
Abstract
Blast-induced traumatic brain injury (bTBI) has been a health concern in both military and civilian populations due to recent military and geopolitical conflicts. Military service members are frequently exposed to repeated bTBI throughout their training and deployment. Our group has previously reported compounding functional deficits as a result of increased number of blast exposures. In this study, we further characterized the decrease in long-term potentiation (LTP) by varying the blast injury severity and the inter-blast interval between two blast exposures. LTP deficits were attenuated with increasing inter-blast intervals. We also investigated changes in microglial activation; expression of CD68 was increased and expression of CD206 was decreased after multiple blast exposures. Expression of macrophage inflammatory protein (MIP)-1α, interleukin (IL)-1β, monocyte chemoattractant protein (MCP)-1, interferon gamma-inducible protein (IP)-10, and regulated on activation, normal T cell expressed and secreted (RANTES) increased, while expression of IL-10 decreased in the acute period after both single and repeated bTBI. By partially depleting microglia prior to injury, LTP deficits after injury were significantly reduced. Treatment with the novel drug, MW-189, prevented LTP deficits when administered immediately following a repeated bTBI and even when administered only for an acute period (24 h) between two blast injuries. These findings could inform the development of therapeutic strategies to treat the neurological deficits of repeated bTBI suggesting that microglia play a major role in functional neuronal deficits and may be a viable therapeutic target to lessen the neurophysiological deficits after bTBI.
Introduction
Since 2000, the United States Department of Defense has recorded approximately 430,000 cases of traumatic brain injury, of which a significant portion is due to blast-induced traumatic brain injury (bTBI), in U.S. service members deployed throughout the world. 1 Some service members, like breachers, are exposed to multiple blasts not only during deployment but also during training. 2 Repeated low-level overpressures experienced by breachers and other military personnel throughout the duration of their training and deployment has been associated with neurological and neurosensory deficits. 3 –6
Previously, we have reported a decrease in long-term potentiation (LTP) in rat organotypic hippocampal slice cultures (OHSCs) following a single mild bTBI. 7,8 A second mild bTBI delivered less than 6 days after the first blast aggravated LTP deficits. 9 The low levels of blast that we have tested in rat OHSCs are comparable to those experienced by military personnel firing shoulder mounted weapons 10 or large caliber guns, 11 or participating in explosive breaching exercises. 12
Following traumatic brain injury (TBI), there is generally an increase in neuroinflammation, which can cause electrophysiological deficits. 13,14 Neuroinflammation following injury has been noted in in vitro cell and brain tissue cultures following injury, in animal models of TBI, and in clinical cases of TBI. 15 –18 OHSCs exposed to bTBI have a more intense activation of microglia than astrocytes. 9 Although microglia may be involved, the neuroinflammatory processes that might lead to electrophysiological deficits following a single and repeated mild bTBI are not well understood. Activated microglia release various pro-inflammatory cytokines, which have been associated with neurological deficits following TBI in multiple injury models. 19 Pro-inflammatory cytokines remain at very low levels in healthy brain tissue 20 ; at low concentrations, cytokines may be needed for physiological functions, including LTP. 21,22 However, following an injury or other pathological insult, the resulting neuroinflammation and changes in cytokine/chemokine concentrations have been associated with neurological deficits. 23,24 Repeated low-level blast injury in rats elicited various changes in cytokines, neurodegenerative protein markers, blood–brain barrier proteins, and mechanotransduction proteins. 25,26 Studies examining military service members exposed to occupational blast overpressures have reported changes in serum cytokines and neuroinflammatory biomarkers, like glial fibrillary acidic protein (GFAP) and ubiquitin carboxyl hydrolase (UCH)–L1. 27 -29 Others have reported that military breachers, who are more likely to be exposed to multiple low-level blasts during the course of their training and deployment, have changes in transcriptional and inflammatory biomarkers. 27,30
We used an in vitro OHSC model of bTBI to characterize changes in LTP after various mild blast exposure levels and inter-blast intervals. We also studied the contribution of microglia, and quantified changes in pro- and anti-inflammatory cytokines and chemokines. By partially depleting microglia in our injury model, functional deficits were markedly improved. Lastly, treatment with a novel drug, MW01-6-189WH (MW-189), prevented LTP deficits. These findings provide promising pathways to explore for reducing functional deficits following repeated, mild bTBI. Additionally, the success of MW-189 in attenuating LTP deficits warrants further evaluation in pre-clinical and clinical studies of repeated bTBI.
Methods
Organotypic hippocampal slice culture
All animal procedures were approved by the Columbia University Institutional Animal Care and Use Committee (IACUC). OHSCs were generated as previously described. 31 In brief, hippocampi, excised from P8-10 Sprague Dawley rat pups, were sectioned into 400-μm thick slices and grown on Millipore Millicell™ cell culture inserts (0.4 μm pore size, Millipore, Billerica, MA) in Neurobasal medium supplemented with 2 mM GlutaMAX, 1x B27 supplement, 10 mM HEPES, and 25 mM D-glucose (Life Technologies, Grand Island, NY). Following culturing, OHSCs were fed every 3-4 days by replacing half of the medium with fresh full-serum medium, containing 50% minimum essential medium, 25% Hank's balanced salt solution, 25% heat inactivated horse serum, 2 mM GlutaMAX, 25 mM D-glucose, and 10 mM HEPES (Sigma). Slices were maintained for at least 10 days prior to experimentation.
Primary blast injury exposure
Blast injury methods have been previously described in detail. 7,8,32,33 Individual culture inserts were sealed in sterile polyethylene bags (Whirl-Pak, Fort Atkins, WI) filled with serum-free medium (75% minimum essential medium, 25% Hank's balanced salt solution, 2 mM GlutaMAX, 25 mM D-glucose, and 10 mM HEPES; Life Technologies) and positioned 85 mm into the fluid-filled receiver, maintained at 37˚C. The receiver was sealed with a polydimethylsiloxane (PDMS) membrane and placed directly at the shock tube exit, with a 5-mm gap. Either compressed helium or nitrogen was used as the driver gas for specific exposures.
