
Abstract
Pediatric traumatic brain injury (TBI) is a major public health issue, and a risk factor for the development of post-traumatic epilepsy that may profoundly impact the quality of life for survivors. As the majority of neurotrauma research is focused on injury to the adult brain, our understanding of the developing brain's response to TBI remains incomplete. Neuroinflammation is an influential pathophysiological mechanism in TBI, and is thought to increase neuronal hyperexcitability, rendering the brain more susceptible to the onset of seizures and/or epileptogenesis. We here hypothesized that peripheral blood-derived macrophages, recruited into the injured brain via C-C motif ligand 2 (CCL2) chemokine/C-C chemokine receptor type 2 (CCR2) signaling, contributes to neuroinflammation and thus seizure susceptibility after experimental pediatric TBI. Using Ccr2 gene-deficient mice in the controlled cortical impact (CCI) model of TBI, in 3-week-old male mice we found that TBI led to an increase in susceptibility to pentylenetetrazol (PTZ)-evoked seizures, associated with considerable cortical tissue loss, a robust cellular neuroinflammatory response, and oxidative stress. Intriguingly, although Ccr2-deficiency increased CCL2 levels in serum, it did not exacerbate seizure susceptibility or the neuroinflammatory cellular response after pediatric TBI. Similarly, acute post-injury treatment with a CCR2 antagonist did not influence seizure susceptibility or the extent of tissue damage in wild-type (WT) mice. Together, our findings suggest that CCR2 is not a crucial driver of epileptogenesis or neuroinflammation after TBI in the developing brain. We propose that age may be an important factor differentiating our findings from previous studies in which targeting CCL2/CCR2 has been reported to be anti-inflammatory, neuroprotective or anti-seizure.
Introduction
Traumatic brain injury (TBI) poses a significant health burden on society, 1 and there is a higher risk of TBI with both a younger age (<4 years) and male sex. 2 –4 Moderate-to-severe TBI in young children often results in long-term disability across a range of functional domains. 5,6 In particular, TBI may lead to the development of chronic disorders such as post-traumatic epilepsy (PTE), defined as spontaneous, recurrent seizures, occurring more than 1 week after a head injury. 7,8
At a molecular and cellular level, one of the key features of TBI in both the adult and pediatric brain is a robust neuroinflammatory response. This response involves the activation of central nervous system (CNS)-resident microglia and astrocytes, which release a plethora of inflammatory cytokines such as tumor necrosis factor (TNF)-α, interleukins (ILs) such as IL-1β, as well as chemokines including C-C motif ligand 2 (CCL2; also known as monocyte chemoattractant protein-1 or MCP-1). These mediators are elevated within hours post-injury in the brain parenchyma, cerebrospinal fluid (CSF), and serum, and they have been attributed roles in both neurodegeneration and repair processes. 9 –15 Blood-derived monocytes and macrophages may infiltrate the acutely injured brain, via blood–brain barrier (BBB) disruption as well as active recruitment via chemotaxis, a multi-step process of transmigration and infiltration into the parenchyma. 16,17 This latter process is largely mediated by the binding of CCL2 to the C-C chemokine receptor type 2 (CCR2), which is highly expressed on monocytes and macrophages. 18 In the adult injured rodent brain, reduction of monocyte/macrophage infiltration by pharmacological depletion, genetic manipulation, or pharmacological blocking of CCL2/CCR2 signaling has demonstrated the contribution of peripherally derived immune cells to secondary brain damage. 19 –22
Accumulating evidence supports a central role for neuroinflammation in neuronal and circuit hyperexcitability, with cytokines in particular implicated as factors that can render the CNS microenvironment more susceptible to seizures, and/or drive the process of epileptogenesis. 23 –25 In particular, CCL2 and CCR2 expression are upregulated in human epileptic brain tissue, leading to the hypothesis that CCL2/CCR2 signaling may play a role in the pathogenesis and progression of seizures or hyperexcitability. 26 –29 This may occur via classical chemokine signaling; for example, increased CCL2 leading to recruitment of CCR2+ peripheral macrophages at the epileptic focus after kainic-acid-induced seizures, 30 and infiltrating CCR2+ macrophages further exacerbating seizure-induced neural damage. 31 Further, CCL2 has been reported to act directly on neurons by modulating ion channel expression and composition, 32 decreasing neuronal inhibition, 33 and impairing synaptic currents, 34 which may promote seizure susceptibility of excitatory circuitry. CCL2 may also indirectly predispose toward seizures via the induction of other pro-inflammatory cytokines such as IL-1β, which themselves have been shown to contribute to neuronal death, neuroinflammation, and increased seizure susceptibility in TBI and/or epilepsy. 35 –37
Together, these studies suggest a plausible association between peripherally derived CCR2+ monocyte accumulation in the brain and subsequent seizure activity. In particular, one study demonstrated that pharmacological inhibition of CCL2/CCR2 signaling to reduce macrophage infiltration was sufficient to suppress lipopolysaccharide (LPS)-induced seizures in mice. 38 However, this potential relationship has not yet been explored in the context of TBI. The current study therefore aimed to define the importance of CCR2+ infiltrating immune cells in the pathogenesis of seizure susceptibility, as a potential mechanism underlying the process of epileptogenesis and PTE after pediatric TBI. We hypothesized that recruitment of peripheral CCR2+ macrophages after an experimental TBI in the pediatric mouse would augment hyperexcitability to evoked seizures by exacerbating neuroinflammation.
Methods
Animals and ethics
Ccr2 knock-out (KO) (B6.129S4-Ccr2 tm1Ifc/J) and littermate (C57Bl/6J) wild type (WT) male mice were obtained from an in-house breeding colony maintained at the Alfred Research Alliance Precinct Animal Services in Melbourne, Australia (breeding license #B/BL-2017 1103IL). The heterozygous breeding colony was derived from breeding pairs generously donated by the Martino Lab at the Australian Regenerative Medicine Institute (Monash University, Clayton, Australia), sourced from The Jackson Laboratory (Bar Harbor, ME, USA), and originally generated by Charo and colleagues by targeted vector deletion of the entire Ccr2 coding region. 39
Genotyping was performed by Transnetyx (Cordova, TN, USA) using real-time polymerase chain reaction on ear tissue samples collected by animal facility staff. Specifically, the forward and reverse primers CTTGTCATAAAACCAGTGTGAAGCA and CCCACAAAACCAAAGATGAATACCA, with the reporter sequence ATCCTGCCTCCACTCTAC, were used to detect the Ccr2 WT sequence; whereas the Ccr2 KO assay was comprised of primers TGACCACAGAATCAAAGGAAATGGA (forward) and GGCTTCTGAGGCGGAAAGA (reverse), with the reporter sequence TCAGTTCATCCACGGCCTC (targeting the junction of the WT insertion region; hence this would not amplify in WT samples). A random sampling of genotyping results were further validated by neomycin assay (Transnetyx). Animals were identified by both ear notches and tail markings.
Following weaning, all mice were group housed in ventilated Optimice® cages (4–6 per cage), with unrestricted access to food and water under controlled humidity and a standard 12-h light/dark cycle. All experiments were conducted with approval from the local Animal Ethics Committee (#E/1831/2018/M) and in accordance with the guidelines provided by the Australian Code for the Care and Use of Animals for Scientific Purposes. Animals were randomized to treatment and injury groups, and researchers were blinded to experimental groups throughout procedures, data collection, and analysis.
Experimental design
Ccr2 KO and WT male littermate mice were genotyped prior to weaning and experimental procedures (Fig. 1). On post-natal day (p) 21 ± 1 day (i.e., day 0), pediatric Ccr2 KO and WT mice received either an experimental TBI or sham surgery. A subset of WT and KO mice was euthanized at 3 days post-surgery to collect serum for detection of inflammatory cytokine-protein levels, and brains were either saline-perfused for flow cytometry analysis to identify immune cell phenotypes, or perfusion-fixed for immunofluorescent (IF) staining and microscopy (brain histology). Another subset of mice underwent a chemoconvulsant challenge with a single dose of pentylenetetrazol (PTZ) at 14 days post-surgery to evaluate evoked seizure susceptibility, prior to perfusion-fixation and collection of brains for immunostaining and subsequent analysis of neuropathology. Mice were randomized to four groups: WT/sham, WT/TBI, Ccr2 KO/sham, and Ccr2 KO/TBI (n = 4–9/group for the 3-day time-point, and n = 5–15/group for the 14-day time-point). An additional subset of C57Bl/6J mice was used to initially optimize the dose of PTZ required at 14 days post-surgery (Supplementary Table S1).

