
Abstract
Acid-sensing ion channel 1a (ASIC1a) is a proton-activated channel that is expressed ubiquitously throughout the central nervous system and in various types of immune cells. Its role in spinal cord injury (SCI) is controversial; inhibition of ASIC1a has been reported to improve SCI pathology in vivo, but conversely, gene ablation increased kainite-mediated excitotoxic cell death in vitro. Here, we re-examined the role of ASIC1a in a mouse model of SCI. First, we observed functional outcomes up to 42 days post-operation (DPO) in SCI mice with a selective genetic ablation of ASIC1a. Mice lacking ASIC1a had significantly worsened locomotor ability and increased lesion size compared with mice possessing the ASIC1a gene. Next, we explored pharmacological antagonism of this ion channel by administering the potent ASIC1a inhibitor, Hi1a. Consistent with a role for ASIC1a to attenuate excitotoxicity, accelerated neuronal cell loss was found at the lesion site in SCI mice treated with Hi1a, but there were no differences in locomotor recovery. Moreover, ASIC1a inhibition did not cause significant alterations to neutrophil migration, microglial density, or blood–spinal cord barrier integrity.
Introduction
Spinal cord injury (SCI) results in a complex cascade of downstream processes. The injury—whether a blunt force or penetrative trauma—causes immediate damage to the macrostructure of the spinal cord, opening the blood–spinal cord barrier (BSCB) to peripheral interference. Immune cells migrate toward the lesion site where they contribute to an inflammatory microenvironment that is non-conducive to healing. 1 -3
Neutrophils are the first peripheral immune cells to arrive in the spinal cord after it is injured, migrating through endothelial barriers within 24 h post-SCI. 4 -6 These neutrophils trigger damage through release of reactive oxygen species (ROS) and elastase at the injury site. 7 –10 Infiltrating macrophages (and resident microglia) also contribute to pathology, peaking at 7 days post-operation (DPO). 4 Here, they exist in a pro-inflammatory state, causing axonal dieback through production of neurotoxic tumor necrosis factor-α and increased astrocyte reactivity, resulting in myelin loss and impaired locomotor recovery. 11 –14 It is proposed that these cells persist in this state at the lesion site, resulting in maintenance of a chronic wound post-SCI. 2
This progressive spread of damage beyond the original insult site is defined as secondary damage. 15,16 Long-term outcomes are linked to the severity of secondary damage, which negatively impacts locomotor recovery, resolution of pain, and restoration of bladder and bowel control. 17 The financial burden is estimated at GBP 1.12 million per patient. 18 The life-changing impacts of SCI upon the individual, in addition to the massive socioeconomic cost, have driven efforts to develop effective neuroprotective treatments to reduce secondary damage after injury.
Central nervous system (CNS) trauma causes acidosis, with tissue pH dropping as low as 6.0 during severe cerebral ischemia. 19 This drop in pH is sufficient to robustly activate acid-sensing ion channel 1a (ASIC1a), which is expressed in—and contributes to the function of—some immune cells. 20 –24 Immune cells currently known to express ASIC1a include astrocytes, microglia, macrophages, dendritic and T-cells, as well as oligodendrocytes. 24 ASIC1a is expressed throughout the CNS and contributes to secondary damage after stroke. 25 –30 ASIC1a is thought to contribute to cell death after ischemic injury via mechanisms including modulation of glutamate receptors and activation of necroptosis. 31 –35
ASIC1a expression has been shown to increase at the periphery of spinal cord lesions, and thus has been investigated for its role in SCI. 36,37 Studies utilizing the selective venom-derived ASIC1a inhibitor PcTx1 and the pan-ASIC blocker amiloride, as well as modulation of ASIC1a gene expression, have revealed neuroprotective effects including improved behavioral outcomes after SCI. 37,38 In contrast, it was later found that ASIC1a is necessary for reducing kainite-induced excitotoxicity. 39 To rationalize these seemingly conflicting reports, we decided to examine the effects of genetic ablation and potent pharmacological inhibition of ASIC1a in a mouse model of SCI. Our data overall suggest that ASIC1a is not a viable therapeutic target for SCI, and that the presence of the gene might aid recovery in mice.
Methods
Animals
All experimental procedures were approved by the University of Queensland (UQ) Animal Ethics Committee (Approval SBMS/SCBM/449/18) and conducted in accordance with relevant policies of the Australian National Health and Medical Research Council.
For pharmacological experiments, we used 8-week-old female C57BL6/J mice obtained from colonies at the UQ Biological Resources Facility (UQBRF). For genetic ablation experiments, we used nine 15-week-old female mice with a specific genetic knockout of ASIC1a, but not the ASIC1b splice isoform, as described previously. 40 ASIC1a knockout mice (ASIC1a–/– , n = 9) and wild-type (WT) littermates (ASIC1a+/+ , n = 5) were bred at the Florey Institute for Neuroscience and Mental Health, Melbourne, Australia. Procedures were performed a minimum of 1 week after arrival to our facility. To supplement the WT littermates, C57BL6/J mice (n = 5) were acquired from UQBRF as the ASIC1a–/– mice are on a C57BL6/J background. We show in Supplementary Figure S1A, S1B, and S1D (spinal cord volume) these controls exhibit the same patterns as the wild type littermates. Mice were maintained in individually ventilated cages under standard conditions on a 12 h light-dark cycle with ad libitum access to food and water.
Spinal cord surgery
Mice were exposed to a contusion spinal cord injury as described previously. 41 Tiletamine/zolazepam (50 mg/kg; Virbac) and xylazine (20 mg/kg; Troy Laboratories), were used to anesthetize mice prior to SCI via intraperitoneal (i.p.) injection. The T9 thoracic vertebra was identified based on anatomical markers, then a dorsal laminectomy was performed. A severe (∼70 dyne) contusion injury was inflicted upon the exposed spinal cord (Infinite Horizon impactor device; Precision Systems and Instrumentation). Then, using 6–0 polyglactin dissolvable sutures (Ethicon), the muscle above the injury was closed. After this, Michel wound clips (Kent Scientific) were used to close the skin. Investigators performing surgery were blinded to the treatment condition of the animals.