OHSCs were exposed to multiple level 1, 2, or 3 injuries (Table 1) as per our previous publication 8 with inter-blast intervals of 1, 3, or 6 days. For comparisons, samples in the sham group were prepared and placed in the receiver, but did not receive a shockwave exposure. Slices were maintained for 4 days following the last of the repeated blast injuries and interrogated for LTP. OHSCs that were partially depleted of microglia and those that were treated with MW-189 were exposed to two repeated level 3 blast injuries with a 1-day inter-blast interval.
Blast Characteristics and Injury Protocol
Blast exposure levels and injury protocol tested in this study (as reported earlier). 8
Cell death measurement
At Day 10 prior to blast injury and immediately before electrophysiology experiments and use in any biochemical assays, OHSCs were incubated in 2.5 μM propidium iodide (PI; Life Technologies) in serum-free medium for 1 h before imaging. Images were acquired using an Olympus IX81 microscope (Olympus America, Center Valley, PA) with 568/24 nm (peak/width) excitation and 610/40 nm emission filters. Cell death was quantified as the percent area of PI staining above a predetermined threshold immediately prior to injury and immediately prior to electrophysiological studies or use in biochemical assays. OHSCs were deemed healthy if there was less than 5% PI staining.
Electrophysiological recordings
Electrophysiological activity was recorded using 60-channel microelectrode arrays (MEAs, 8 × 8 electrode grid without the corners, 30-μm electrode diameter, 200-μm electrode spacing, 60MEA200/30iR-Ti-gr, Multi-Channel Systems, Reutlingen, Germany). The OHSCs were perfused with artificial cerebral spinal fluid (aCSF) containing 125.0 mM NaCl, 3.5 mM KCl, 26.0 mM NaHCO3, 1.2 mM KH2PO4, 2.4 mM CaCl2, 1.3 mM MgCl2, 10.0 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), and 10.0 mM D-glucose (pH = 7.4), which was bubbled with 5% CO2/95% O2 warmed to 37°C. 34 Recordings were acquired at 20 kHz with a 6 kHz analog, anti-aliasing filter and further filtered in MATLAB™ (Mathworks, Natick, MA) using an eighth-order, digital, low-pass (1000 Hz) and a fourth-order, digital, high-pass (0.2 Hz) Butterworth filter.
To induce LTP, the OHSCs were stimulated in the Schaffer Collateral (SC). First, stimulus-response curves were generated by applying a constant current, biphasic, bipolar stimulus of increasing magnitude (0-100 μA in 10 μA increments). Peak-to-peak evoked responses were fit to a sigmoidal curve, and three parameters were quantified: Rmax, the maximum amplitude of the evoked response; m, the slope of the sigmoidal fit; and I50, the current needed to elicit a half maximal response. 7 This I50 was used to stimulate the electrodes in the SC pathway once a minute for 30 min to establish baseline recordings. LTP was induced by stimulating the same electrodes in the SC pathway with a high frequency stimulus train (three trains of 100 Hz applied for 1 sec with each train separated by 10 sec). Post-LTP responses were evoked by stimulating at I50 every minute for 60 min. LTP induction was calculated as the percent change between the average peak-to-peak responses in the last 10 min of the baseline and post-LTP recordings of the electrodes in the CA1 region, normalized to the baseline response.
To ensure stable responses were included for analysis, electrodes were discounted if the coefficient of variance (in the average of the peak-to-peak responses of the last 10 min of either baseline or post-LTP signals) was greater than 20%. Electrodes were also discounted if the average of the peak-to-peak responses in the last 10 min of the baseline response was not within the range of 35-65% of the Rmax. This range was chosen to accommodate the electrodes that would approximately lie in the linear range of the sigmoidal stimulus-response curve. After applying these criteria, slices were also excluded if there were less than five electrodes to average in the CA1 region.
Drug treatment
To determine the role of microglia in contributing to LTP deficits following repeated bTBI, a group of OHSCs were treated with liposomal clodronate (Clodrosome, Encapsula NanoSciences, Brentwood, TN) or PLX3397 (pexidartinib, Selleckchem, Houston, TX) to deplete microglia from the slices. Immediately after culturing the OHSCs, the slices were treated with either 25 μg/mL of Encapsome (the liposomal constructs without the encapsulated clodronate) or either 10 or 25 μg/mL of Clodrosome in Neurobasal medium. After 24 h of exposure, the culture medium was replaced with fresh Neurobasal medium. The OHSCs were then maintained with the same feeding protocol as described above. A subset of OHSCs was treated with either 1 μM pexidartinib (dissolved in 0.05% dimethyl sulfoxide [DMSO]) or vehicle (DMSO, 0.05%) from Days in vitro (DIV) 6-10. As a positive control for microglial activation, OHSCs were treated with 100 ng/mL of lipopolysaccharide (LPS, L4516, Sigma Aldrich, St. Louis, MO) following PI imaging on DIV 10. Stock solution of MW-189 (dissolved in 0.9% saline), kindly gifted by Dr. Linda Van Eldik (University of Kentucky), was diluted to 30 μM in fresh, full serum medium.
Multiplex enzyme-linked immunosorbent assay
OHSCs exposed to a single level 3 blast, two repeated level 3 blasts, or two repeated sham injuries were collected 3 h and 24 h after the final blast or sham exposure. On ice, four OHSCs were lysed in Milliplex MAP Lysis Buffer (Millipore Sigma, St. Louis, MO) supplemented with 2 mM PMSF, 1 mM sodium orthovanadate, and a protease inhibitor cocktail (Santa Cruz Biotechnologies, Dallas, TX). The tissue was mechanically lysed with a 27-gauge needle and syringe 20 times, centrifuged for 15 min at 13,000 rpm, and the supernatant collected. The concentration of protein in the collected supernatant was determined by the bicinchoninic acid (BCA) assay according to the manufacturer's instructions (Thermo Fisher Scientific, Waltham, MA). All samples were standardized to the same concentration of total protein.
