
Abstract
Mild traumatic brain injury (mTBI) produces subtle cerebrovascular impairments that persist over time and promote increased ischemic stroke vulnerability. We recently established a role for vascular impairments in exacerbating stroke outcomes 1 week after TBI, but there is a lack of research regarding long-term impacts of mTBI-induced vascular dysfunction, as well as a significant need to understand how mTBI promotes stroke vulnerability in both males and females. Here, we present data using a mild closed head TBI model and an experimental stroke occurring either 7 or 28 days later in both male and female mice. We report that mTBI induces larger stroke volumes 7 days after injury, however, this increased vulnerability to stroke persists out to 28 days in female but not male mice. Importantly, mTBI-induced changes in blood–brain barrier permeability, intravascular coagulation, angiogenic factors, total vascular area, and glial expression were differentially altered across time and by sex. Taken together, these data suggest that mTBI can result in persistent cerebrovascular dysfunction and increased susceptibility to worsened ischemic outcomes, although these dysfunctions occur differently in male and female mice.
Introduction
Traumatic brain injury (TBI) can alter both the incidence of—and outcomes from—ischemic stroke. 1 –5 Our lab recently showed that even a mild TBI (mTBI) exacerbates ischemic stroke outcomes in male mice 6 and may do so via impairments in cerebrovascular function. Specifically, we showed that vascular reperfusion following experimental ischemic stroke was much poorer in mice that had previously experienced an mTBI.
Our previous research used a 1-week delay between mTBI and experimental stroke to demonstrate proof of the concept that TBI renders animals more vulnerable to ischemic injury. 6 Importantly, however, the epidemiological evidence suggests that enhanced risk of stroke persists long after brain injury. 1,3,5 In fact, some clinical studies have reported that an increased risk of stroke after brain injury persists 5 years post-TBI. 7 This time course is in line with post-injury vascular remodeling and rarefaction that takes significant time to develop after injury. 8 The timing of remodeling and recovery after TBI has been explored extensively in basic animal research, with even mild injuries leading to lasting immune responses and vascular breakdown for months and years after injury 9 -11 ; however, the window of vulnerability during which mTBI continues to exacerbate subsequent ischemia remains understudied.
Here, we propose that glial and cerebrovascular dysfunction mediate TBI-induced vulnerability to ischemia. Glial cells rapidly respond to cerebrovascular injury, including brain injuries and strokes. 12 –16 These types of injuries affect blood vessels and oxygen levels to promote microglial activation and production of factors that promote vascular repair. 17,18 After ischemia, microglia migrate to infarcted tissue and phagocytose dead cells and vessels. 19 Astrocytes form tight junctions with the cerebral blood vessels and help to control water and solute flow through the vessels via aquaporins, as well as contributing to the blood–brain barrier (BBB). 20,21 Additionally, after ischemia, astrocytes elongate their processes and help to form glial scars to protect against additional tissue damage and facilitate repair of the vessels and neurovascular units. 22,23 However, although acute glial responses to mTBI can promote tissue repair, prolonged activation leads to phagocytosis and reactive oxygen species production, and cytokine storms may result in exacerbation of a subsequent ischemic incident.
Post-stroke reperfusion deficits and the resultant impairments in infarct volume after an injury may also be driven by BBB dysfunction after ischemic stroke and TBI. 24 -26 Leakiness of the BBB after an injury contributes to cerebral edema and increased intravascular coagulation, 27 –31 both of which could be key contributing factors to the observed increases in infarct volume and serum protein accumulations after injury. Thus, the current study was designed to investigate the hypothesis that an experimental mTBI would lead to lasting cerebrovascular deficits that would in turn result in persistent susceptibility to worsened ischemic stroke outcomes. Given that cerebrovascular damage and changes in intravascular coagulation cascades are common TBI sequelae, 32 -34 we reasoned that TBI-induced alterations in cerebrovascular, glial, and BBB function could mediate this phenomenon.