At 1 h post-surgery, mice were either given purified recombinant Hi1a (25 μg; provided by Dr Natalie Saez, UQ Institute for Molecular Bioscience) in a maximum of 200 μL phosphate buffered saline (PBS), or a PBS control, via the femoral vein (intravenous [i.v.]). The 25-μg dose of Hi1a was based on a previous study that used the related ASIC1a inhibitor PcTx1 in a rat model of SCI. 38 As animals recovered from surgery, they were administered a single subcutaneous (s.c.) injection of buprenorphine (0.5 mg/kg) in Hartmann's sodium lactate solution for analgesia. After surgeries, mice were provided with Necta Gel Water Sachets and mashed food pellets for nutrition and hydration. For 5 DPO, with surgery being 0 DPO, animals were also given s.c. injections of 1.0 mg/kg gentamicin. Bladder voiding was performed twice a day until the experimental end-points.
Assessment of functional recovery
For initial experiments comparing recovery of saline and Hi1a-treated mice, the actual applied force was 72.7 ± 0.6 kdyne (mean ± standard error of the mean [SEM]; n = 12) and 72.3 ± 1.0 kdyne (n = 12), with an average tissue displacement of 558.2 ± 17.8 μm and 559.8 ± 17.9 μm, respectively. In studies utilizing WT and ASIC1a−/− mice, the applied force was 71.0 ± 0.4 kdyne (n = 10) and 72.2 ± 0.6 kdyne (n = 9), with an average tissue displacement of 578.1 ± 26.1 μm and 650.3 ± 51.3 μm, respectively. There were no significant differences in injury parameters between experimental groups (p > 0.05). Hindlimb recovery was assessed using Basso Mouse Scale (BMS) scoring. 42 Using the 9-point scale, mice were assessed at 1, 3, 5, 7, 14, 21, 28, 35, and 42 DPO. For BMS scoring, two to three trained investigators who were blinded to the mouse genotype and treatment regimen, assessed open field locomotion for 3 min.
Tissue processing for histology
At either 1 DPO, 7 DPO, or 42 DPO, depending on the experiments described below, mice were anesthetized using an i.p. injection of sodium pentobarbitone (100 mg/kg, Virbac), then transcardially perfused with 20 mL of saline (0.9% NaCl) containing 10 IU/mL heparin (Pfizer) and 2% NaNO3, followed by phosphate-buffered Zamboni's fixative (2% picric acid, 2% formaldehyde, pH 7.2–7.4). The vertebral column was harvested and placed in Zamboni's fixative for 24 h at 4°C. Subsequently, the spinal cord was dissected out and, for cryoprotection, placed in 10% sucrose for 24 h and then 30% sucrose for 24 h. After all organs had sunk in 30% sucrose, tissues were cryopreserved in Optimal Cooling Temperature (OCT) compound (ProSciTech) through snap-freezing in dry ice-cooled isopentane, then stored at -80°C until cryosectioned (Leica CM3050-S). Coronal sections (20 μm thick) were collected on Superfrost Plus slides (1:5 series; Lomb Scientific) and stored at -80°C until staining.
General staining procedures
Antibodies
Neurons were stained using rabbit monoclonal NeuN antibody (1:1000 dilution; Abcam ab177487) and secondary horseradish peroxidase (HRP)-conjugated antibody (Goat Anti-Rabbit IgG [immunoglobulin G] H&L HRP, Abcam ab6721, diluted 1:1000 in BB). To quantify neutrophils, the primary antibody was rat anti-mouse Ly6b.2 (1:400 dilution; AbD Serotec) and goat anti-rat 594 (1:200 dilution, Invitrogen) was used as secondary antibody. For microglia/macrophages, the primary antibody was rabbit anti-mouse Iba1 (1:400 dilution; #019-19741; Fujifilm Cellular Dynamics, Inc.) and goat anti-rabbit IgG-AlexaFluor-488 (1:200; Invitrogen) was the secondary antibody. To detect myelin, FluoroMyelin™ Red Fluorescent Myelin Stain (1:150, ThermoFisher Scientific) was used. Endogenous IgG was visualized using a mouse secondary IgG antibody (anti-mouse 488, 1:150). In all fluorescent panels, nuclei were also stained during application of secondary antibodies (Hoechst 33342, 1:1000 dilution, Sigma-Aldrich). For fluorescent antibodies: all were diluted in a blocking buffer (BB) recipe utilized for fluorescent antibody stability (0.2% Triton X-100, Sigma-Aldrich; 2% bovine serum albumin, Sigma-Aldrich; in PBS). For the HRP-conjugated antibody, a recipe to reduce background staining (0.1% Triton X-100, Sigma-Aldrich; 10% FBS, Sigma-Aldrich; in PBS) was used.
All slides were defrosted at room temperature (RT) for 30 min and incubation steps were performed in the dark. For NeuN/3,3′-diaminobenzidine (DAB) staining, slides were first placed in antigen retrieval solution (Revealit Antigen) in PELCO BioWave® Pro 36500-230 (550 WT, no vacuum, 50°C max. temp, 5 min) and then rinsed twice with distilled H2O and once with PBS. All slides were washed with PBS on a shaker (3 × 10 min). Dry liquid block pen was used to outline the perimeter of the sections. BB was applied to the area and the slide was left in a humidified chamber for 1 h. The BB was then poured off prior to staining. Primary antibodies were applied and slides left in a humidified chamber overnight at 4°C. To remove unbound antibodies, slides were washed the next morning with PBS (3 × 10 min). Next, for all fluorescent antibody experiments, secondaries were applied and slides incubated for 1 h, RT. Slides were again washed with PBS (3 × 10 min). To perform DAB visualization upon HRP-conjugated antibody for NeuN+ cells, we used the manufacturer's instructions (DAB Substrate Kit, ab64238, Abcam) and followed with hematoxylin and eosin (H&E) staining. 43 All slides were mounted with coverslips in either Dako fluorescent mounting medium (Sigma-Aldrich; for IF) or DPX mountant (Merck; for IHC).