The levels of cytokines and chemokines from the brain tissue supernatant were measured by multiplex immunoassay using Milliplex MAP Rat Cytokine/Chemokine magnetic panel according to the manufacturer's instructions (Millipore Sigma, St. Louis, MO). Analytes included interleukin (IL)-2, IL-5, IL-18, IL-12 (p70), IL1-α, IL-1β, IL-17A, IL-4, IL-6, IL-13, IL-10, tumor necrosis factor-α, granulocyte colony stimulating factor, monocyte chemoattractant protein (MCP)-1, macrophage inflammatory protein (MIP)-1α, LPS-induced CXC chemokine (LIX), fractalkine, vascular endothelial growth factor, regulated on activation, normal T cell expressed and secreted (RANTES), granulocyte-macrophage colony-stimulating factor, eotaxin, MIP-2, leptin, epidermal growth factor, interferon γ, GRO/KC/CINC-1, and interferon gamma-inducible protein (IP)-10.
Western blotting
Following injury or microglial depletion, six OHSCs were pooled together, flash frozen, and stored at -80°C until tissue lysis. On ice, the pooled tissues were lysed with a standard radioimmunoprecipitation assay lysis buffer kit (Santa Cruz Biotechnologies, Dallas, TX), supplemented with 2 mM PMSF, 1 mM sodium orthovanadate, and a protease inhibitor cocktail. The tissue was mechanically lysed with a 27-gauge needle and syringe 20 times, sonicated (Misonix XL 2000 Ultrasonic Cell Disruptor, IL, USA), and centrifuged for 15 min at 13,000 rpm. The concentration of protein in the collected supernatant was determined by the BCA assay. Samples probed for Iba1 were denatured and reduced with NuPAGE™ LDS Sample Buffer (4 ×) (Invitrogen, Waltham, MA) and NuPAGE™ Reducing Agent (Invitrogen, Waltham, MA) and boiled at 95°C for 15 min. Samples probed for CD68 and CD206 were only denatured with NuPAGE™ LDS Sample Buffer (4 × ) and boiled at 95°C for 15 min. A total of 50 μg protein of each sample was loaded onto a 4-12% Bis-Tris gel (Life Technologies, Carlsbad, CA) and separated at 150 V for 60 min using mPAGE™ MES SDS running buffer (EMD Millipore, Burlington, MA).
Proteins were transferred to a nitrocellulose membrane (Life Technologies), blocked in Tris-buffered saline (TBS; pH 7.4) with 3% bovine serum albumin (BSA) and 3% donkey serum for 1 h. Blots were incubated overnight at 4°C with primary antibodies (Iba1 [Abcam, #ab5076, 1:1000], β-tubulin [Invitrogen, #32-2600, 1:2000], CD68 [Abcam, #ab125212, 1:1000], CD206 [Abcam, #ab64693, 1:500]) in TBS with Tween (TBS-T, 0.1% Tween-20, pH 7.4) with 1% BSA and 1% donkey serum. Membranes were washed and then incubated with corresponding secondary antibodies (donkey anti-goat Alexa Fluor 647 [Abcam, #ab150131, 1:1000], donkey anti-rabbit Alexa Fluor 647 [Abcam, #ab150073, 1:1000], and donkey anti-mouse Alexa Fluor 488 [Abcam, ab150105, 1:2000]) at room temperature for 2 h. After washing, the blot was imaged using a CRi Maestro 2 Imaging System (Perkin Elmer, Waltham, MA).
Immunohistochemistry
OHSCs that were sham or blast injured and OHSCs that were depleted of microglia using either Clodronate or pexidartinib were fixed in 10% formalin, incubated with primary antibodies (Iba1 [Abcam, #ab5076, 1:1000], CD68 [Abcam, #ab125212, 1:1000], or CD206 [Abcam, #ab64693, 1:1000]) at room temperature for 1 h, followed by incubation with secondary antibodies (donkey anti-goat Alexa Fluor 647 [Abcam, #ab150131, 1:1000], donkey anti-rabbit Alexa Fluor 647 [Abcam, #ab150073, 1:1000]) at room temperature for 1 h. Hoechst (Thermo Fisher Scientific, Waltham, MA) was used to label the nuclei. At the completion of staining, OHSCs were cleared with the CUBIC (Clear, Unobstructed Brain/Body Imaging Cocktails and Computational) method. 35 OHSCs were imaged with a Zeiss LS700 confocal microscope (Oberkochen, Germany).
Statistical analysis
Long-term potentiation deficits following repeated mild bTBI and Western blots evaluating microglial activation and microglial depletion were analyzed by analysis of variance (ANOVA), followed by a Dunnett post hoc analysis. For the studies with Clodrosome, an ANOVA was performed with a Bonferroni post hoc test. For pexidartinib and MW-189 studies, treated groups were compared with control groups with a Student's t-test. Statistical significance was set as p < 0.05 for all of these studies. For multiplex enzyme-linked immunosorbent assay (ELISA) studies, to correct for the large number of measurements, p values were adjusted with the Benjamini-Hochberg (BH) procedure and a false discovery rate determined to yield less than one false positive result. 36,37 Blast injured groups that were deemed to be statistically significant following the BH procedure were analyzed by a Tukey post hoc test with statistical significance set as p < 0.05. Groups treated with MW-189 were analyzed with a Student's t-test followed by the BH procedure.
Results
Increasing the interval between two repeated blast exposures attenuated LTP deficits
Compared with sham cultures, LTP was significantly reduced after exposure to two level 3 blasts at both the 1-day and 3-day inter-blast intervals (Fig. 1). As the inter-blast interval increased, post-injury LTP increased, suggesting that the compounding effects of repeated blast on LTP deficits were lessened as the interval between blast injuries increased. Although not statistically significant, LTP was slightly decreased after level 1 or 2 exposures at all intervals tested. To explore the mechanism by which repeated mild bTBI might be compounding neurological deficits, we focused on the injury exposure that yielded the greatest LTP deficits (two level 3 blast injuries with a 1-day interval) as well as a shorter recovery time (2 days following injury).