Methods
Animals
Swiss Webster mice were purchased from Charles River (Wilmington, MA) and bred at West Virginia University (WVU). Swiss Webster mice were used because their skulls are much less likely to fracture with our injury device than other strains. Pups were weaned at 21 days of age and were housed in a 12:12 light cycle with ad libitum access to food (HarlanTeklad #8640) and filtered tap water. All procedures were approved by the WVU Institutional Animal Care and Use Committee and were conducted in accordance with National Institute of Health guidelines.
A total of 272 mice were utilized in this study, and a total of 19 deaths occurred across all experiments, including one non-stroke mouse and 18 middle cerebral artery occlusion (MCAO) mice. The mortality rate in our study is consistent with a 10-15% published mortality rate for MCAO in mice. 35,36 In all experiments, animals were randomly assigned to groups (injury, time after injury, and ischemia) and all observations were conducted by researchers blind to experimental conditions.
Surgical procedures
A single closed-head mTBI was conducted on post-pubertal male and female mice (6–8 weeks of age) as previously reported. 37 Beginning 7 or 28 days after mTBI, some cohorts underwent a single transient unilateral MCAO, while others were processed for histological markers of neuropathology (see Fig. 1 for experimental timeline). All mice experienced similar total amount of time under anesthesia, thus controlling for any potential neuroprotective effects of repeated isoflurane exposure.

Experimental timeline. Cohort 1: Middle cerebral artery occlusion (MCAO) was conducted 7 or 28 days after mild traumatic brain injury (mTBI) and tissue collected for 2,3,5-triphenyltetrazolium chloride (TTC) staining 5 days later. Cohort 2: intravenous dextran was injected 7 or 28 days following mTBI and tissue was immediately collected for blood–brain barrier (BBB) permeability analysis. Cohort 3: mice were transcardially perfused 7 or 28 days following mTBI for immunohistochemistry analyses. Cohort 4: MCAO was conducted 7 or 28 days following mTBI and mice were perfused 1 hour later for immunohistochemistry analyses.
Traumatic brain injury
Mice were randomly assigned to an experimental group and then placed into a stereotaxic frame under inhaled isoflurane anesthesia (3% induction, 1.5% maintenance). The injury was induced with an Impact One device (Leica Biosystems, Buffalo Grove, IL). A 3-mm diameter impactor tip was retracted and driven into the skull along the midline (−2 mm AP relative to bregma) to a depth of 1.2 mm at a rate of 3 mm/sec (dwell time: 30 msec). Local analgesia was provided with subcutaneous bupivacaine (1.5 mg/kg) at the incision site. The skin was sutured with 6/0 nylon suture and mice were returned to their cages. The control procedure was performed identically but the impactor was not driven into the skull (just placed on the surface and retracted). This mTBI procedure is highly reproducible and does not produce skull fracture or ear damage. 6,38,39
Middle cerebral artery occlusion
Briefly, following a 7-day or 28-day recovery period after TBI, mice in cohorts 1 and 4 (Fig. 1) were reanesthetized with inhaled isoflurane (3% induction, 1.5% maintenance) and a unilateral MCAO was achieved by insertion of a 0.23 mm occluder (Doccol) into the right middle cerebral artery via the internal carotid artery and extending 6 mm beyond the internal carotid-pterygopalatine artery bifurcation. The occluder was secured and the mouse was returned to its home cage for 1 h. Following the 1-h occlusion period, the mouse was re-anesthetized, and reperfusion initiated by removal of the occluder. Local analgesia was provided with subcutaneous bupivacaine (1.5 mg/kg) at the incision site. MCAO mice were included in the study if a measurable infarct was present.
Determination of infarct volume
Five days after MCAO, mice were euthanized by rapid decapitation; brains were sectioned into four 2-mm thick coronal sections through the forebrain and incubated in 1% TTC (2,3,5-triphenyltetrazolium chloride in saline) for 10 min at 37°C. TTC produces a pink formazan product in the presence of live mitochondria, thus ischemic lesions are visualized as unstained white tissue. Slices were post-fixed in 4% paraformaldehyde overnight, photographed, and infarct area was outlined on both sides of each slice using Fiji imaging software. 40 Infarct size was determined after correcting for edema and reported as percent of the contralateral hemisphere using the formula: (1-(total ipsilateral hemisphere – infarct)/total contralateral hemisphere) *100.