Experimental timeline. Genotyping confirmed Ccr2 KO and littermate WT male mice before weaning, prior to experimental procedures commenced. Pediatric (p21) Ccr2 KO and littermate WT mice either received an experimental TBI or sham surgery at day 0. A subset of mice was culled at day 3 post-surgery followed by serum collection for cytokines analysis, saline-perfused brains collection for identifying immune cells via flow cytometry, and perfused-fixed brain collection to perform IF and histological analysis. Another subset of mice underwent PTZ challenge at day 14 post-surgery (p35), followed by perfused-fixed brain collection for IF analysis. Created with
A final subset utilized C57Bl/6J mice to assess whether pharmacological blockage of CCR2 would impact seizure susceptibility after pediatric TBI (Fig. 2A). Mice were randomized to four groups: vehicle/sham, vehicle/TBI, drug/sham, and drug/TBI; n = 10 mice/group. No mortality was reported throughout these studies before the anticipated terminal time-points.

Ccr2 gene deletion had no effect on brain volume loss after pediatric TBI. Representative cresyl violet-stained images at 14 days post-surgery
Controlled cortical impact
Controlled cortical impact (CCI) was used to induce an experimental TBI in Ccr2 KO and littermate WT mice at p21 (± 1 day), as previously described. 35,40 After anesthetizing pups with 4% isoflurane in oxygen in an induction chamber, mice were maintained at 1.5% isoflurane via a nose cone throughout the surgery duration; skin was sterilized with an antiseptic solution (chlorhexidine gluconate [0.5% w/v] in 70% alcohol). The head was stabilized in a stereotaxic frame, then a midline incision was made and an approximately 3-mm diameter craniotomy was performed on the left parietal bone to expose the intact dura, at −1 mm Bregma, 1 mm left of the midline sagittal suture, and 1 mm anterior to Lambda. 41 A moderate TBI was induced in the left parietal cortex using a CCI device (eCCI-6.3; Custom Design and Fabrication Inc., Sandston, VA, USA), with an impact velocity of 4.5 m/sec, penetrating depth of 1.5 mm and dwell time of 150 msec. Sham surgery was similarly performed, except animals did not receive the impact. The skin was then sutured closed, and the surgery site was cleaned with an antiseptic solution. Mice were injected with up to 0.5-mL sterile saline subcutaneously for rehydration, removed from anesthetic, and placed individually in heated cages for recovery. Self-righting and mobility behaviors were observed before returning mice to group housing with littermates (mix of Ccr2 KO and WT, and TBI and sham per cage).
PTZ administration
To determine how pediatric TBI and CCR2 manipulations influenced seizure susceptibility at 14 days post-surgery, individual mice were administered with a single dose of PTZ (Sigma-Aldrich, Burlington, MA, USA; 40 mg/kg i.p.), a pro-convulsant agent commonly used to reveal seizure susceptibility as a surrogate indicator of epileptogenesis. 42,43 This dose was chosen based on a dose-dependent pilot experiment in a separate cohort of mice in which three different doses were tested (30, 40, and 50 mg/kg i.p.). After PTZ administration, individual mice were placed in a fresh clean cage and video-recorded for behavioral responses for a 15-min period, whereby quantification of evoked seizures in response to PTZ reflect changes in brain network excitability. 35 A modified Seizure Severity Score, adapted from the original Racine score, 44 was used to evaluate overall behavior responses to PTZ-evoked seizures, defined as 0 = no response; 1 = behavioral arrest, loss of posture, or immobility; 2 = isolated and focal twitches; 3 = forelimb clonus and head nodding; 4 = rearing; 5 = generalized convulsive seizure (loss of posture, rearing then falling); 6 = repeated and/or extended generalized seizures; and 7 = status epilepticus resulting in death. 35 The latency to develop the generalized convulsive seizure from the time of PTZ injection, and the duration of the seizure(s), were evaluated (n = 10–15/group).
Tissue collection
Mice were euthanized either at 3 days post-surgery, or immediately following the PTZ challenge at 14 days post-surgery, with a single i.p. overdose of sodium pentobarbitone (Lethabarb; Virbac, Australia). Cardiac blood was collected, centrifuged (10,000g for 10 min, then 17,000g for 10 min, at 4°C) to isolate serum (n = 4/group). Transcardial perfusion with ice-cold sterile saline (0.9% NaCl w/v) was followed by 4% paraformaldehyde (PFA) to fix the brain tissue for histology and IF staining. Brains were post-fixed in 4% PFA overnight at 4°C, then transferred to 30% sucrose for 3–5 days prior to embedding in optimal cutting temperature compound, and storage at −80°C before sectioning (n = 5–7/group).
Serum cytokines multiplex
Cytokine concentrations in serum were assessed using a Bio-Plex Pro™ mouse cytokine standard 23-plex (Bio- Rad, Hercules, CA, USA) kit as per the manufacturers' protocol, and run on a Luminex® 200 detection system at 360Biolabs (Melbourne, Australia). Samples were analyzed in duplicate, and results were normalized to standards for quantification in pg/mL (n = 4/group).
Immunofluorescence
Perfusion-fixed brains were cryosectioned to 12-μm thick coronal sections collected onto Superfrost® Plus slides (Thermo Fisher Scientific, Waltham, MA, USA). Following tissue rehydration and blocking of non-specific binding, primary antibodies against the astrocyte marker glial fibrillary acidic protein (GFAP; rabbit polyclonal, Dako, Z0334; 1:1000), microglial marker ionized calcium binding adaptor molecule 1 (IBA-1; goat polyclonal, Abcam, AB5076; 1:500), and CD68 (rat anti-mouse CD68, MCA1957GA, Bio-Rad; 1:500) were incubated simultaneously, overnight at 4°C. Subsequent application of secondary antibodies (Invitrogen, Waltham, MA, USA)—donkey anti-rabbit AF594 (1:250), donkey anti-goat AF488 (1:250), and donkey anti-rat AF488, (1:250) to detect GFAP, IBA-1, and CD68, respectively—were then incubated for 1 h at room temperature, followed by counterstaining with 4′,6-diamidino-2-phenylindole (DAPI; 1:10,000).
A final subset was stained against primary antibody heme-oxygenase-1 (HO-1) (polyclonal rabbit HO-1, #106-08950, Sapphire Biosciences/Enzo Life Sciences, New South Wales, Australia; 1:2000) incubated overnight at 4°C, followed by secondary antibody (donkey anti-rabbit AF 594, Thermo Fisher Scientific, Waltham, MA, USA; 1:250) incubation for 1 h at room temperature then counterstained using Hoechst stain (33342, Thermo Fisher Scientific; 1:5000). Fluorescent mounting media (Dako, Carpinteria, CA, USA) was used to apply glass coverslips on these three different subsets of slides prior to imaging.
Fluorescent images were captured using a Nikon Ti-E inverted fluorescence motorized microscope (Monash Micro Imaging Facility, Monash University, Melbourne) under consistent exposure times, at either 20 × magnification (GFAP/IBA-1/DAPI and CD68/DAPI) or 10 × magnification (HO-1/Hoechst). For GFAP/IBA-1/DAPI and CD68/DAPI staining, the peri-lesional cortex was captured from six equidistant sections per brain (approximately 420 μm apart) through the injury site, as well as CA1, CA3, and dentate gyrus (DG) regions of the dorsal hippocampus from three equidistant sections per brain (approximately 420 μm apart). For HO-1/Hoechst staining, 3 × 2 stitched large image files were captured to encompass the entire dorsal cortex and hippocampus, from four equidistant sections per brain (approximately 420 μm apart). ImageJ (FIJI) software (National Institutes of Health, USA) was then used to quantify fluorescent intensity of staining for GFAP (pseudo-colored to red), IBA-1 (pseudo-colored to green), CD68 (pseudo-colored to green), and HO-1 (pseudo-colored to yellow), following thresholding. An increase in fluorescence intensity for any marker was considered to reflect an increased inflammatory response, with data presented as a ratio of percentage area (ipsilateral to contralateral to the injury site) for GFAP, IBA-1, and CD68 markers; whereas HO-1 data are presented as fluorescence intensity in the dorsal ipsilateral cortex.