Long-term potentiation (LTP) following repeated blast-induced traumatic brain injury to organotypic hippocampal slice cultures (OHSCs).
Repeated bTBI exposure induced microglial activation
CD68 expression significantly increased after two level 3 blasts, indicating an increase in microglial activation (Fig. 2D). Interestingly, repeated level 3 bTBI markedly decreased CD206 expression (Fig. 2E), indicating a decrease in anti-inflammatory microglial phenotype. Iba1 expression, a surrogate for total number of microglia, did not change (Fig. 2F) compared with sham-exposed OHSCs.
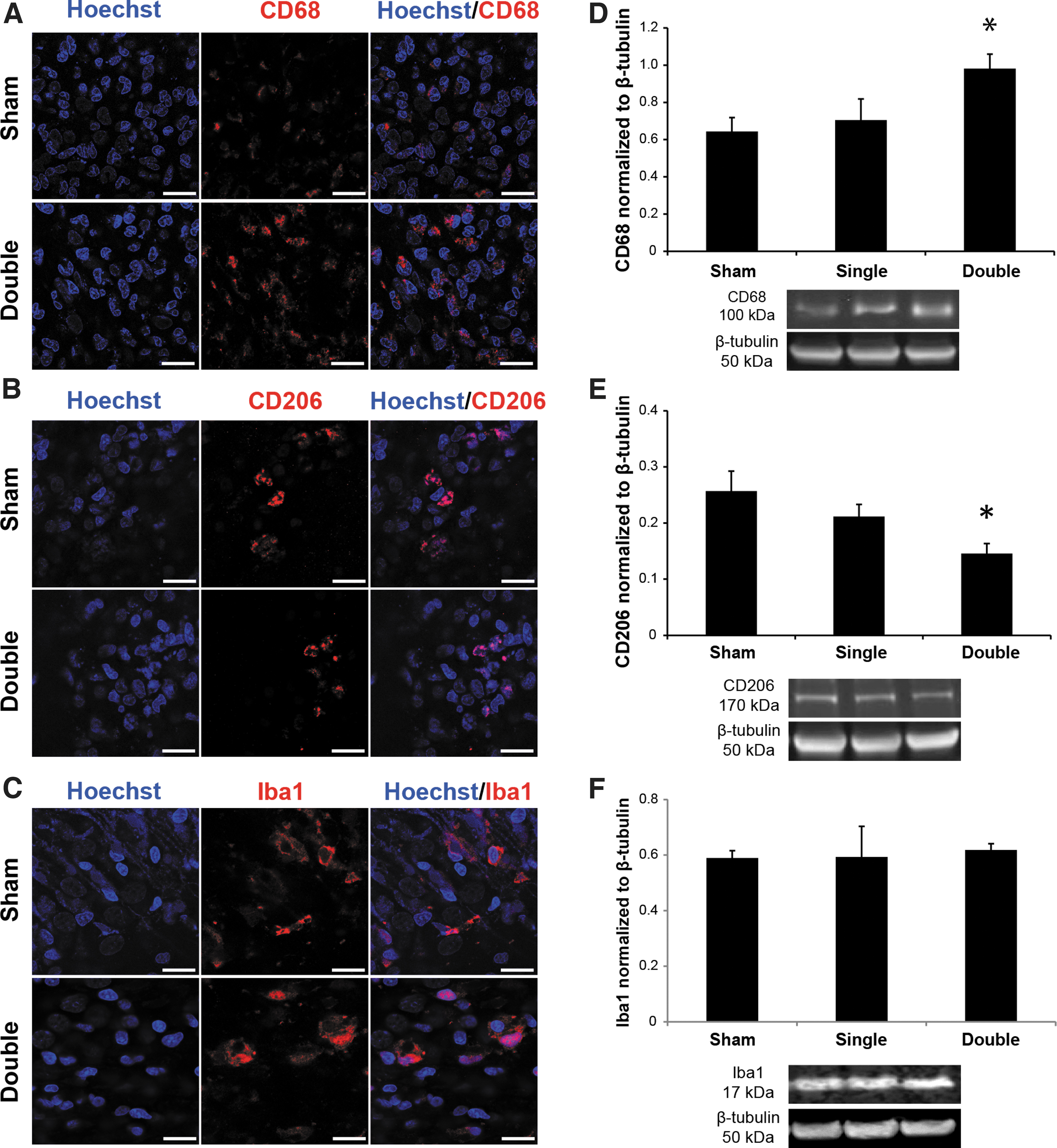
Microglial activation following blast-induced traumatic brain injury (bTBI). Representative immunohistochemical staining of Hoechst (nuclei),
Clodrosome and pexidartinib treatments partially depleted microglia
To determine the role of activated microglia following repeated bTBI, we used two different pharmaceutical methods to partially deplete microglia. Clodrosome, a clodronate encapsulated liposome construct, is selectively engulfed by microglia, causing apoptosis. 38 Pexidartinib, a colony stimulating factor 1 receptor (CSF1R) inhibitor, is commonly used to deplete microglia in vivo and in vitro. 39 To verify that microglia were in fact depleted following either Clodrosome or pexidartinib administration, LPS was used to fully activate any remaining microglia. 40,41 Compared with Encapsome treated controls, Clodrosome (10 or 25 μg/mL) treatment significantly decreased Iba1 expression (Fig. 3C). Similarly, compared with vehicle treatment, pexidartinib treatment significantly decreased Iba1 expression. Both treatments resulted in marked depletion of microglia.
