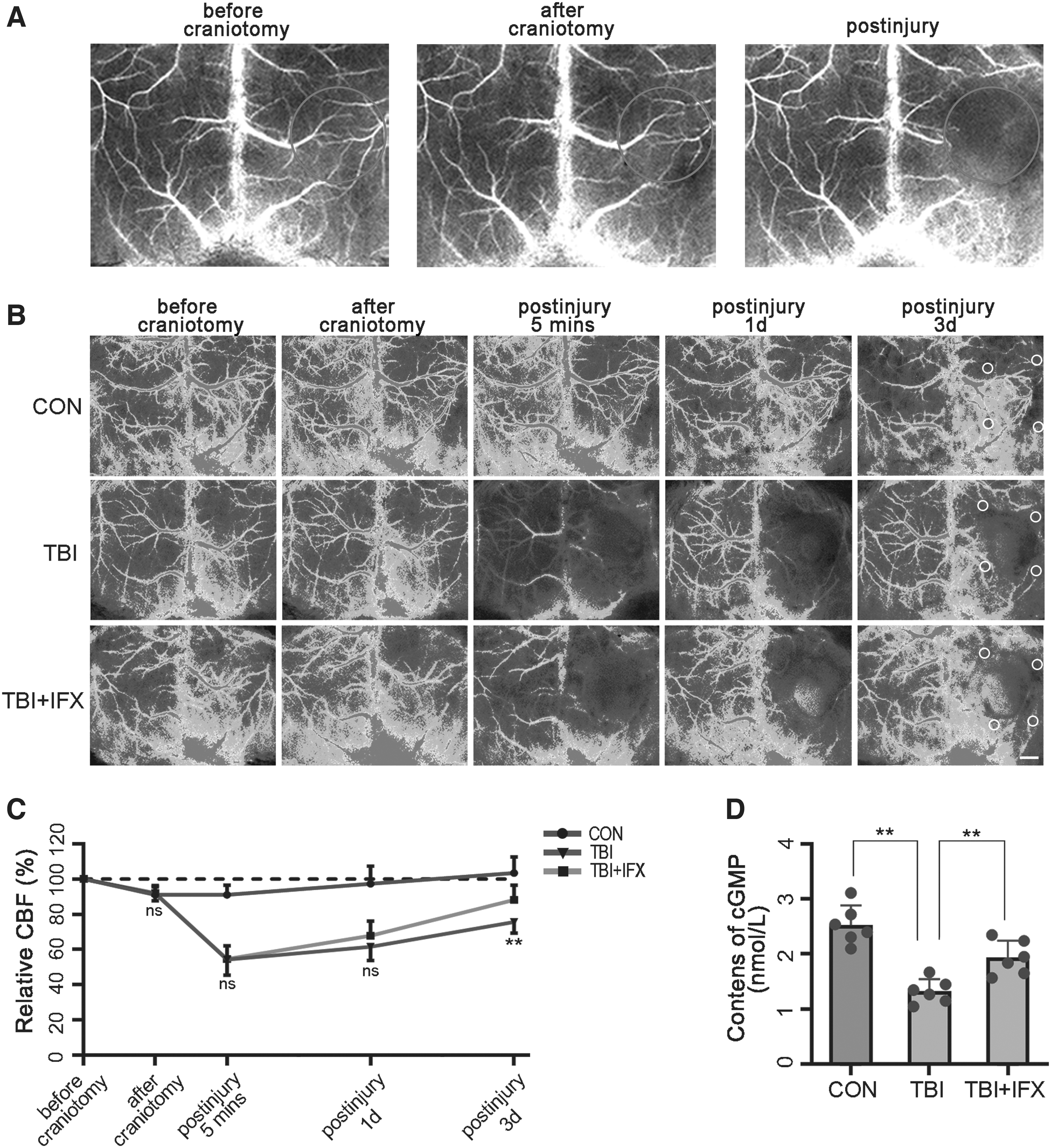
Pericyte dysfunction affects microcirculatory blood flow after traumatic brain injury (TBI).
Discussion
Microcirculation disturbance mediated by pericytes is an important factor of secondary brain injury after TBI, which is closely related to pathological processes such as neuroinflammation and oxidative stress. 1,48 Our study corroborated the importance of neuroinflammation and oxidative stress in the secondary injury following TBI. Inhibition of the inflammatory factor TNF-α can improve the prognosis of TBI. Meanwhile, we explored the effects of neuroinflammation and oxidative stress on pericyte-mediated microcirculation by assessing the TNF-α/NF-κB/iNOS pathway. We clarified that the TNF-α/NF-κB/iNOS axis in microglia affects the microcirculation by regulating pericyte function and then plays a vital role after TBI (Fig. 8).