Flow cytometry
To quantify resident microglia and peripheral cell infiltration, saline-perfused brains were collected from Ccr2 KO and littermate WT mice at 3 days post-surgery. Isolated brains were divided medially into ipsilateral and contralateral hemispheres, then homogenized using a Gentle MACs tissue dissector (Miltenyi Biotec, Bergisch Gladbach, Germany) in digestion buffer containing RPMI medium (Thermo Fisher Scientific), 1% fetal calf serum (produced in-house), 2.5% Liberase™ (Sigma-Aldrich), 1% HEPES (Thermo Fisher Scientific), and 1% DNAse I (Sigma-Aldrich), for 30 min at 37°C. Digested brain tissue was filtered through a 70-μm nylon mesh and isolated single cells were centrifuged at 300g for 10 min at 4°C then resuspended in cold phosphate-buffered saline (PBS). Samples were mixed with Debris Removal Solution (Miltenyi Biotec) and cold PBS was overlaid onto the cell suspension, followed by centrifugation at 3000g for 10 min at 4°C to remove fatty debris. Rat anti-mouse CD16/CD32 (Mouse BD Fc Block) was incubated for 20 min at 4°C to block non-specific binding of fluorescent antibodies.
Cells were then incubated for 20 min at 4°C with antibodies against CD45, CD11b, TMEM119, Ly6G, and Ly6C to identify subsets of different immune cell phenotypes in the samples (Table 1). Before acquisition of the samples on the BD LSR Fortessa X-20 Cell Analyzer within the Flow Cytometry Core Facility (AMREPFlow) (Alfred Research Alliance, Monash University, Melbourne), cells were incubated with Flurogold to distinguish between live and dead cells, and Calibrate 2 Color Beads (BD Biosciences, Franklin Lakes, NJ, USA) per manufacturer instructions to calibrate the cell numbers in the samples. Data were analyzed using FlowJo software (v. 10.6). Data were compensated using single color controls, and a gating strategy was employed to identify live (Flurogold) macrophages (CD45HiCD11b+), microglia (CD45MidCD11b+TMEM119+), monocytes (CD45+CD11b+TMEM119-LY6C+), and neutrophils (CD45+CD11b+TMEM119-LY6G+), with data expressed as absolute cell counts. Both ipsilateral and contralateral hemispheres were assessed; however, no group differences were observed in the contralateral hemisphere, so graphical data from only the ipsilateral hemisphere are presented.
Antibodies Used for Flow Cytometry Analysis of Brain Samples
APC, allophycocyanin; BV, brilliant violet; CD, cluster of differentiation molecule; FITC, fluorescein isothiocyanate; Ly6, lymphocyte antigen-6; PE, phycoerythrin; TMEM-119, transmembrane protein 119.
Cresyl violet staining
For quantification of tissue volume atrophy at 3 days or 14 days post-surgery, cresyl violet acetate (0.25% w/v) was used to stain six equidistant sections (420 μm apart; approximately −1.2 to −3.4 mm bregma), as previously described. 45 Slides were incubated in cresyl violet for 20 min followed by differentiation in descending concentrations of ethanol, cleared in xylene, then cover-slipped and dried before imaging on a Leica Aperio AT Turbo Brightfield slide scanner (Monash Histology Platform, Monash University, Australia). Images were exported to FIJI/ImageJ for analysis, using the unbiased Cavalieri method to estimate the volume of the intact dorsal cortex and dorsal hippocampus of each hemisphere (ipsilateral and contralateral to the injury). Each grid point represented 100 μm2 and sections were sampled at a frequency of 7. Measurements were restricted to the dorsal portion of each region of interest (ROI) as previously described. 45 Group means are expressed as ratio of ipsilateral to contralateral volumes.
CCR2 antagonist (RS102895) administration
The commercially available CCR2 antagonist RS102895 (Sigma-Aldrich) was dissolved in 100% dimethyl-sulfoxide (DMSO) hybri-max™ (Sigma-Aldrich), then diluted in 0.9% saline to reach the desired dose of 5 mg/kg RS102895 in 4% DMSO to be administered to C57Bl/6J mice. A 4% DMSO solution in 0.9% saline was used as a vehicle control. As RS102895 has a reported half-life of 1 h, 46 C57Bl/6J mice received three doses of either drug or vehicle following sham or TBI surgery at p21, starting at immediately prior to surgery (0 h) followed by 1.5 h post-surgery and 6 h post-surgery (Fig. 2A). Mice were then group housed in their home cage (mix of vehicle or drug treated, and TBI and sham per cage) until PTZ administration to evaluate seizure susceptibility at 14 days post-surgery as described above. The timing and dose of RS102895 was based upon previous evidence that CCL2 is released robustly and acutely post-injury, 11 and recent studies in which a similar acute time course was shown to successfully reduce the inflammatory response, the extent of brain damage, and/or seizures after hypoxic-ischemia in p10 mouse pups, 47 after LPS in chronically epileptic animals, 38 and after TBI in adult mice. 48
Statistical analysis
GraphPad Prism v. 9 (GraphPad Software, San Diego, CA, USA) was used to perform all statistical analyses, with statistical significance reported as p < 0.05. Two-way analysis of variance (ANOVA) tests were performed to examine factors of injury and genotype (or CCR2 antagonist treatment). Fischer's exact test was performed to compare sham versus TBI groups for PTZ dose-dependent analysis as well as for mice with PTZ-evoked seizures analysis. Post hoc multiple comparison tests were conducted where significant interactions were observed. For analyses in which pathology or cellular responses were evaluated both ipsilateral and contralateral to the injury site, where there were not group differences detected in the contralateral values, data were expressed as ipsilateral relative to contralateral. Group sizes were determined based on our previous work using similar outcomes. 35,73 All results are expressed as mean ± standard error of the mean (SEM).
Results
Ccr2 gene deletion altered pro-inflammatory cytokine levels in serum
Serum samples were extracted from terminal blood collected at 3 days post-surgery, to evaluate inflammatory cytokine protein levels using a multi-plex assay. Ccr2 KO mice showed a five-fold increase in CCL2 protein levels (two-way ANOVA, effect of genotype: F1, 12 = 68.18, p < 0.0001; Fig. 3A). Conversely, TNF-α protein levels were decreased in Ccr2 KO mice (effect of genotype: F1, 12 = 8.23, p < 0.05; Fig. 3B), whereas neither CCL2 nor TNF-α were significantly altered by TBI at this time. Serum levels of GM-CSF, IL-6, and IL-10 were unaffected by TBI or Ccr2 deficiency (Supplementary Fig. S1).