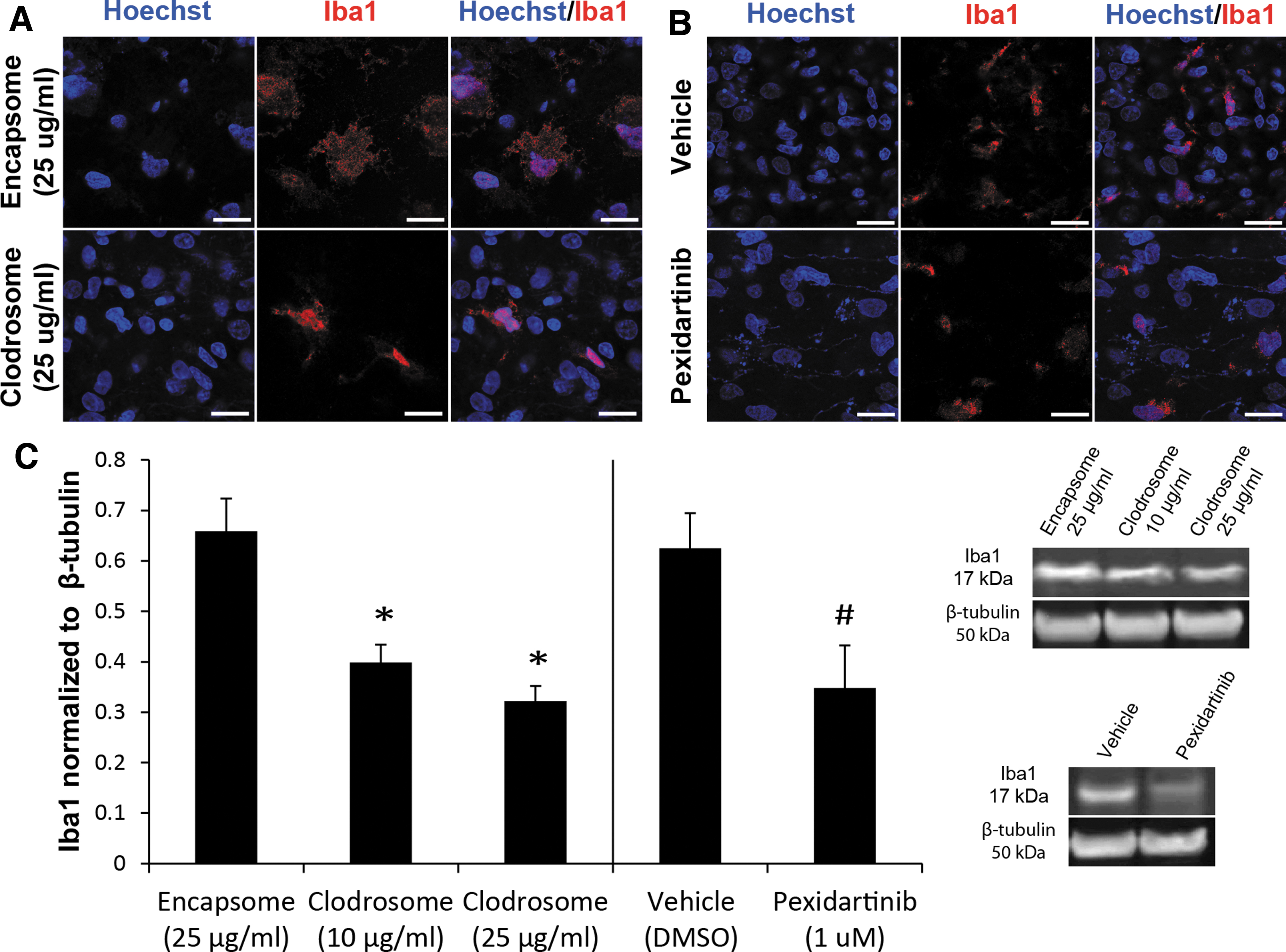
Microglial depletion following Clodrosome or pexidartinib administration. Representative immunohistochemical staining of Hoechst (nuclei) or Iba1 following either
Partial depletion of microglia prevented LTP deficits after repeated blast injury
Partial depletion of microglia from OHSCs using either Clodrosome or pexidartinib yielded viable slice cultures on DIV 10 as evidenced by very low PI staining (data not shown). Treatment with 25 μg/mL Clodrosome non-significantly decreased LTP (Fig. 4), compared with vehicle-treated OHSCs. Following repeated level 3 blast injury, LTP was significantly decreased in vehicle-treated OHSCs, whereas Clodrosome treatment significantly reduced the LTP deficit compared with the injured vehicle group.