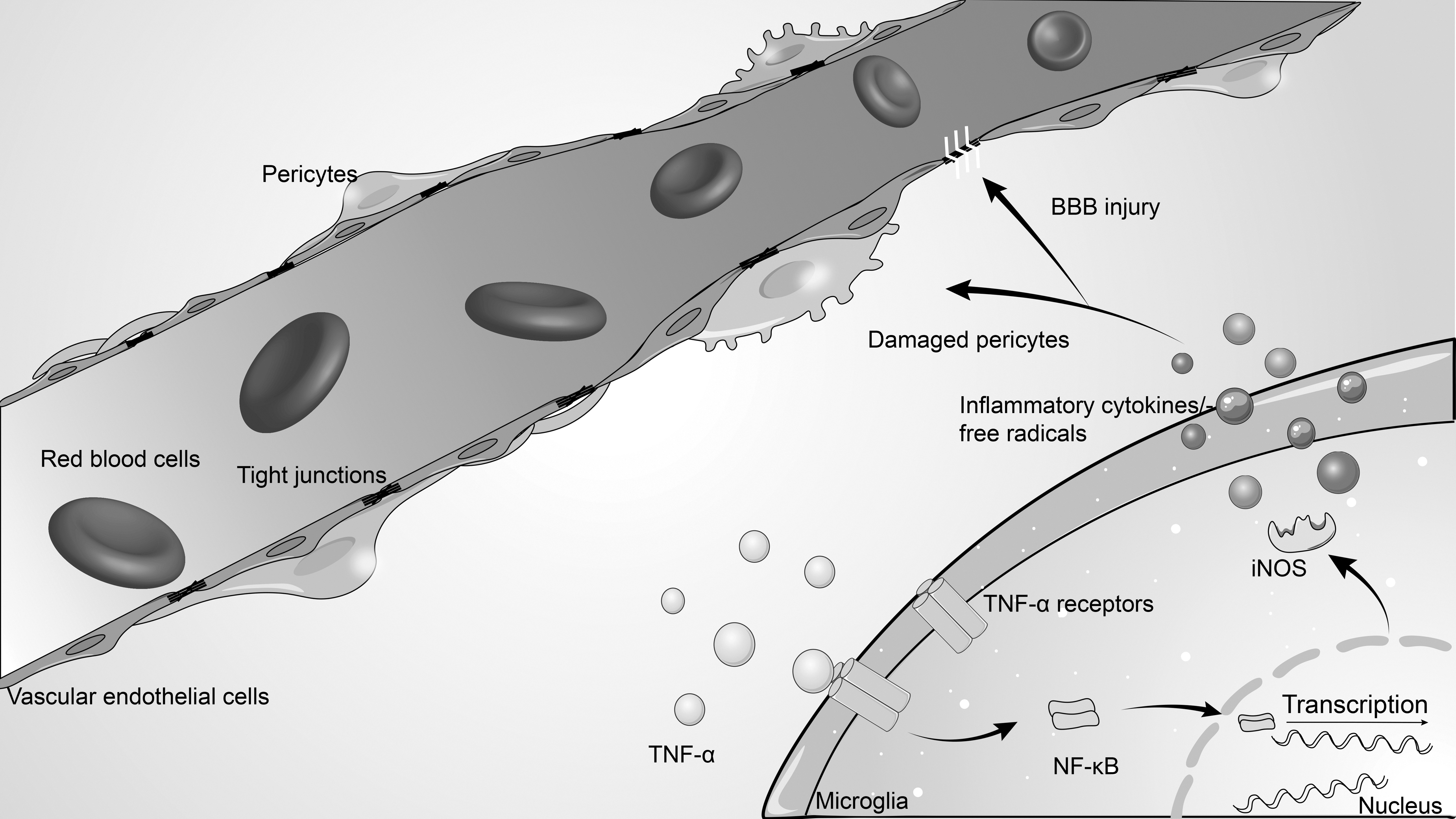
Schematic illustrating of possible mechanisms of pericyte-mediated microcirculation disturbance after traumatic brain injury (TBI). As illustrated, tumor necrosis factor α induces intracellular nuclear factor-κB/inducible nitric oxide synthase signaling activation by binding to surface receptors of microglia after TBI, which in turn promotes the release of a large number of inflammatory cytokines and free radicals. Excessively activated neuroinflammation and oxidative stress act on adjacent capillaries, resulting in pericyte dysfunction and damage, which impairs local blood perfusion and blood–brain barrier integrity, resulting in microcirculation disturbance.
Pericytes are vascular mural cells in the NVU that play a major role in the regulation of the microcirculation. 13,14,46 Studies in adult and aging brains demonstrated that pericytes are required for BBB integrity, capillary perfusion. 17 To investigate the effects of TBI on pericytes, we used a combination of pericyte markers including PDGFRβ, α-SMA, and NG2 to visualize pericyte changes. At 3 days after TBI, the expression levels of pericyte markers were significantly decreased compared with those in the control group, suggesting the degeneration and loss of pericytes. It was also accompanied by disruption of TJ proteins, which suggested damage to the BBB. Loss of pericyte-endothelium integrity has been shown to increase transendothelial fluid flow and paracellular transport as a result of reduced TJ protein expression, both causing BBB disruption. 17 Further studies showed that the leakage of EB in brain tissue increased in the TBI group, accompanied by the increase of AQP4 expression and brain water content. These results confirmed the damage of the BBB. To evaluate the changes of pericyte-mediated microcirculation perfusion after TBI, we used LSCI to monitor CBF. The results showed that the perfusion of the microcirculation around the injury areas was significantly inhibited after trauma, and gradually recovered with time.