Pro-inflammatory cytokine levels were altered in serum from Ccr2 KO mice at 3 days post-surgery. CCL2 protein levels were increased
The cellular neuroinflammatory response was independently altered by pediatric TBI or Ccr2 gene deletion, acutely post-surgery
Phagocytic-macrophages/microglia (CD68+), astrocytes (GFAP+), and both resting and activated microglia (IBA-1+) were evaluated by quantification of IF intensity in the cortex and DG of the dorsal hippocampus at 3 days post-surgery (Fig. 4A). In the cortex, all three markers were increased in TBI mice compared with sham controls, including CD68 (two-way ANOVA, effect of injury: F1, 20 = 34.23, p < 0.0001; Fig. 4B), GFAP (F1, 20 = 11.75, p < 0.01; Fig. 4C), and IBA-1 levels (F1, 20 = 53.33, p < 0.0001; Fig. 4D). This inflammatory response was extended into the DG, where TBI likewise increased the levels of CD68 (F1, 20 = 56.90, p < 0.0001; Fig. 4E), GFAP (F1, 20 = 101.9, p < 0.0001; Fig. 4F) and IBA-1 (F1, 20 = 45.75, p < 0.0001; Fig. 4G) compared with sham groups. Surprisingly, Ccr2 gene deficiency had no effect on the level of these markers in the cortex (two-way ANOVAs, p > 0.05 effect of genotype). In the DG region, however, a significant genotype × injury interaction was observed (F1,20 = 5.42, p < 0.05), as GFAP levels were increased in Ccr2 KO-TBI mice compared with WT-TBI (post hoc Tukey's test; p < 0.05; Fig. 4F); whereas IBA-1 expression was increased in Ccr2 KO compared with WT (effect of genotype: F1, 20 = 4.9, p < 0.05; Fig. 4G). The CA1 and CA3 regions of the dorsal hippocampus were also quantified for all three markers, with findings mirroring those seen in the DG (Supplementary Fig. S2).
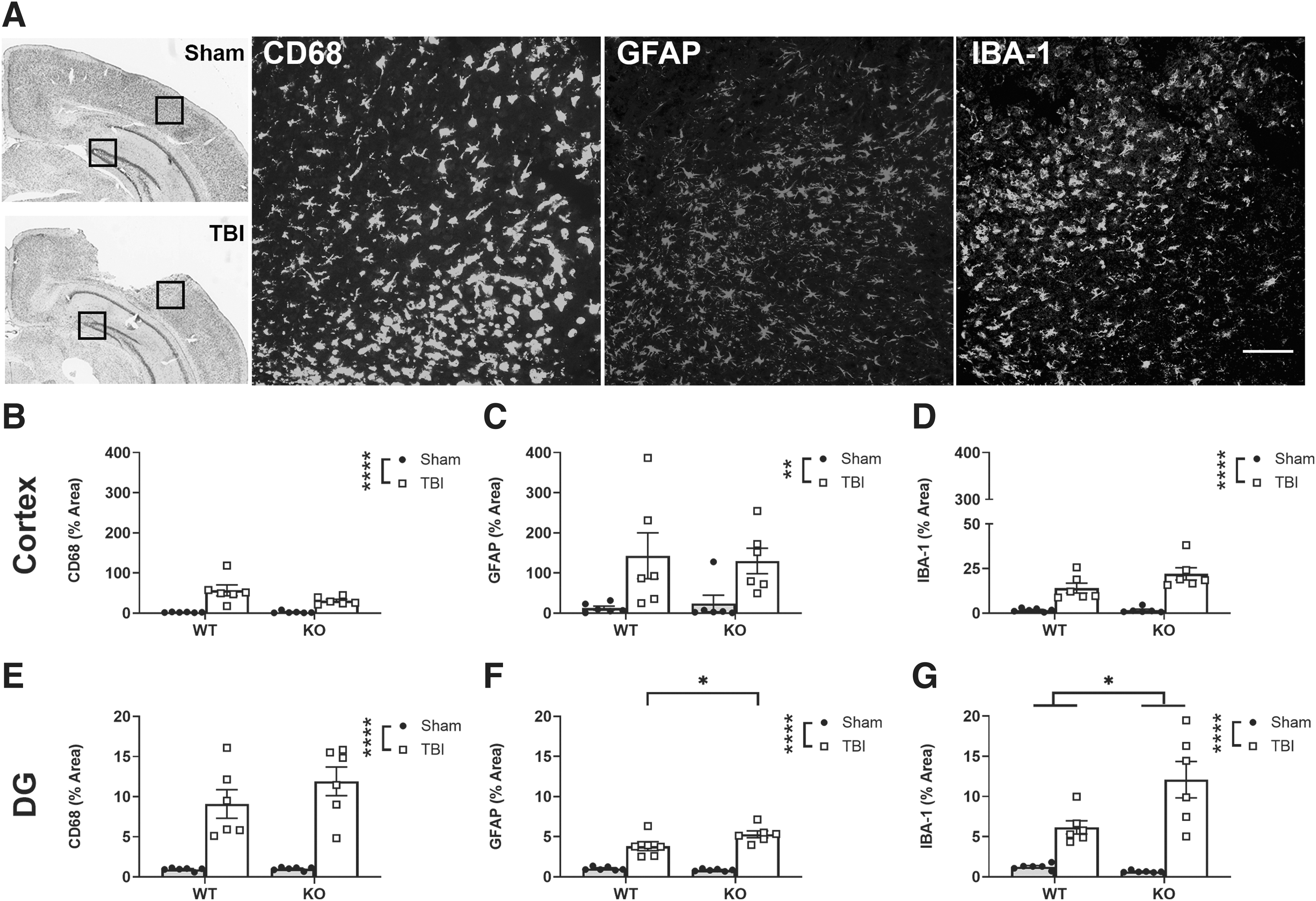
Pediatric TBI and Ccr2 gene deletion independently influenced brain immune cell populations. IF staining for CD68+, GFAP+, and IBA-1+ cells was quantified as % area fluorescent intensity in the cortex and DG (data shown here as the ratio of ipsilateral to contralateral), at 3 days post-surgery (black boxes indicate regions of interest on representative cresyl-violet-stained sections) and representative IF images from TBI cortex are shown in
Ccr2 gene deletion did not affect immune cell phenotypes in brain at 3 days after TBI
Brain samples were isolated at 3 days post-TBI, to identify immune cell phenotypes using flow cytometry analysis (Fig. 5A). Total CD45+ cells (Fig. 5B), macrophages (Fig. 5C), microglia (Fig. 5D), monocytes (Fig. 5E), and neutrophils (Fig. 5F) proportions were evaluated from the ipsilateral hemisphere of WT and Ccr2 KO mice. We found that Ccr2 gene deletion had no impact on immune cellular profile after 3 days post-TBI, compared with WT controls (two-tailed unpaired t test; p > 0.05).