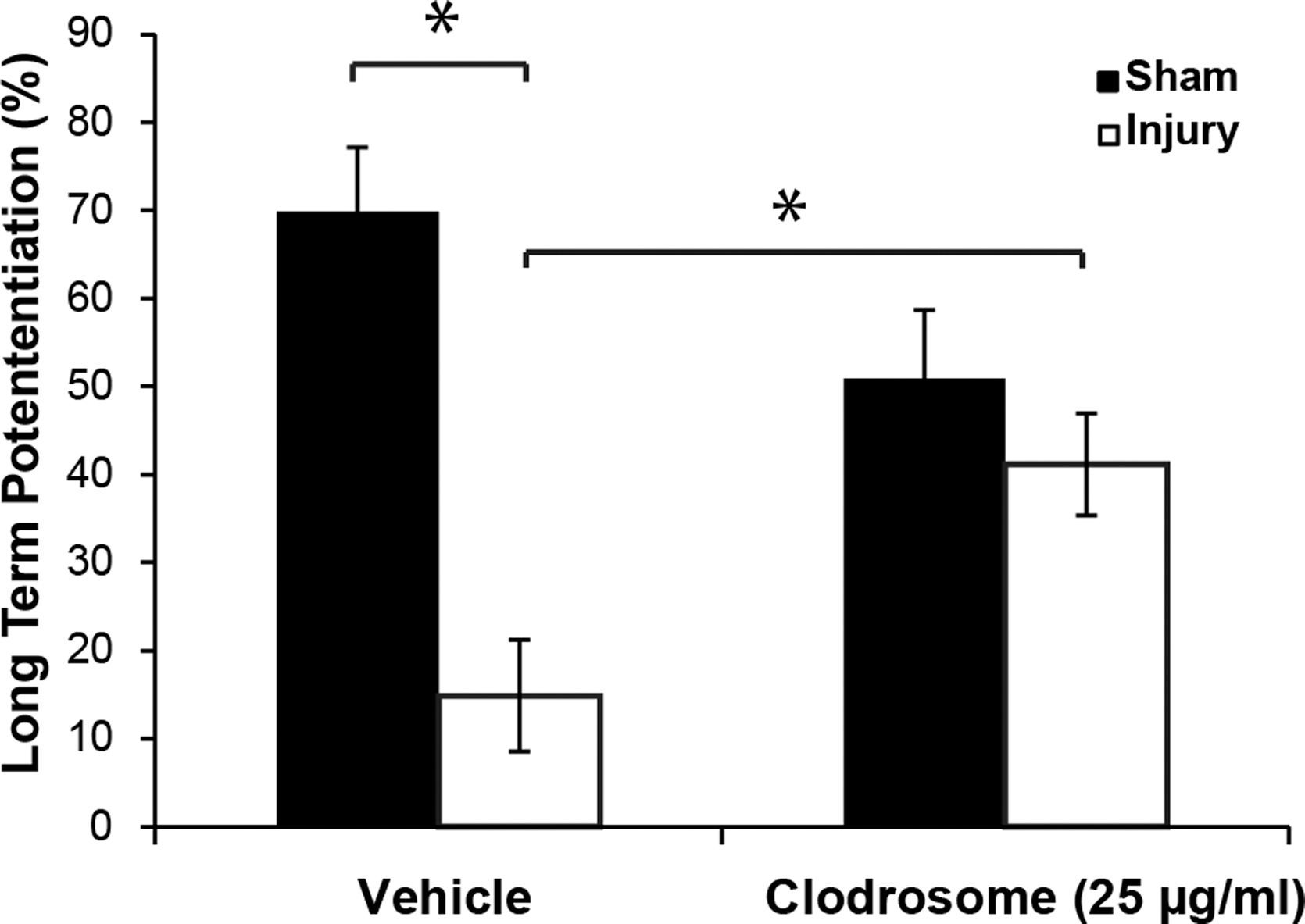
Long-term potentiation (LTP) in organotypic hippocampal slice cultures (OHSCs) treated with either vehicle or Clodrosome (25 μg/mL) before and after repeated mild blast-induced traumatic brain injury (bTBI). LTP was significantly decreased after injury in vehicle treated OHSCs. Clodrosome significantly improved LTP following repeated bTBI. Mean ± standard error of the mean; n ≥ 8; *p < 0.05.
Similarly, pexidartinib (1 μM) also prevented LTP deficits (Fig. 5) following repeated blast exposures. After injury, LTP significantly improved after partial depletion of microglia with pexidartinib treatment from DIV 6-10 and throughout the post-injury period. There were still significant functional improvements even when injured slices were only pretreated with pexidartinib from DIV 6-10. LTP also improved in OHSCs that were treated with pexidartinib only following blast injury.

Effects of microglial depletion on long-term potentiation (LTP) in organotypic hippocampal slice cultures (OHSCs) treated with either vehicle or pexidartinib (1 μM) before and/or after repeated mild blast-induced traumatic brain injury. Pexidartinib treatment of sham OHSCs did not significantly decrease LTP. LTP was significantly improved after blast by pexidartinib treatment before and after injury, only after injury, or only before injury. Mean ± standard error of the mean; n ≥ 7; *p < 0.05, #p < 0.05, §p < 0.05 compared with vehicle treated OHSCs.
After repeated bTBI, LTP recovered following treatment with MW-189
LTP significantly improved after treatment with MW-189 immediately following the second of two repeated level 3 bTBIs (Fig. 6A). Unlike repeated level 3 blasts, a single level 3 blast injury did not cause significant LTP deficits (sham: 63.06% ± 8.10 [mean ± standard error of the mean] vs. injured: 59.01% ± 5.47; n = 8). In addition, treatment of OHSCs only during the interval between two level 3 blasts also significantly improved LTP deficits 48 h after a second blast exposure.

Effects of MW-189 treatment on long-term potentiation (LTP) in organotypic hippocampal slice cultures following repeated blast-induced traumatic brain injury (bTBI).
Multiplex analysis of cytokines/chemokines
The levels of 27 cytokines/chemokines in OHSCs at 3 and 24 h following single and repeated bTBI were quantified by multiplex ELISA (Supplementary Table S1). At 3 h following a single bTBI, concentrations of MIP-1α, IL-1β, MCP-1, and IP-10 were increased (Fig. 7). Repeated bTBI increased concentrations of IL-1β, IP-10, MIP-1α, and RANTES 3 h after the final injury. At 24 h following either a single or repeated bTBI, compared with the sham group, IL-10 was significantly decreased (Fig. 7F). No other analytes were significantly altered 24 h following injury.

Changes in cytokine and chemokine expression following a single or repeated blast-induced traumatic brain injury (bTBI).
MW-189 treatment did not significantly alter the expression of any analytes (Supplementary Table S2). Although not statistically significant following corrections for multiple comparisons, there was an increase in IL-10 expression (p = 0.0287) at 3 h and a decrease in IP-10 expression (p = 0.032) at 24 h after repeated bTBI and treatment with MW-189.