Analysis
Slides were visualized on a Zeiss ApoTome.2 microscope at 20 × magnification (Binning 1 × 1, Resolution: 2752 × 2208 pixels), using ZEN 2 (blue edition) software. For immunohistochemistry (IHC), brightfield was used with the apotome removed. For immunofluorescence (IF), the widefield monochrome setting was applied and the apotome was used. Tiles were stitched in ZEN 2. Images were taken 1.5-2.0 mm rostral and caudal of the lesion epicenter and analyzed using FIJI (Schindelin et al., 2012). At 7 DPO, Ly6B.2+ cells were manually counted. Manual counting of cells was performed using the Cell Count macro written by Dr. Kurt De Vos. Counts were performed as whole cell counts over the entire slice, for every slice that was imaged and analyzed. At 1 DPO for Ly6B.2+, and both 1 and 7 DPO for nuclei, cells were counted using a specifically developed macro, described in the Supplementary Methods.
To measure area, slices within the pia mater were outlined in FIJI using the manual hand selection tool; dorsal root ganglion (DRG) regions were not included. Limit to threshold function was utilized to measure stained area versus total area as follows. To assess astrogliosis with glial fibrillary acidic protein (GFAP)+, total stained area was measured multiplied by section thickness and the number of slices in series (1:5). This protocol was repeated when exploring white matter loss with FluoroMyelin staining for myelinated regions, but here only the dorsal, lateral and ventral funiculi were outlined. Additionally, a protocol utilizing threshold values was applied to calculate the ratio of the stained area to the total outlined areas for GFAP+, FluoroMyelin and Iba1+ positive regions where values are given as percentages (%). 44 These arbitrary values were used to compare cohorts. In their respective analyses, lesion epicenter was defined as the section with the most neutrophils, least FluoroMyelin Red or GFAP+ comparative to section area.
Statistical analysis
GraphPad Prism was used for statistical analyses and data visualization. At 42 DPO, BMS data were analyzed by ordinary two-way repeated-measures analysis of variance (ANOVA) and Bonferroni's post hoc tests. For BMS data from the Hi1a experiments, due to differing cohort sizes after tissue collection at two time-points (1 DPO: n = 12; 7 DPO: n = 6), the mixed-effects model (REML) with Šídák's multiple comparisons tests was used. When exploring histological data across the 1600 μm (lesion size, percentage coverage of cells, immune cell and neuronal counts) the mixed-effects model was used with post hoc Šídák's multiple comparisons tests. When exploring neuron quantification, at the epicenter, across time-points as well as treatment groups, we used ordinary one-way ANOVA with Tukey's multiple comparisons. To compare volume measurements at the lesion epicenter, two-tailed t-tests were used. All tests are detailed in figure legends. The data exploring ASIC1a ablation are presented as mean ± SEM. At acute time-points in the pharmacological experiments utilizing Hi1a, missing values from lost sections impacted row-column combination comparisons. Therefore, the graphs plotted from rostral to caudal (at 1 and 7 DPO), utilized section averages over 400-μm slices from the lesion epicenter. Statistical significance was set at p < 0.05.
Results
Genetic ablation of ASIC1a is detrimental to recovery after SCI
We first explored how genetic ablation of ASIC1a impacted SCI outcomes, using BMS scoring to assess functional recovery and post-mortem immunostaining for histopathology. Comparable levels of near-complete hindlimb paralysis were observed between WT and ASIC1a–/– animals acutely post-SCI (1 DPO), followed by a gradual return of some locomotor function in both genotypes over the following weeks. Overall, BMS scores for ASIC1a–/– mice were not different to their WT counterparts (two-way ANOVA, p = 0.0539). The interaction between DPO and mouse cohort was significantly different (two-way ANOVA, p = 0.0039), due to the significant impact of post-injury time on BMS score (two-way ANOVA, p < 0.0001), which is to be expected from the well-established spontaneous recovery of mice over time after SCI. BMS scores were significantly different at 35 DPO (Bonferroni's multiple comparisons, p = 0.0498; Fig. 1A). The worsened functional recovery from SCI for ASIC1a–/– mice was corroborated by a significant increase in the cross-sectional lesion volume at the epicenter (mixed-effects model, p = 0.0163; Fig. 1C) and significant differences were found using Šídák's multiple comparisons tests at the lesion epicenter (p = 0.0157) and both 300 μm (p = 0.0124) and 400 μm (0.0063) rostral from the epicenter. Mixed effect analyses found that overall proportional area of GFAP+ was significantly less in the ASIC1a–/– group [GFAP area (%); p = 0.0105]. Using Šídák's multiple comparisons, significance was seen at the lesion epicenter (p < 0.0001) and notably, 200 μm (p < 0.0001) and 100 μm rostral (p = 0.0050) as well as 100- μm (p = 0.0029), 300-μm (p < 0.0001), and 400-μm (p = 0.0312) caudal from the epicenter (Fig. 1B). Comparison of lesion size and BMS scores in WT and ASIC1a–/– mice at the study end-point not only segregated these genotypes (Fig. 1A-1C), we also show this when comparing the two control groups to the ASIC1a–/– cohort (Supplementary Fig. S1A, S1B), despite significant differences in weight throughout the experimental timeline (Supplementary Fig. S1D). Due to this issue, we compared spinal cord area across cohorts, which revealed no significant differences (Supplementary Fig. S1E).