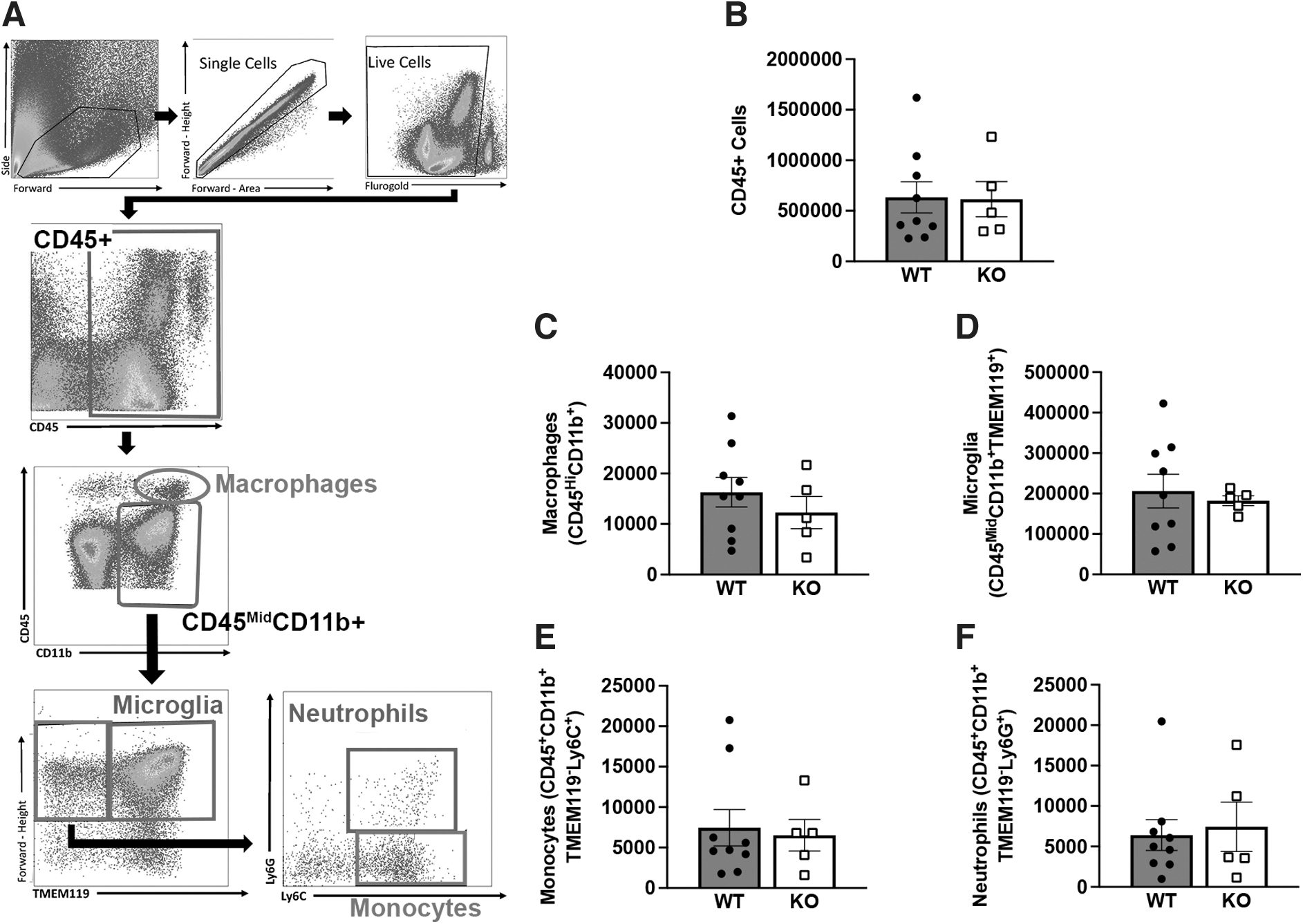
Flow cytometry analysis revealed no differences in the brain immune cell profile of Ccr2 KO and WT mice at 3 days post-TBI. The gating strategy
TBI increased neuroinflammatory HO-1 expression at 3 days post-surgery
In addition to the cellular inflammatory response, we next quantified HO-1+ IF at 3 days post-surgery as an indicator of the oxidative stress response (Fig. 6A). TBI induced a robust increase in HO-1 in the ipsilateral dorsal cortex (two-way ANOVA, effect of injury: F1, 20 = 81.18, p < 0.0001; Fig. 6B). However, Ccr2 gene ablation did not significantly influence HO-1 levels (effect of genotype: F1, 20 = 3.65, p = 0.07).
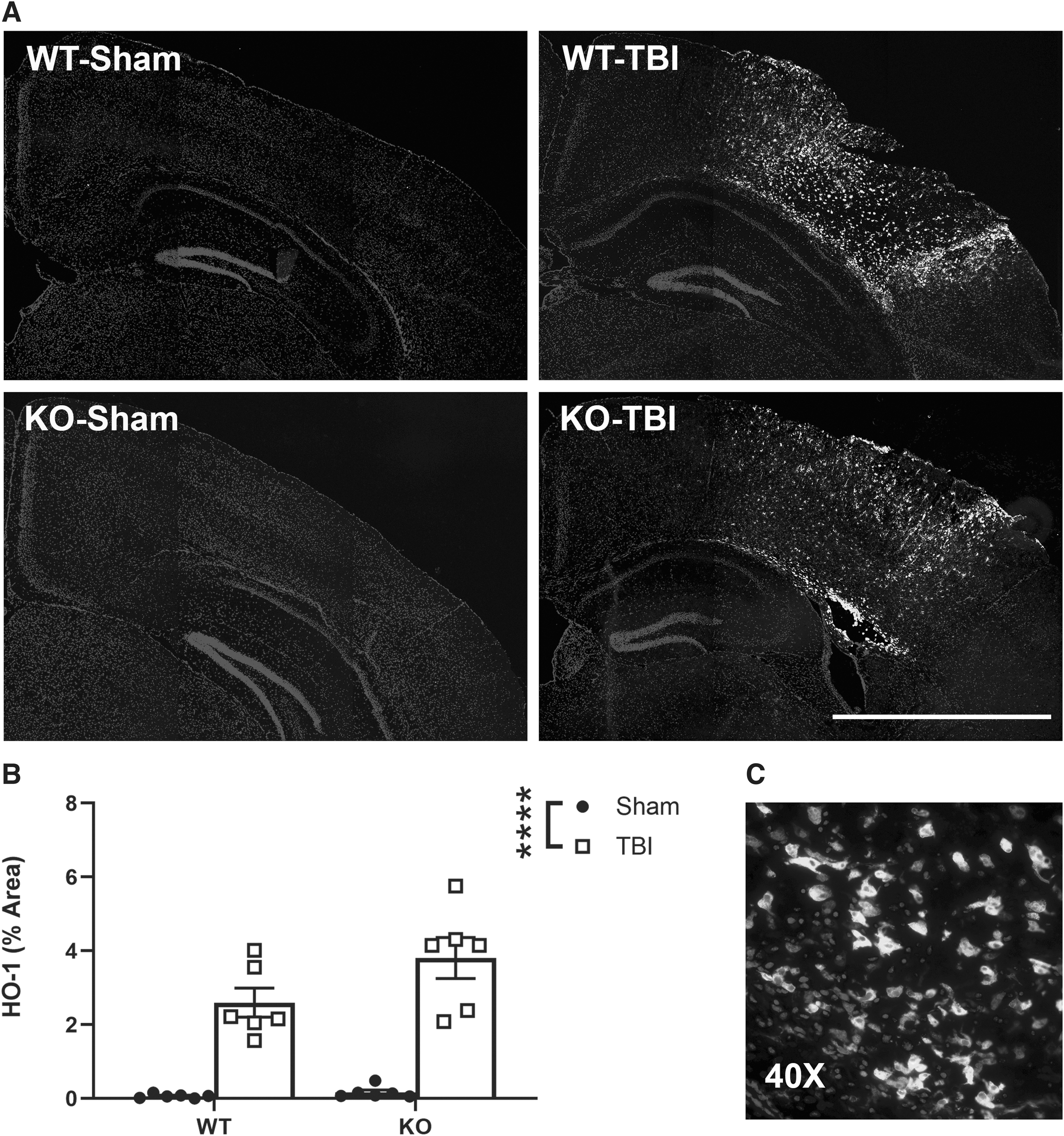
Pediatric TBI triggered a robust increase in HO-1 indicative of oxidative stress in the ipsilateral cortex. Representative images illustrate the localization and cellular morphology of HO-1+ cells, detected almost exclusively in the brains of TBI animals
Susceptibility to PTZ-evoked seizures was increased due to pediatric TBI
Considering our initial hypothesis regarding the CCR2-driven cellular inflammatory response and post-traumatic seizure susceptibility, we next sought to evaluate how Ccr2 gene deficient mice responded to the pro-convulsive agent PTZ, as an indicator of a change in brain network excitability. First, a dose-dependent study was conducted in pediatric C57Bl/6J mice to optimize the PTZ when administered i.p. at 14 days post-injury. In TBI mice, 33.34%, 75%, and 100% of mice showed evoked seizures when challenged with PTZ at 30 mg/kg, 40 mg/kg, and 50 mg/kg, respectively (Fig. 7A). However, with a highest dose of PTZ (50 mg/kg), in addition to 100% TBI mice, approximately two-thirds (67%) of sham mice also developed evoked seizures (Fisher's exact test, p < 0.0001). On the other hand, the lowest dose of PTZ (30 mg/kg) evoked seizures in only one-third of C57Bl/6J TBI mice (Fisher's exact test, p < 0.0001). The intermediate dose of 40 mg/kg of PTZ resulted in evoked seizures in 75% of TBI mice, with minimal effects in sham animals (Fisher's exact test, p < 0.0001). Similarly, this dose yielded the greatest contrast between sham and TBI mice in terms of their behavioral response to PTZ (Seizure Severity Score; two-way ANOVA, effect of injury: F1, 14 = 8.33, p < 0.05; effect of dose: F2, 14 = 6.3, p < 0.05; Fig. 7B). A dose of 40 mg/kg i.p. was therefore chosen as the most appropriate for this age and injury model.