Discussion
We previously reported that a single mild primary blast induced a period of heightened vulnerability to a subsequent primary blast injury. 9 In this study, we expanded upon our previous findings by varying the blast severity and the inter-blast interval, while keeping the recovery period constant. In doing so, we found improvements in LTP deficits with increasing blast intervals, suggesting that longer time periods between blasts resulted in better functional outcomes—findings in line with previous studies. 9,42 Herein, we report that repeated mild bTBI induced changes in microglial activation with a differentiated response in pro- and anti-inflammatory microglial markers. Depleting microglia with two different drugs, each of which has a unique mechanism of action, improved LTP after repeated blast exposure. Administration of the novel drug, MW-189, markedly improved LTP following repeated injury.
Several studies have suggested that microglia are activated after bTBI. 43 –46 Our group previously reported that mild bTBI induced a change in microglial morphology to a more amoeboid type. 9 To better understand the state of microglial activation following repeated bTBI, we quantified the expression of the pro-inflammatory microglial marker, CD68, and the expression of the anti-inflammatory microglial marker, CD206. Recent studies have concluded that Iba1 is not considered a pan-microglial marker, as it was traditionally thought to be. 47 A study utilizing an experimental diabetic retinopathy model also concluded that Iba1 was not a sensitive marker for microglial activation. 48 CD68 is specific to microglia that are activated in response to tissue damage 49 and is associated with microglia that have an amoeboid morphology. 47 CD206 is generally specific to microglia that are anti-inflammatory. 50,51 Forty-eight hours after two repeated bTBIs, expression of CD68 was significantly increased, and expression of CD206 was significantly decreased.
In the current study, there were no increases in the expression of Iba1 following single and repeated level 3 bTBI. This lack of increase in the expression of Iba1 following bTBI may be due to the already ubiquitous expression of Iba1 in the days following culturing, 52 the relatively low level of injury that was used for these studies, 46 or the absence of the infiltration of other systemic macrophages and immune cells otherwise attributed to blood–brain barrier permeability in vivo. 53,54 The presence of horse serum in our culture media may also be responsible for increasing Iba1 expression in slice cultures. 55 These data suggest that microglia were activated following repeated bTBI. Our findings are in contrast with one study that reported no significant neuroinflammation or microglial activation following repeated low-level bTBI in the absence of focal hemorrhage. 56 Unlike that study, our current study focused on more acute time-points following bTBI, which may explain the contrasting findings.
As evidenced by the increase in CD68 expression and the decrease in CD206, more pro-inflammatory microglia were prevalent than anti-inflammatory microglia after injury, similar to other studies at acute and chronic time-points after brain trauma. 57 -59 However, on the contrary, some models of TBI have reported a transient increase of anti-inflammatory microglia during acute periods following injury and a later predominance of pro-inflammatory phenotypes. 57,60 Unlike our study, those studies all utilized mice models with a controlled cortical impact (CCI) TBI. The biomechanics of tissue damage from bTBI differs from CCI injury as blast injuries induce lower strains at higher strain rates, which (as in the current studies) induced microglial activation. 61 Additionally, CCI injuries induce significant cell death and neurodegeneration of the brain parenchyma, which is not observed in our bTBI model. 8,9 Additionally, those studies all examined microglial activation in the cortex or the whole brain following injury, and so their results may not account for the heterogeneity of microglial populations in different parts of the brain, like the hippocampus. 62 Lastly, our studies are limited in drawing broader conclusions about the mix in activation states of microglia across different time-points as we only quantified changes in CD68 and CD206 at one time-point (48 h) after injury.
Expression of CD68 was positively correlated with the number of injuries while CD206 expression was negatively correlated. This difference in expression suggests a more predominant pro-inflammatory milieu at the time-point observed. The LTP deficits observed following repeated blast injury exposure may be correlated with increased microglial activation. 9 Although there are slight but non-significant changes in CD68 and CD206 following only a single injury, the lack of functional deficits following a single level 3 bTBI suggests a threshold of microglial activation must be crossed prior to loss of neuronal function, like LTP. The compounding of LTP deficits following a repeated bTBI could be explained by an increasing amount of activated microglia with each subsequent blast injury, which other studies support. 63,64
Although there was a dose-response for the CD68 and CD206 expression as the number of blast injuries increased, there was no such correlation with most of the cytokines and chemokines that were significantly altered. Despite the increase in several analytes, such as IL-1β, MCP-1, IP-10, and MIP-1α 3 h following a single blast injury, those analytes generally remained at the same levels following a second blast injury. Since there were no statistically significant LTP deficits following only a single blast injury, these analytes may not be responsible for LTP deficits after repeated blast exposure.
IL-10 expression was decreased in a dose-dependent manner with an increasing number of blast exposures. Studies evaluating mechanical injury of primary rat microglia have also reported decreased expression of IL-10. 65 Contrary to our findings, other studies have reported elevated levels of IL-10 in cerebrospinal fluid following severe TBI 66 and have associated increased IL-10 levels with mortality after severe TBI. 67,68 However, unlike our studies in OHSCs following mild bTBI, these studies were all conducted in human subjects who had experienced severe TBI injuries, which resulted in severe neurodegeneration and cell death.
RANTES expression also was upregulated following repeated bTBI after 3 h. Several studies have found a significant correlation between RANTES levels in the plasma of TBI patients upon admission and poor outcome. 69,70 One study evaluating pro-inflammatory cytokines in mice following repetitive mild TBI induced by a weight drop have reported increased concentrations of RANTES; RANTES was also localized with neurons, suggesting that perhaps neuronal damage may upregulate RANTES expression. 71 Although RANTES was upregulated in our studies with an increasing number of injuries, it may not be responsible for the LTP deficits that are observed following repeated injury. In a knock-out mouse model, RANTES was found to be necessary for recovery from mild TBI. 72 Another RANTES knock-out mouse model displayed impaired learning and cognition that was restored following re-expression of RANTES. 73 In mild TBI cases, increasing RANTES may be a possible therapeutic intervention to prevent neurological deficits. Of the analytes that we quantified, RANTES may be considered a biomarker for injury severity.