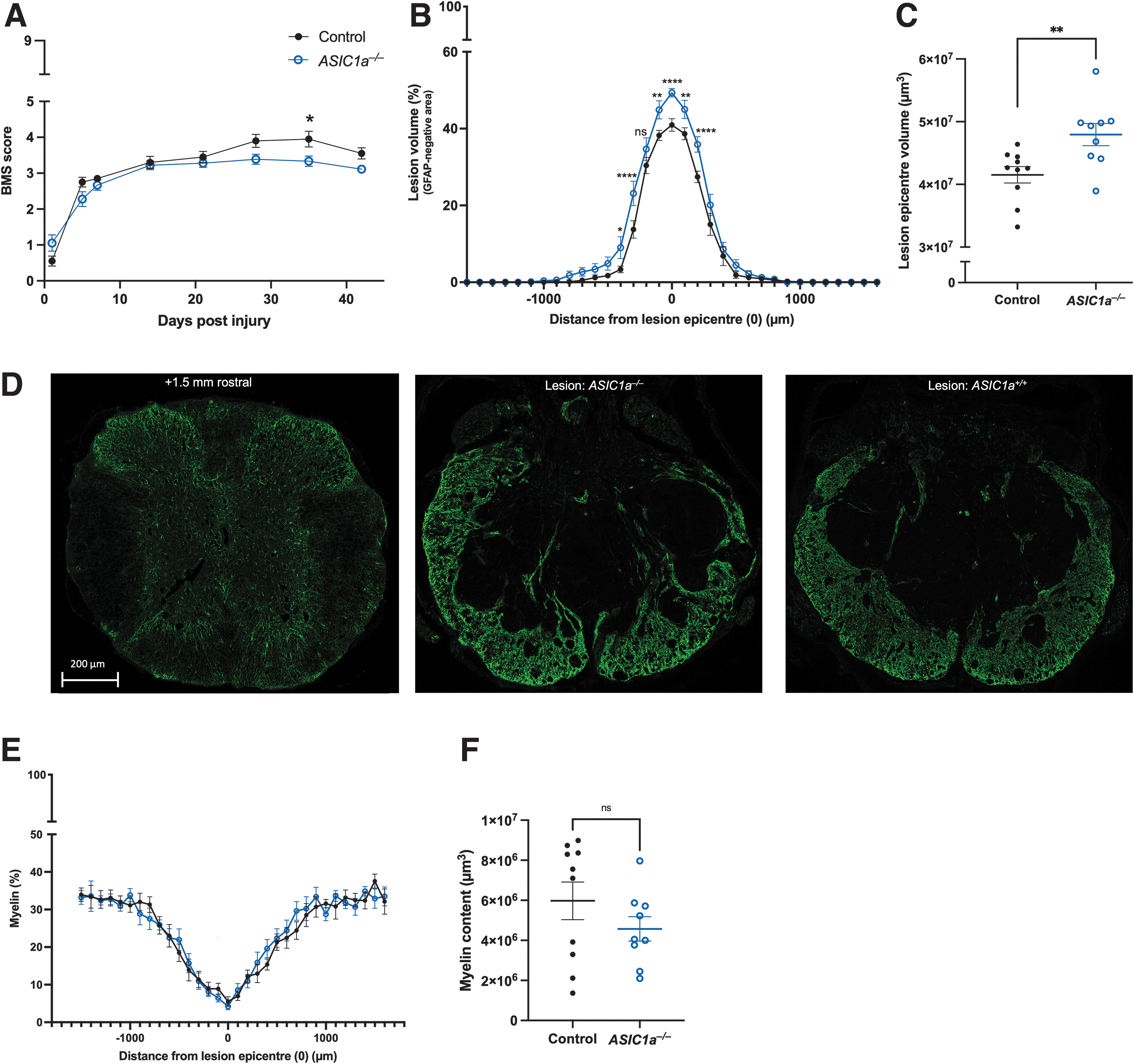
Comparison of ASIC1a–/–
mice (n = 9, unfilled blue circle) and ASIC1a+/+
mice (“control”; n = 10, black circle) following a severe T9 contusion spinal cord injury at 42 days post-operation (DPO).
Lastly, ASIC1a expression in oligodendrocytes is thought to render them more susceptible to acid-induced injury, although previous studies have reported varied effects of ASIC1a inhibition on myelin sparing and/or content. 37,38,45 Using mixed effects analyses, we found that genetic ablation of ASIC1a had no overt impact on myelin sparing after SCI when comparing myelin content (p = 0.7800), with no significant difference between cohorts revealed at any distance from the lesion epicenter with Šídák's multiple comparisons (Fig. 1E, 1F).
Pharmacological inhibition of ASIC1a accelerates neuronal death and does not improve functional outcomes
To understand mechanistically how loss of ASIC1a function may negatively influence outcomes, we next performed a series of pharmacological experiments that focused specifically on acute neuronal loss and intraspinal inflammation. Here, we used SCI mice receiving treatment with peptide Hi1a, the most potent known inhibitor of ASIC1a (IC50 ∼500 pM). 46 A dose of 25 μg Hi1a per mouse (i.v.) was used here to account for clearance and loss of peptide in the periphery. The rationale for this dosage was based on several factors. In previous experiments that employed venom peptide inhibitors of ASIC1a in rodent models of stroke, an intracerebroventricular dose of 2 ng/kg was used. 46 In a contusion SCI study, 48 μg of PcTx1 was used per rat and delivered using a combination of i.p. injection and s.c. pump. 38 We also showed a lack of effect, but also no peripheral toxicity in a pilot study at this low dosage (2 ng/kg; Supplementary Fig. S1F). Given a mouse blood volume of 2 mL, body weight of 20–25 g, and Hi1a molecular mass of 8723 Da, the i.v. dose we used (25 μg) should yield a peak serum concentration of ∼1.1 μM, which is 2000-fold higher than the IC50 for Hi1a inhibition of ASIC1a. 46 Assuming good permeability of Hi1a across the BSCB after SCI, as based on previous data, 38 this dose of Hi1a should inhibit the majority of ASIC1a channels at the injury site in the spinal cord.