Susceptibility to PTZ-evoked seizures was increased after pediatric TBI
WT and Ccr2 gene deficient mice were then administered 40 mg/kg PTZ at 14 days post-TBI/sham surgery. PTZ evoked seizures in 10% of mice in the WT/sham group (i.e., 1/10 mice), whereas WT/TBI mice showed a heightened response with 86% mice (12/14 mice) developing seizures (Fisher's exact test, p < 0.001). Similarly, 10% of Ccr2 KO/sham mice (1/10 mice) had a seizure, compared with 53% of mice in the Ccr2 KO/TBI group (8/15 mice) (p < 0.05; Fig. 7C). Direct comparison of WT/TBI and KO/TBI responses found no significant impact of Ccr2 gene deletion on susceptibility to PTZ-evoked seizures (Fisher's exact test, p > 0.05).
Assessing the behavioral response to PTZ, a higher Seizure Severity Score was assigned to TBI mice indicating a more severe response compared with sham controls (two-way ANOVA, effect of injury: F1, 45 = 29.55, p < 0.0001; Fig. 7D). Ccr2 gene deletion did not affect this (p > 0.05, effect of genotype). In addition, among those animals that did develop seizures, neither seizure latency nor seizure duration were affected by either TBI or genotype (data not shown).
Pediatric TBI but not Ccr2 deficiency drives cellular neuroinflammation by 14 days post-surgery
The magnitude of the CD68+, GFAP+, and IBA-1+ cellular response in the brain was next evaluated at 14 days post-surgery (Fig. 8A). In the cortex, only CD68+ expression was significantly increased due to TBI compared with sham controls (two-way ANOVA, effect of injury: F1, 22 = 6.8, p < 0.05; Fig. 8B). However, in the DG region, persistent activation was noted for all three cellular markers in TBI animals, with TBI increasing CD68 (effect of injury: F1, 22 = 12.65, p < 0.01; Fig. 8E), GFAP (F1, 22 = 20.74, p < 0.001; Fig. 8F) and IBA-1 (F1, 22 = 12.73, p < 0.01; Fig. 8G) compared with sham groups. Intriguingly, there was no effect of Ccr2 gene deficiency in the cortex or DG for any marker at this subacute time-point. CA1 and CA3 regions were also quantified, and mirrored responses were observed in the DG (Supplementary Figs. S2 and S3).
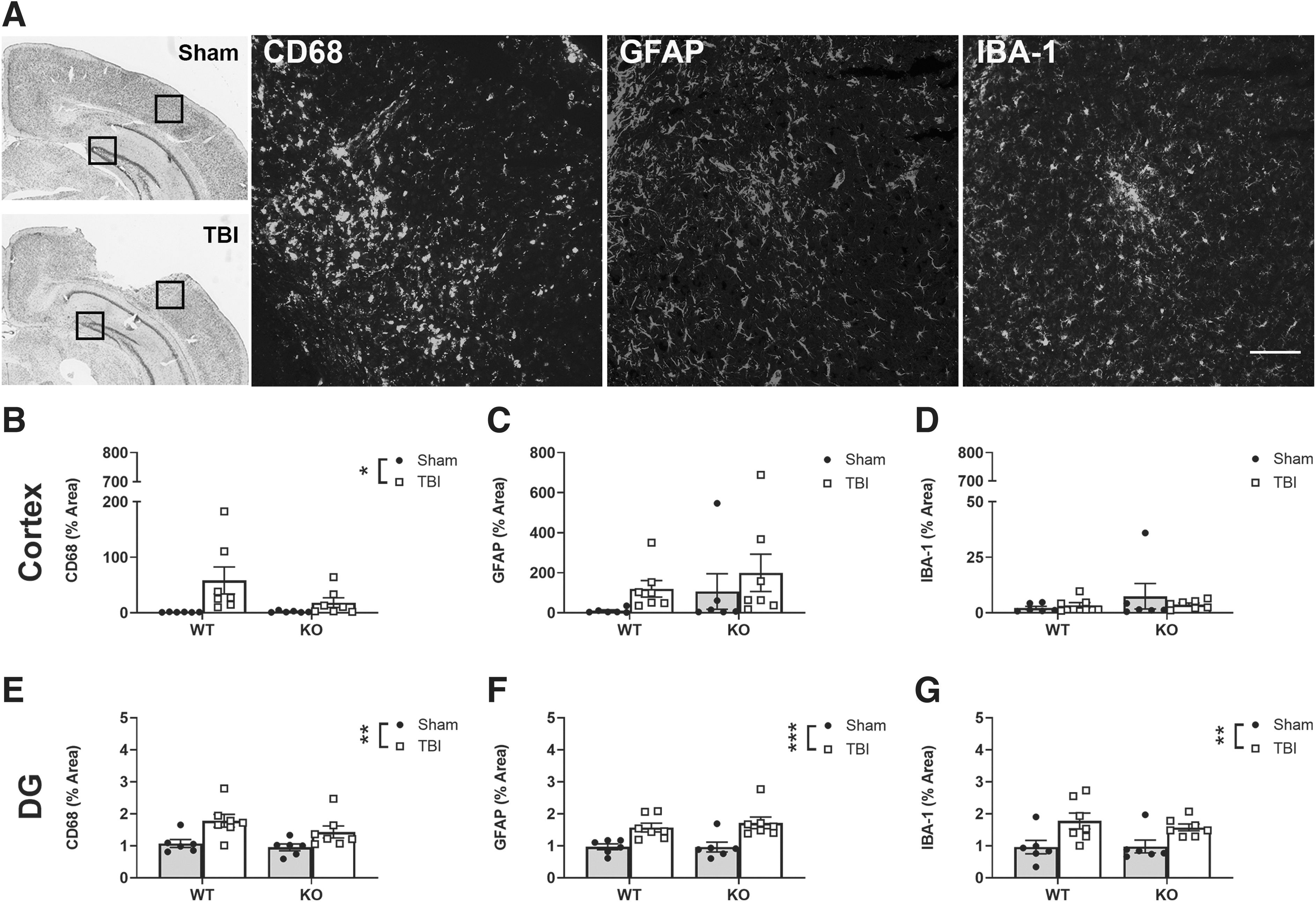
Pediatric TBI increased the presence of immune cells in the brain at 14 days post-injury, whereas no effects of Ccr2 deficiency were observed. Representative images of CD68, GFAP, and IBA-1 from TBI ipsilateral cortex are illustrated, with regions of interest (black box) depicted on cresyl-violet-stained images for quantification of % area fluorescent intensity in the cortex and DG (data shown here as the ratio of ipsilateral to contralateral), at 14 days post-surgery
Pediatric TBI resulted in brain volume loss independent of Ccr2 genotype
Lesion volume analysis was performed on cresyl violet-stained sections from 3 days and 14 days post-surgery to evaluate the extent of tissue atrophy (Fig. 2A). Pediatric TBI resulted in volume loss of the dorsal cortex compared with sham controls at both 3 days (two-way ANOVA, effect of injury: F1, 20 = 53.47, p < 0.0001; Supplementary Fig. S4A) and 14 days (F1, 21 = 34.46, p < 0.0001; Fig. 2B). TBI also induced volume loss of the dorsal hippocampus 3 days post-surgery (effect of injury: F1, 20 = 7.74, p < 0.05; Supplementary Fig. 4B); although this appeared to be resolved by 14 days post-surgery (Fig. 2C). Of note, Ccr2 gene deletion did not influence the extent of tissue atrophy at either time-point.