Because microglial activation may be responsible for the LTP deficits we observed, we hypothesized that the depletion of microglia from the slice cultures would prevent such deficits following repeated injury. Clodronate encapsulated liposomes, and pexidartinib, have both been previously used to prevent kainic acid induced neuronal cell death. 74 Pexidartinib has been used extensively to deplete microglia in mice without causing any significant behavioral or cognitive deficits. 39 Although LTP deficits were significantly attenuated following partial microglial depletion with Clodrosome or pexidartinib after repeated bTBI, there was a slight, but non- significant decrease in LTP compared with sham cultures treated with a vehicle. These non-significant LTP deficits following partial microglial depletion could be partially explained by the role that microglia have in maintaining and regulating LTP 75,76 and functional synapses. 77
There are conflicting reports on the efficacy of microglial depletion in attenuating deficits associated with TBI. One pre-clinical study administered PLX5622, a similar CSF1R inhibitor, to adult rats one month after a CCI injury to deplete chronically activated microglia. 78 Following a period of drug withdrawal and microglial repopulation, the rats that underwent TBI had limited neuropathological changes 3 months after injury compared with rats that did not receive the PLX5622 treatment. Another group using PLX5622 to deplete microglia prior to TBI with a midline fluid percussion injury (FPI) in mice found that elimination of microglia reduced acute and chronic inflammatory responses. 13,79 Treatment of mice with PLX3397 prior to a moderate FPI improved post-traumatic neurite outgrowth, preserved dendritic spines, and reduced cell death. 80 In non-TBI models of neurodegeneration, like hippocampal lesions via the neuronal expression of diphtheria toxin A-chain, 81 Alzheimer's disease, 82,83 and leukoencephalopathy, 84 CSF1R inhibitors have improved functional outcomes via microglial depletion. However, some other studies have suggested that microglial depletion results in more severe brain injury 85 and neuronal network dysfunction. 86 Depletion of microglia in neonatal rats using liposomes containing clodronate increased neurodegeneration following a closed head injury model of TBI. 87 In another recent study, depleting microglia only prior to a CCI injury in mice did not improve spatial learning or memory, but allowing for the microglia to repopulate attenuated TBI-induced deficits through the stimulation of interleukin-6 mediated neurogenesis. 88 Microglial depletion in models of epilepsy 89 -91 and inflammation-induced sickness 92 increased neurological deficits. Given the conflicting reports of microglial depletion in attenuating TBI induced deficits, microglial depletion may not be a viable therapeutic strategy. However, our studies suggest that microglial activation may be responsible for neurological deficits following mild bTBI.
To further test whether microglia are responsible for causing LTP deficits, we utilized a novel drug, MW-189, that modulates the release of cytokines and chemokines from activated microglia. 93 The administration of MW-189 following repeated bTBI did not prevent bTBI associated increases in pro-inflammatory cytokines and chemokines. However, treatment with MW-189 did increase the anti-inflammatory cytokine, IL-10, a result consistent with a phase 1 clinical trial that reported increased plasma levels of IL-10 following dosing with MW-189. 94 Increased expression of IL-10 may explain why LTP improved in the current study; other studies have reported improvements in LTP, learning, and memory with increased expression of IL-10. 95 -97
MW-189 administration only during the period between the repeated bTBIs also prevented LTP deficits. Although IL-10 decreased 3 h after a single blast injury, the short treatment with MW-189 during this acute time-point following an initial bTBI may have prevented the decrease in IL-10, thereby providing protection against a subsequent injury that otherwise could induce functional deficits.
There are several limitations to consider in interpreting the results of this study. The functional deficits in our in vitro model of bTBI may not directly translate to the behavioral and functional deficits observed in vivo. 98 However, LTP is considered to be a neuronal correlate for learning and memory. 99,100 The identification of microglial populations using CD68 and CD206 provided a biomarker for understanding the changes in microglial activation following injury. For future studies, combinations of several microglial markers can be used with fluorescence-activated cell sorting and other types of single cell sequencing to characterize the different microglial phenotypes that arise after activation. 47 Such studies may identify microglial populations with distinct gene signatures responsible for regulating homeostatic pathways following injury. 101 Improved characterization of the microglial population after an injury may provide more insight into which specific microglial phenotypes may be involved in causing neurological deficits and could offer strategies for more targeted therapeutic interventions.
Although partial depletion of microglia with Clodrosome and pexidartinib protected LTP following injury, the drugs may be maintaining LTP via some other non-microglial mechanism. However, having used two different drugs, each of which has a unique mechanism of action, provides more confidence that any improvements in LTP were a result of partial microglial depletion. Although we gained insight into changes in various pro- and anti-inflammatory cytokines and chemokines, the multiplex ELISA does not provide enough information to determine which cell types were responsible for the observed changes. Future studies could injure monocultures of neurons, microglia, and astrocytes to better understand the contribution of each cell-type to the overall neuroinflammatory profile. MW-189 has been found to be safe in phase 1 clinical trials, but has not been tested as a therapeutic intervention following bTBI. 94,92 Further in vitro validation of MW-189 in treating functional deficits of bTBI may help advance the drug into clinical trials for bTBI. In this study, we have better elucidated the contribution of microglial activation to the LTP deficits observed following bTBI. Microglia may be an attractive therapeutic target for the attenuation of neurological deficits following blast injury.
Footnotes
Acknowledgments
The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. MW-189 was a kind gift from Dr. Linda Van Eldik, University of Kentucky.
Authors' Contributions
N.V.: conceptualization (lead), writing – original draft (lead), formal analysis (lead); writing – review and editing (equal), methodology (equal), visualization (equal). B.M.: writing – review and editing (equal), resources (lead), project administration (lead), investigation (lead), funding acquisition (lead), methodology (equal).
Funding Information
This work was supported by DEVCOM Army Research Laboratory and the Paul G. Allen Frontiers Group, grant 12347 as well as the Biomarkers Core Laboratory at the Irving Institute for Clinical and Translational Research, home to Columbia University's Clinical and Translational Science Award Program hub. This publication was supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant Number UL1TR001873.
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.