We first explored whether Hi1a treatment would impact locomotor function/recovery at the acute stage (1–7 DPO) after SCI (Fig. 1A), as significant detriment to gait has been previously noted at 7 DPO upon depletion of microglia. 47 Behavioral assessment was analyzed using two-way ANOVA, and consistent with the stabilized neuron count at 7 DPO, we found no overall significant difference in relation to locomotor function in Hi1a-treated animals compared with vehicle-treated controls (p = 0.3044). When comparing separate time-points, no significance was seen with Bonferroni's multiple comparisons test at 1 DPO (p = 0.8107), 5 DPO (p > 0.9999), or 7 DPO (p = 0.8107). Therefore, Hi1a-treatment did not incur any locomotive benefit or detriment at the acute stage of SCI.
We also investigated the impacts of Hi1a at a histopathological level, first using quantitative analysis of neurons by NeuN+ staining (Fig. 2B). At the lesion epicenter across groups there were several significant differences. Overall, the cohorts were significantly different with ordinary one-way ANOVA (p < 0.0001). Using Tukey's multiple comparisons tests, we see significant differences between the number of neurons at 1 DPO in Hi1a-treated versus vehicle-treated animals (p = 0.0012). This suggests that at 1 DPO, there are fewer neurons after treatment with Hi1a. Interestingly, no further major loss during the subacute phase was seen in the Hi1a-treated cohort (p = 0.5035), whereas we discovered a significant decrease in the number of neurons between 1 DPO and 7 DPO in the vehicle-treated cohort (p < 0.0001). We also found significant loss between the 1 DPO vehicle-treated cohort, and in the 7 DPO Hi1a-treated cohort (p < 0.0001). These data suggest that, with Hi1a-treatment, there is increased cell loss at the epicenter at 1 DPO. They do not suggest any differences to neuron number at 7 DPO between groups.
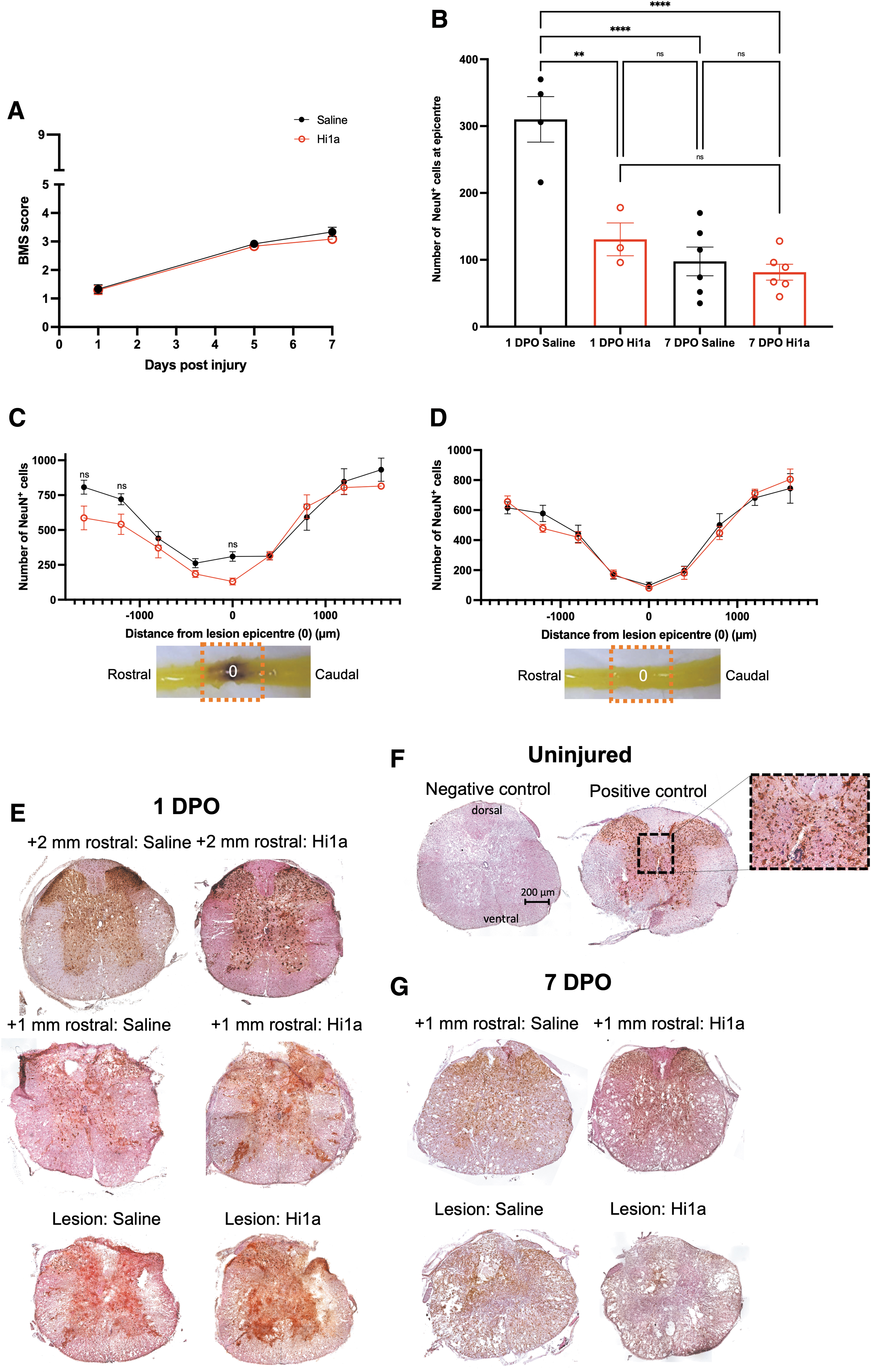
Behavioral and tissue comparisons between saline (“saline”; black circles) and Hi1a-treated (25 μg i.v.; “Hi1a”, unfilled red circles) mice following a severe T9 contusion spinal cord injury at time-points up to 7 days post-operation (DPO).