Pharmacological CCR2 antagonist treatment had no effect on seizure susceptibility after pediatric TBI
Next, we tested whether pharmacological blocking of CCR2 receptors in C57Bl/6J mice could alter seizure susceptibility to PTZ after pediatric TBI. The CCR2 antagonist drug RS102895 (5 mg/kg, i.p.) was administered acutely post-injury/sham surgery, then responses to PTZ were evaluated at 14 days (Fig. 9A). Pediatric TBI was again found to increase hyperexcitability to evoked seizures compared with sham controls (Fisher's exact tests: vehicle/TBI [60%] versus vehicle/sham [0%], p < 0.0001; drug/TBI [60%] versus drug/sham [30%]; p < 0.0001; Fig. 9B). Further, the mean Seizure Severity Score was increased in both TBI groups compared with sham groups (two-way ANOVA, effect of injury: F1, 36 = 11.96, p < 0.01; Fig. 9C). Intriguingly, mirroring our findings in Ccr2 gene deficient mice, CCR2 antagonist treatment had no impact on seizure susceptibility after pediatric TBI. Finally, we quantified the extent of tissue volume loss in the injured cortex (unpaired t test, p > 0.05; Fig. 9D) and hippocampus (unpaired t test, p > 0.05; Fig. 9E), which confirmed that CCR2 antagonism did not affect tissue integrity after pediatric TBI.
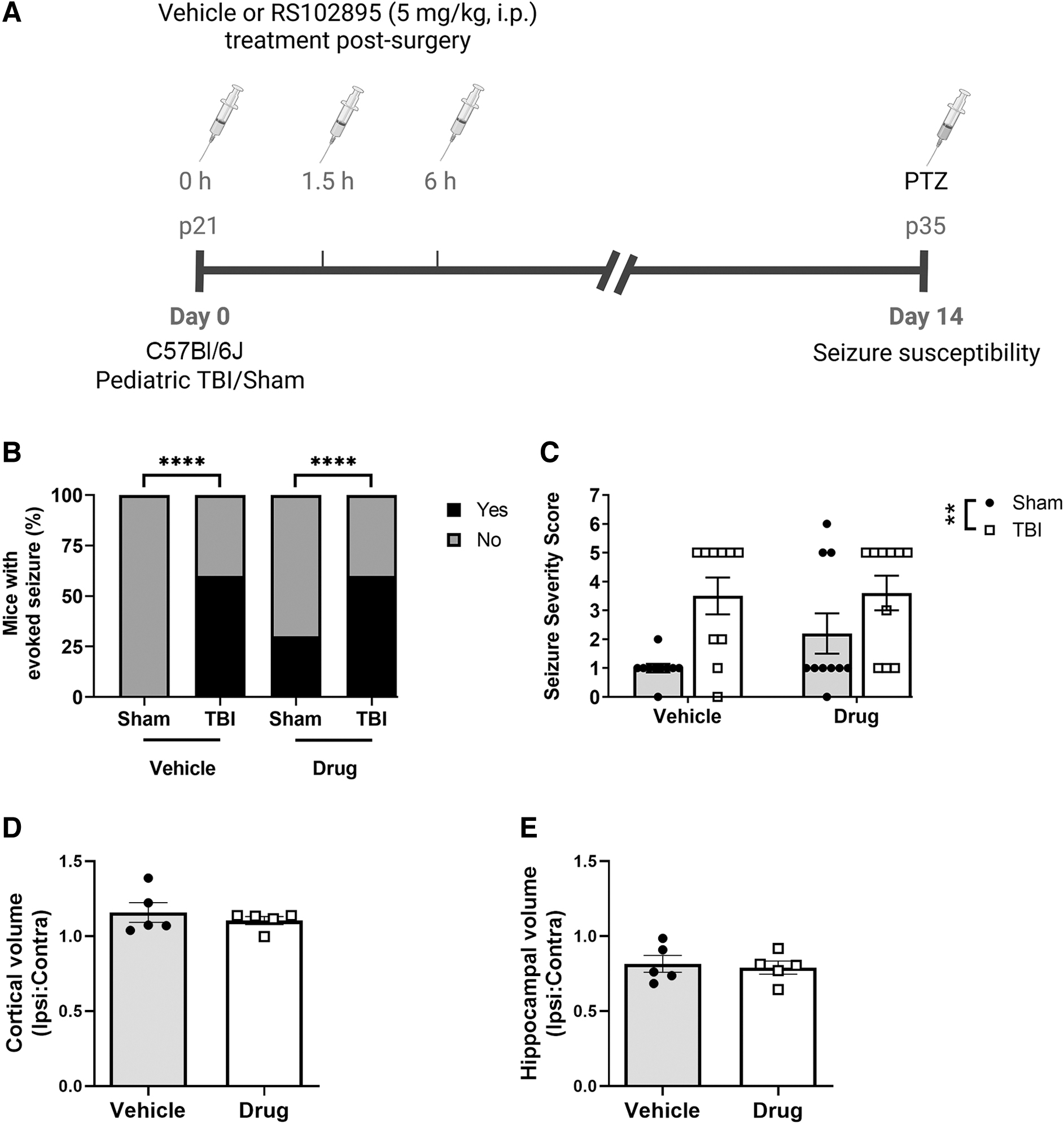
Acute post-injury treatment with a CCR2 antagonist did not influence subacute seizure susceptibility after pediatric TBI. TBI or sham surgery was performed on pediatric C57Bl6/J mice on day 0, followed by three doses of the CCR2 antagonist RS102895 (5 mg/kg, i.p.) or vehicle (4 % DMSO). At 14 days post-surgery, seizure susceptibility to PTZ (40 mg/kg, i.p.) was evaluated
Discussion
To evaluate the role of CCL2/CCR2 signaling pathway in hyperexcitability after pediatric TBI, we compared the responses of Ccr2 gene deficient mice to their WT littermates, as well as C57Bl/6J mice treated acutely with a CCR2 antagonist to block CCL2/CCR2 signaling. Overall, although we confirmed that pediatric TBI induced an increase in seizure susceptibility, associated with considerable loss of brain tissue volume and a robust neuroinflammatory cellular response, we did not observe any effects of CCR2 manipulations on these outcome measures.
These results were surprising on several levels. First, other studies have shown that the Ccr2 deficient mouse model is characterized by reduced monocyte/macrophage recruitment in pre-clinical models of neurological injury. For example, in the context of adult experimental TBI, CCR2+ macrophages were reportedly reduced by 80–90% in the brains of Ccr2 KO mice at 4 days after TBI, when compared with non-littermate WT mice. 49 In addition, Ccr2 deficiency reduced monocyte infiltration along with diminished lesion volume and reduced axonal damage after mild TBI, also in the adult mouse brain. 50 Similarly, adult Ccr2 KO mice exhibit limited neuropathology, reduced peripheral immune cell infiltration, and improved functional outcomes after ischemic stroke. 51 –53
In our hands, however, although we observed a robust increase in CD68 and IBA-1 immunoreactivity after pediatric TBI, this response occurred to a similar degree in the injured brains of WT and Ccr2 KO mice. CD68 is thought to represent phagocytic cells, as it is predominantly expressed on intracellular lysosomes of tissue resident-immune cells, the majority of which would be macrophages in this context. 54 –58 However, as the distinction between peripherally derived macrophages recruited into the brain, versus resident, activated microglia, is challenging by IF staining methods, 59 we also employed flow cytometry to assess immune cell profiles in the brains of the two strains acutely after pediatric TBI. Here, we similarly observed a lack of effect of Ccr2 deficiency on the numbers of infiltrating peripherally derived immune cells including macrophages, monocytes, and neutrophils, or on the number of TMEM119+ microglial cells, compared with WT mice.
Despite the lack of an expected phenotype in our current study, we importantly did observe a robust elevation of CCL2 protein levels in the serum of Ccr2 KO mice, collected at 3 days post-TBI/sham surgery, confirming their expected genotype and suggesting perhaps a compensatory response to Ccr2 gene deficiency. This finding, in line with published literature, 60,61 may explain the robust immune cell infiltration observed in this model even in the absence of CCR2, although this is unlikely as CCL2 appears to affect leukocyte recruitment almost exclusively via CCR2. 62 However, additional acute time-points (within hours after injury) would have been insightful to better capture the acute peak of cytokine expression, and evaluate potential changes in Ccr2 KO animals compared with WT.