TNF-α/NF-κB signaling is the most important pathway in the inflammatory response. 49 NF-κB is associated with the expression of numerous cytokines and is involved in the regulation of the inflammatory response, oxidative stress, apoptosis, and other pathological processes. 21,50,51 The NF-κB family consists of five structurally related subunits including P50, p52, p65, RelB, and c-Rel. LPS can stimulate c-Rel activation, enhance the binding of c-Rel to the NF-κB site in the iNOS promoter and thus induce iNOS expression. 35,36 Previous studies have suggested that the activation of the TNF-α/NF-κB/iNOS axis is related to the inflammatory response and oxidative stress. 45,52 Our findings suggested that inflammation-associated microglia were activated following trauma, accompanied by increased NF-κB phosphorylation and nuclear translocation, and upregulate iNOS expression. Further experiments in vitro also confirmed the relationship between the activation of the TNF-α/NF-κB/iNOS axis and inflammation in BV2 cells. These results are consistent with those of a previous study. 53
To verify the role of TNF-α in TBI, we utilized IFX to antagonize TNF-α. IFX is a monoclonal antibody that binds to TNF-α, and its protective effects including anti-inflammation, anti-oxidative stress, and anti-apoptosis. 27 We found that IFX inhibited the expression of TNF-α in microglia and downregulated NF-κB/iNOS pathway activity. In addition, the expression of inflammatory cytokines, free radicals, and 3-NT decreased in the brain, and neuronal injury and apoptosis were reduced, as well as neurological deficits. These results indicated the critical role of TNF-α in the secondary injury following TBI, which was associated with the NF-κB/iNOS axis.
A previous report has shown that NF-κB/iNOS pathway activation was involved in mediating pericyte apoptosis. 54 In our study, we found that inhibition of the TNF-α/NF-κB/iNOS axis could significantly ameliorate pericyte and TJ protein loss, thus contributing to the preservation of the integrity of BBB structure and function. We also measured the content of cGMP in brain tissue, which is a mediator of NO-mediated vasodilation. 33,47 Cyclic guanosine monophosphate–related signaling is one of the pivotal mechanisms by which pericytes regulate blood flow. 55,56 Through IFX treatment, we found that microcirculation perfusion was improved to some extent at 3 days after TBI, and cGMP levels were also increased accordingly. What is interesting is that our experimental results are contrary to Foley's conclusion. Lesley's study based on iNOS knockout mice claims that iNOS plays a positive role in the recovery of CBF after TBI. 57 We speculate that the contradictory conclusions may be related to different means of blood flow measurement, while iNOS knockout may carry other unanticipated effects. Because NO has a complex role, both neuroprotective and neurotoxic, an appropriate concentration of NO can be of benefit in maintaining blood perfusion.
Nevertheless, this study had limitations. First, we explored the impact on pericytes after activation of the TNF-α/NF-κB/iNOS axis in microglia, but we ignored the effect of other NVU components, such as astrocytes. Astrocytes, as one of the major cells in the NVU with their end-feet in direct contact with the vessel surface, may be more spatially related to pericytes. However, considering the dominant role of microglia in neuroinflammation, we preferred to explore the dialogue between microglia and pericytes after TBI. Second, vascular endothelial cells are also involved in the regulation of the microcirculation, and secondary injury after TBI may also destroy the microcirculation by damaging the endothelium. In addition, it remains unclear by which pathway the TNF-α/NF-κB/iNOS axis damaged pericytes, and whether pericytes underwent some specific phenotypic transformation in this process. At the same time, the effective therapeutic time window of IFX and its influences on the ultimate functional outcome in TBI mice remains questionable, as our study was relatively short-term and IFX was given quite early after TBI.
In conclusion, our study demonstrated that pericyte degeneration after TBI mediated microcirculatory disturbance, which was associated with the TNF-α/NF-κB/iNOS signaling axis activation in microglia. The inhibition of TNF-α affected the activity of this signaling axis, thus reducing neuroinflammation and oxidative damage, ultimately protecting cerebral microcirculatory perfusion and BBB integrity and promoting neurological functional repair.
Footnotes
Acknowledgments
Sincere appreciation is given to Dr. Jianhe Zhang from the Affiliated Hospital of Putian University and our colleagues from the 900th Hospital, who participated in this study with great cooperation.
We thank International Science Editing for editing this manuscript.
Funding Information
This work was supported by grants from Dr. Xiangrong Chen's Young and middle-aged backbone talent foundation of Fujian Provincial Commission of Health Construction (2020GGA058), Joint Funds for the innovation of science and Technology, Fujian province (2020Y9033), Dr. Shaorui Zheng's Fujian Provincial Health Technology Project (2019-ZQN-95), and Dr. Shousen Wang's Fujian Joint Fund for Innovation Project (2019Y9045).
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.