Given these changes to neuronal counts across both cohorts and time-points, we explored changes rostral and caudal to the lesion epicenter (Fig. 1C–D). Between cohorts, at 1 DPO, there were no changes to overall cell counts using mixed effects analysis (P = 0.1794), and despite the significance in Figure 1B, Šídák's multiple comparisons tests revealed no significant differences across regions. There also was no difference in the number of neurons overall between groups at 7 DPO (mixed effects analysis; p = 0.7805) or post hoc Šídák's multiple comparisons tests. Using Hoechst staining, we also compared changes to nucleated cell numbers between cohorts (Supplementary Fig. S1E, S1F). At 1 DPO, we found a decrease in nucleated cell numbers from spinal cords of Hi1a-treated animals with an overall significance with mixed effects analysis (p = 0.0215); however, multiple analyses revealed fewer nuclei only 2000 μm rostral from the lesion epicenter with Šídák's multiple comparisons test (Supplementary Fig. S1G; p = 0.0181). Mixed effects analyses also found no difference to nuclei counts at 7 DPO (Supplementary Fig. S1H; p = 0.039), nor at any depth in the spinal cord with Šídák's multiple comparisons tests between groups. Therefore, postmortem quantitative analysis of NeuN+ staining at and around the lesion epicenter revealed that acute ASIC1a inhibition appears to accelerate the loss of neurons at the spinal cord lesion instead of simply increasing cell death overall (Fig. 2B-2D).
Pharmacological inhibition of ASIC1a does not alter immune cell number or blood–spinal cord barrier
Since early-stage inflammatory processes are a key prognostic indicator of long-term SCI recovery in anatomically incomplete lesions, we sought to quantify any changes to the immune response after Hi1a-treatment. We specifically focused on myeloid cells, as they dominate the acute response. Neutrophils—the first major population of peripheral immune cells infiltrating the lesioned spinal cord—were quantified using immunofluorescent staining with Ly6B2+ antibody at 1 and 7 DPO (Fig. 3A, 3B). As seen in Fig. 3C, 3D, and consistent with previous studies, the number of neutrophils is greatest at the lesion epicenter at both 1 DPO and 7 DPO, but significantly higher at the early time-point. 6,48 Importantly, using mixed effects analysis (1 DPO: p = 0.7377; 7 DPO: p = 0.4857) with Šídák's multiple comparisons post hoc testing revealed no significant difference in cell numbers along the rostro-caudal extent of the lesion between conditions (Fig. 3C, 3D). Therefore, ASIC1a inhibition does not alter neutrophil migration to, or presence at the lesion site during both the acute (1 DPO) and subacute (7 DPO) stages of SCI.
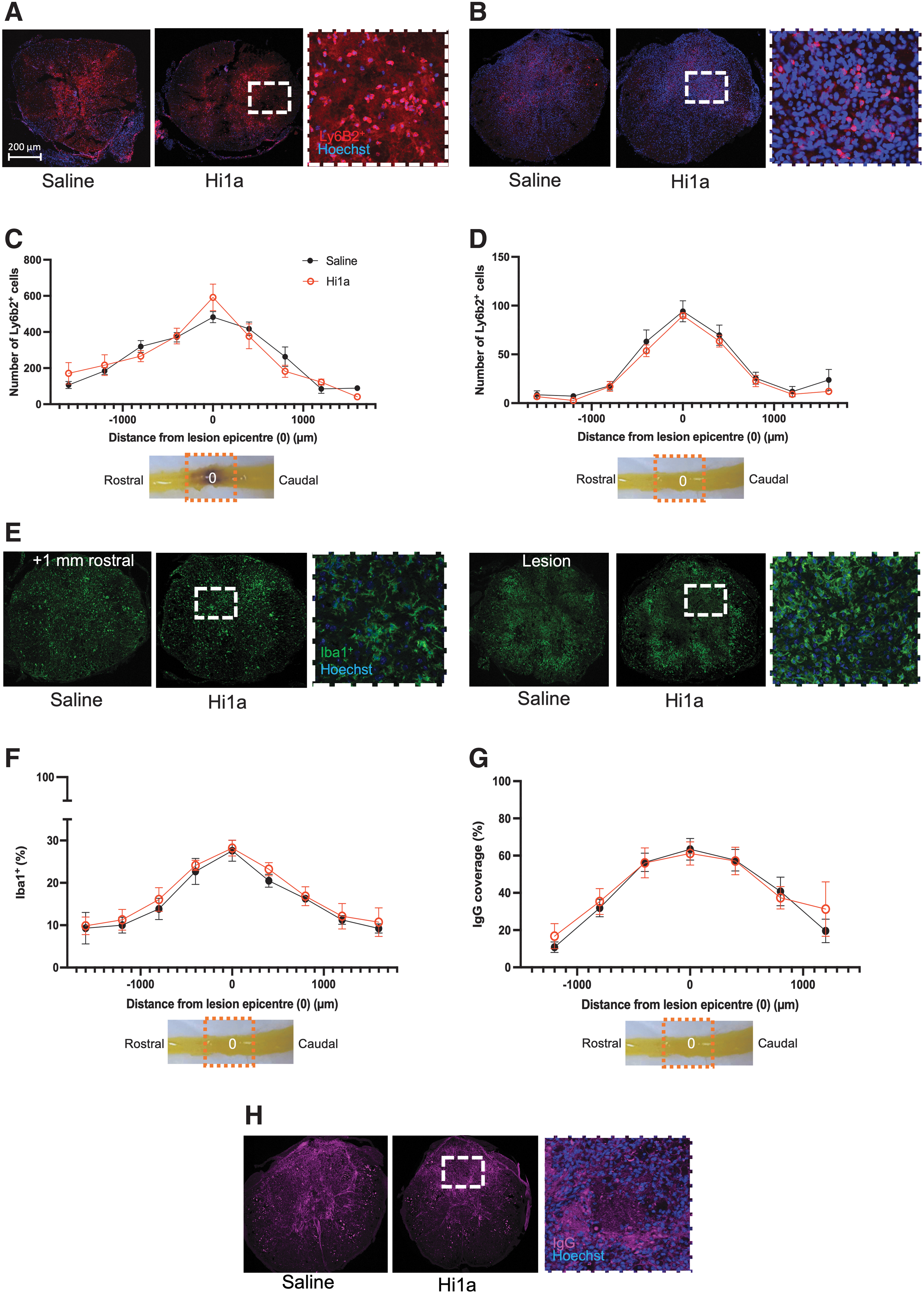
Immune response in the spinal cord between saline (“saline”, black circles) and Hi1a-treated (25 μg i.v.; “Hi1a,” unfilled red circles) mice following a severe T9 contusion spinal cord injury at time-points up to 7 days post-operation (DPO). Stains: blue (Hoechst, nuclei), green (Iba1+, microglia and macrophages), red (Ly6B2+, neutrophils and subset of activated macrophages) and endogenous immunoglobulin G (IgG; magenta).