Coming back to our initial hypothesis, we examined seizure susceptibility to the chemoconvulsant PTZ, which reduces inhibitory neurotransmission and increases hyperexcitability to decrease the threshold for seizure onset. 42,63 Previous studies in experimental TBI models have indicated that responsiveness to PTZ and other such compounds provides insight into underlying changes in brain network excitability associated with an increased propensity for the subsequent development of spontaneous, recurrent seizures, or epilepsy. 35,64,65 Although epilepsy may take several months to develop after experimental TBI, and many years in humans, 66,67 examining the response to PTZ acts as a surrogate indicator of epileptogenesis at earlier post-injury time-points.
Here, at 2 weeks post-TBI, we found that a higher proportion of TBI mice developed PTZ-evoked seizures with a worse behavioral seizure phenotype compared with sham control mice, consistent with other rodent studies in experimental TBI models. 35,68 Pharmacological targeting of CCR2 in WT mice, using RS102895 hydrochloride to bind specifically to the predominant β subunit of the CCR2 receptor, 69 –71 did not alter susceptibility to PTZ-evoked seizures. Although 53% of Ccr2 KO mice exhibited seizures compared with 83% of WT mice, this difference was not statistically significant, so we must also conclude a lack of effect of Ccr2 deficiency. Nonetheless, this subtle potential difference might be more overt at a more chronic time-point, or if we had examined the incidence of spontaneous unprovoked seizures (i.e., PTE).
In a study by Cerri and colleagues, a key driver of our hypothesis, the investigators used a CCL2 transcription inhibitor (Bindarit), the CCR2 antagonist RS102895, or anti-CCL2 antibodies to demonstrate that targeting CCL2/CCR2 signaling in a status epilepticus mouse model is crucial for seizure induction and epileptiform activity. 38 However, our current findings instead support those of Foresti and colleagues, who recently reported that CCR2 antagonist treatment did not influence spontaneous seizures or PTZ-evoked seizure susceptibility in adult rats at 30 days after status epilepticus. 72 Together, our findings indicate that the manipulation of CCR2 either genetically or pharmacologically does not influence brain hyperexcitability after pediatric TBI.
Developmental age is known to impact TBI outcomes. 73 –76 It is possible that the pediatric nature of this TBI model yields a differential response whereby CCR2 is less critical to the neuroinflammatory response compared with what has been reported in the adult brain to date. At the other end of the lifespan, the peripherally derived, CCR2-driven monocyte/macrophage response was exacerbated in aged animals compared with young adult animals, and contributed to the progression of TBI pathophysiology. 77 In the pediatric injured brain, accumulating evidence indicates a microenvironment that is more vulnerable to BBB damage, edema, neutrophil infiltration, and inflammatory cytokine responses compared with the adult brain. 78 –81 As the current study is, to the best of our knowledge, the first to examine Ccr2 gene deficient mice in the context of pediatric TBI, further investigation is required to determine whether age at the time at injury is indeed a determining factor in how these animals respond by incorporating both pediatric and adult-age animals in the same experiments.
A lack of difference in terms of the cellular inflammatory response and extent of tissue damage observed in our current study, indicating equivalent peripheral macrophage influx and secondary injury in WT and Ccr2 KO mice, suggests that CCR2-independent mechanisms of monocyte recruitment into the brain may be occurring here. For example, several other chemokine receptors such as CCR5, CCR6, and CCR1 have been shown to be involved in monocyte/macrophage recruitment in inflammatory diseases such as atherosclerosis and multiple sclerosis. 82,83 Others have reported CCR2-independent transmigration of monocytes into the brain in response to interferon-gamma, even in the absence of chemokine signaling. 84 Our findings are also in alignment with reports that CCR2 antagonism does not prevent monocyte recruitment (or influence secondary injury outcomes) after permanent focal cerebral ischemia in rodent models. 85 This is in stark contrast to CCR2 antagonist-mediated neuroprotection observed after transient cerebral ischemia, 86 suggesting that different mechanisms of monocyte recruitment may be involved depending on the context.
Considering the GFAP astrocytic and IBA-1 microglial responses, we hypothesized that these would be increased by pediatric TBI but may be reduced in Ccr2 KO mice compared with WT, via indirect mechanisms. For example, the anticipated reduction in CCR2-driven monocyte/macrophage influx was expected to yield a microenvironment with reduced pro-inflammatory cytokines, reactive oxygen species, and damage-associated molecular patterns, which would typically promote astrocyte and microglial activation. 87 –89 However, one study found that although Ccr2 gene deletion reduced the infiltration of peripheral macrophages into the adult injured brain at 3 days post-TBI, a pronounced microglial response was unaffected. 50 In contrast, Somebang and colleagues recently reported that Ccr2 gene deficiency in adult brain-injured mice triggered the alternative activation of different microglial phenotypes, suggesting an intimate relationship between resident and infiltrated immune cells. 90 Future studies could utilize bone marrow chimera mice or CCR2 fluorescently tagged reporter mice to better distinguish between these cell populations.
Similarly, we hypothesized that the levels of HO-1, a hallmark of the oxidative stress response, would be aligned with the magnitude of neuroinflammation. HO-1 is expressed by microglia, macrophages, astrocytes, and neurons after CNS insult, 91,92 and although its levels have been correlated by some investigators with neuroinflammation driving toward unfavorable neurological outcomes after TBI, 93 others have considered HO-1 upregulation to be a neuroprotective measure. 94 –96 A lack of difference in HO-1 between the genotypes after pediatric TBI was unsurprising given their comparable extent of immune cell activation (CD68, IBA-1, and GFAP).
Regarding the extent of tissue damage after pediatric TBI, previous studies investigating Ccr2 gene deficient adult mice in experimental TBI models have reported conflicting findings, of either no effect on regional brain tissue volumes, 49,97 or neuroprotection afforded to Ccr2 KO mice. 50 Here, we observed an equivalent extent of cortical tissue damage up to 2 weeks post-injury in both WT and Ccr2 KO mice, consistent with previous studies in this model, 11,81 and perhaps reflecting the comparable cellular neuroinflammatory response that we observed in the two strains. It is worth noting, however, that we cannot rule out potential strain differences in tissue function, and future studies could perform electrophysiological experiments to probe the health of this remaining tissue and assess potential neuroplasticity in this context.
Conclusions
Due to the lack of effect of Ccr2 deficiency on cellular inflammation in the injured pediatric brain, we were unable to adequately address our initial hypothesis—to determine whether reducing CCR2-dependent peripheral immune cell infiltration, either by genetic manipulation or pharmacologically, suppresses epileptogenesis. As pre-clinical studies in adult TBI models have demonstrated neuroprotection afforded by blocking or impairing the infiltration of CCR2+ macrophages into the injured brain, 49,52,76,98 this hypothesis still warrants further investigation. Future studies may employ other strategies to further address this question, such as clodronate liposomes, 99 transgenic CD11b-diphtheria toxin receptor mice, 97 or the cre-loxP system 100 (e.g., Cxcr4-CreER mice) to specifically deplete hematopoietic stem-cell-derived monocytes. Understanding the biological mechanisms underlying seizure propensity after pediatric TBI has important implications for the design of novel therapeutics aiming to prevent the development of post-traumatic epilepsy.
Footnotes
Acknowledgments
The authors acknowledge technical assistance and use of services from the Monash Histology Facility and Monash Micro Imaging Facility, Monash University (Alfred and Hudson nodes), as well as the Flow Cytometry Core Facility (AMREPFlow).
Authors' Contributions
The authors contributed as follows: conceptualization: RS, SRS, BDS, TOB; experimental investigation: RS, EC, LKD, PCE, AS, AZ; methodologies and analysis: RS, EC, LKD, PCE, AS; resources: BDS, TOB, SRS; supervision, project administration, funding acquisition: BDS, SRS, PCE; manuscript writing: RS, EC, BDS; manuscript editing: all authors. All authors confirm that they read and approved the final manuscript.
Funding Information
The authors are supported by grants from the National Health and Medical Research Council of Australia (APP1141347 to BDS; APP1176426 to TOB; APP1087172 to PCE; APP1159645 to SRS), a Co-Funded Monash Graduate Scholarship (RS), Establishment Grants from Monash University's Central Clinical School (BDS and SRS), a Veski Near-Miss Award (BDS), and an Idea Development Award from the U.S. Department of Defense (W81XWH2010848 to BDS).
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.