Iba1+ staining was used to explore microglia/macrophage density at 7 DPO, which was again highest at the lesion epicenter and with an activated appearance (Fig. 3E; compare left vs. right insets). Coverage of Iba1+ cells did not differ between cohorts based on mixed effects analysis, (3 = 0.3882) or post hoc Šídák's multiple comparisons. We thus conclude that, similar to neutrophils, the density of macrophages/microglia at the site of SCI is not altered by pharmacological inhibition of ASIC1a. Finally, the degree of IgG staining was used as a proxy for leakage of the blood-spinal cord barrier (BSCB) after SCI at 7 DPO (Fig. 3G, 3H). Again, no significant effect of Hi1a treatment was observed between cohorts (mixed effects analysis, p = 0.7917) nor at any distance from the lesion using Šídák's multiple comparison tests.
Discussion
ASIC1a inhibitors have proven beneficial in protecting brain neurons after stroke and other ischemic insults. 29,30,46 ASIC1a is expressed in most laminae of the spinal cord 36 and previous research by Hu and colleagues 37 and Koehn and colleagues 38 suggested that inhibition of ASIC1a may aid locomotor and tissue recovery after spinal cord injury. In striking contrast, Mazzone and colleagues 39 reported that channel ablation results in increased excitotoxic cell death. Our data add an additional perspective to the role of ASIC1a in spinal cord injury and recovery. First, we showed that genetic ablation of ASIC1a appears detrimental to SCI recovery. Second, we found that acute pharmacological blockade of ASIC1a via peripheral administration of a bolus dose of a potent ASIC1a-inhibiting peptide that provides neuronal sparing in the brain after ischemic stroke does not have the same ability to spare spinal cord neurons after SCI in mice.
These combined efforts highlight an important caveat to consider when developing neuroprotective treatments—drugs that protect brain neurons may not have similar therapeutic efficacy in the spinal cord, and outcomes may depend on a number of factors that we discuss below.
Complete loss of ASIC1a is detrimental to SCI recovery
Ablation of the ASIC1a gene has been shown to have minimal adverse phenotypic consequences. 46,49 Nevertheless, the conservation of a gene ubiquitously expressed throughout the CNS in vertebrates suggests that ASIC1a has an important functional role. 50 Indeed, it has been reported to be important for seizure termination 51 and excitatory synaptic function, with loss of the channel resulting in decreased function of N-methyl-D-aspartate receptors (NMDARs), which are believed to underpin motor task-associated memory. 52 Earlier research revealed that ASIC1a contributes to NMDAR function, for example, through increasing the amplitude of NMDAR-driven excitatory postsynaptic currents. 25,53 A previous in vivo study by Hu and colleagues 37 revealed that a reduction, rather than complete ablation, of ASIC1a expression improved outcomes after SCI (in rats). Conversely, another group suggested that increased ASIC1a expression and activation after traumatic SCI might be beneficial, since the presence of ASIC1a reduced neuronal death after kainite-induced excitation in an in vitro model of SCI. 37,39 Our study showed conclusively, for the first time, that total ablation of ASIC1a gene expression in vivo worsens outcomes after SCI, causing a slight decrease in locomotor behavior and increased lesion size.
Acute channel inhibition results in no measurable benefits in SCI
One caveat of genetic ablation of a single subtype of a family of ion channels (e.g., ASIC1a) is that there can be compensatory upregulation of sister subtypes (e.g., ASIC1b, 2a, and 3). Thus, to obviate such a possibility, we examined the effect of acute pharmacological inhibition of ASIC1a on recovery after SCI.
Amiloride been used in many studies for pharmacological inhibition of ASIC1a, but this is problematic as this pan-ASIC blocker is not selective for the ASIC1a subtype. 24 Some studies have employed the spider-venom peptide PcTx1, which is highly selective and more potent than amiloride (IC50 ∼1 nM for rodent ASIC1a). PcTx1 acts like a “super agonist” that promotes and stabilizes a desensitized state of ASIC1a, thereby inhibiting the channel. Hi1a is even more potent than PcTx1 (IC50 ∼400 pM for rodent ASIC1a), and in contrast to PcTx1, it inhibits activation of the channel rather than promoting a desensitized state, and it has a significantly slower off-rate. 46 Two previous in vivo studies have suggested that ASIC1a blockade is beneficial post-SCI. 37,38 Koehn and colleagues 38 found that peripheral administration of PcTx1 improved behavioral recovery after SCI, but tissue sparing was only observed in the dorsolateral white matter of the spinal cord, and there was no impact on total myelin. Hu and colleagues 37 reported that twice daily intrathecal administration of crude Psalmopoeus cambridgei venom (which contains ∼0.4% PcTx1) yielded behavioral improvements and significant reduction in lesion size after contusion SCI. 37,54 An important caveat of this latter study is that Psalmopoeus cambridgei venom is a complex cocktail of hundreds of biological compounds, most of which are expected to target neuronal receptors and ion channels. Therefore, the venom is likely to have extensive and disparate effects in the CNS.
Surprisingly, we found that Hi1a causes a transient increase in neuronal loss 24 h after SCI, which stabilized at 7 DPO. This suggests that inhibition of ASIC1a causes neurons to be cleared or die faster after SCI, which became more apparent when we compared the results of different time-points across cohorts: vehicle-treated animals exhibited a significant loss of neurons between 1-7 DPO, whereas the Hi1a-treated animals did not. Notable differences in BMS scoring have been reported previously within the acute-to-subacute timeframe (1-7 DPO), where microglial depletion resulted in a significant decrease in BMS scores at 7 DPO. 47 Here, despite the transient decrease in neuronal number at 1 DPO, we found no difference in BMS scoring for mice treated with Hi1a.
Due to the potential of masking effects on behavior from spinal shock post-SCI, further exploration of histological data can be especially informative at the acute stage of recovery. 55 Therefore, we analyzed infiltration of IgG into the spinal cord, which can contribute to paralysis in SCI as a result of BSCB barrier disruption. We found no indication from IgG staining that Hi1a treatment affects the extent of BSCB disruption. 56,57 Another useful metric to define BSCB disruption—as well as immune cell communication—are changes to neutrophil migration from the periphery to the CNS. Here, we found no difference between cohorts. These data therefore support the conclusion that, at acute time-points post-SCI, ASIC1a inhibition has no discernible effect on the BSCB. Finally, ASIC1a is notably expressed in macrophages and microglia where inhibition results in decreased expression of inflammatory cytokines and reduced phagocytosis. 27,58 Prevention of microglial and macrophage activation is known to lower the reactivity of astrocytes and reduce lesion volume in SCI, a response that peaks at 7 DPO in the rat. 11,12 Our data did not reveal any changes to the density of microglial/macrophage cells with Hi1a treatment.
It is instructive to consider possible reasons for the different conclusions reached in the current study and the study by Koehn and colleagues 38 with respect to the benefits of acute pharmacological inhibition of ASIC1a on SCI. While we observed no benefit with Hi1a, Koehn and colleagues reported moderate sparing of the dorsolateral white matter without a reduction in overall lesion size with PcTx1. 38 One important difference between the two studies is that we used a mouse model of SCI whereas Koehn's group used rats; although rats and mice have many similarities, there are differences in recovery and, simply due to spinal cord size, the pathology differs. A second difference is that we delivered Hi1a in the current study via bolus i.v. injection whereas Koehn and colleagues delivered PcTx by i.p. bolus followed by continuous infusion to maintain serum levels of the peptide.
However, we do not think that the differences in methods of drug administration would dramatically impact exposure of the spinal cord population of ASIC1a to these pharmacological inhibitors. Hi1a has exceptional stability in human plasma, with <5% degradation over 5 days. 40 Thus, infusion to maintain plasma drug levels, which was considered important for PcTx1, was not considered necessary for Hi1a in the current study. Moreover, as we noted above, the i.v. dose of Hi1a that we used should yield a peak serum concentration that is 2000-fold higher than the IC50 for Hi1a inhibition of ASIC1a. Nevertheless, it is possible that differences in drug metabolism and/or excretion may account for some of the differences observed in the two studies.
Conclusion
Combining previous research with our own findings highlights the complexity of defining the role of ASIC1a in SCI; detrimental impacts of ASIC1a could even be expression dependent, given the improved outcomes observed by Hu and colleagues when ASIC1a expression was reduced rather than completely ablated. 37 Importantly, however, our research revealed that complete loss of the ASIC1a gene after SCI can impede recovery. One last point to consider in future research is the unknown role of heterotrimeric ASIC1a/ASIC2a channels in SCI, which will not be present after ASIC1a ablation. 59
Footnotes
Acknowledgments
The authors wish to express their gratitude to Dr. James Springfield, Dr. Nicholas Condon, and Dr. Deborah Barkauskas at the Institute for Molecular Bioscience for support with the Fluoro2 microscope, and University of Queensland Biological Resources staff for mouse colony maintenance and animal husbandry. We thank Professor Steven Petrou at the Florey Institute for Neuroscience and Mental Health, Melbourne, Australia, for kindly providing ASIC1a knockout mice.
Authors' Contributions
VSF developed and wrote this manuscript and performed all experiments and analyses. TJ, ERG, HWL, WJ, and CR assisted with mouse work, image analysis and intellectual support. HWL and WJ performed SCI surgeries. NJS generated purified Hi1a for experiments and advice on dosage and administration. SM provided ASIC1a KO mice and information on mouse strains during experiments. GFK and MJR provided supervisory and intellectual support and contributed to the writing of this manuscript.
Funding Information
Work in the laboratory of MJR is supported by SpinalCure Australia, the Wings for Life Spinal Cord Research Foundation, and National Health and Medical Research Council of Australia (NHMRC; project grants 1060538 and 1163835). VSF, TJ, ERG, WJ, and HWL were additionally supported by Research Training Program Scholarships (Australian Government). We acknowledge funding from the Australian Research Council (Centre of Excellence Grant CE200100012 to GFK), Australian National Health and Medical Research Council (Principal Research Fellowship APP1136889 to GFK), and the University of Queensland (Research Higher Degree Scholarship to VSF).
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Methods
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.