Immunohistochemistry
Following tissue imaging for TTC in cohort 1, the paraformaldehyde-fixed tissue slices were cryopreserved, frozen, and further sectioned coronally on a cryostat at 40 μm throughout the forebrain. Mice in cohorts 3 and 4 were transcardially perfused and the brains were post-fixed, cryoprotected, and frozen before being cut on a cryostat at 40 μm.
For immunofluorescent staining, slices were washed with phosphate-buffered saline with 0.01% Triton-X (PBS-T) and blocked in 1% normal horse serum. After blocking, slices were incubated overnight in primary antibodies for each stain: CD31 (1:200; RNDSystems, Minneapolis, MN) and glial fibrillary acidic protein (GFAP; 1:250; Agilent, Santa Clara, CA), CD31 and IBA1 (1:250; Abcam, Cambridge, UK), or vascular endothelial growth factor A (1:300; VEGF-A; Abcam), fibrin(ogen) (1:100; Agilent), tomato lectin (1:200; Vector Laboratories, Burlingame, CA), GFAP (1:200; Cell Signaling Technologies, Danvers, MA), and aquaporin-4 (AQP4; 1:200; Cell Signaling Technologies) at room temperature. The next day, sections were washed with PBS-T and incubated with fluorescent secondary antibodies and mounted with Fluoromount mounting medium (Invitrogen, Carlsbad, CA).
Imaging and analysis
All immunohistochemistry (between bregma -1.5 and -2.0) was imaged and stitched on an Olympus VS-120 microscope with a 10 × Plan S Apo/0.40 NA objective using a Monochrome XM10 camera (1376 × 1032 imaging array, 6.45 × 6.45-μm pixel size, and 14-bit digitization). Unless otherwise noted, all images and subsequent analyses were performed in the corticostriatal region (representative of the middle cerebral artery territory) using Nikon Elements software. Three images were taken from adjacent brain slices and data were averaged across all three images unless otherwise indicated. For immunoglobulin G (IgG)/CD31/GFAP histology, we assessed total accumulation of IgG within the CD31+ cell area.
AQP4 expression on GFAP-positive astrocytes was assessed by measuring the area and intensity of the pixels expressing AQP4 in a standardized region of interest (ROI; 2880 × 2470 pixels). Colocalization of fibrinogen and tomato lectin is reported as a percent of colocalization normalized to total expression of tomato lectin. IBA1 and CD31 stains were used to measure the total amount of microglia and vasculature in the corticostriatal region, as well as the area of vascular associated microglia (VAM) and vascular contacting microglia.
VEGF-A and tomato lectin immunohistochemistry was assessed in the corticostriatal region (averaged two images for analysis) and corpus callosum (averaged two images for analysis) by identifying and thresholding the vessel positive area and the VEGF positive area. Once thresholding was conducted, we measured the total levels of VEGF in standardized ROIs (3800 × 2575 pixels in corticostriatal, 2800 × 2200 pixels in corpus callosum) in each set of images, as well as the amount of VEGF expression both in colocalization with the vasculature and separately from the vasculature.
Blood–brain barrier breakdown assessment
Mice in cohort 2 underwent an mTBI or control procedure, then 7 or 28 days later were injected intraperitoneally with 200 μL of a fluorescent tetramethylrhodamine (TMR)-coupled dextran (Life Technologies Corporation, Eugene, OR) in 1X phosphate buffered saline (PBS). Twelve minutes post-injection, mice were perfused transcardially with 20 mL of 0.1 M PBS and one hemisphere was extracted and flash-frozen in Tissue-Tek OCT Compound for later slicing and immunohistochemical staining. Hemispheres were sliced on a cryostat at 10 μm and mounted on gelatin-coated slides, fixed in 4% paraformaldehyde for 10 min, rinsed with PBS, and the fluorescent dye was immunohistochemically co-labeled with CD31 as described above. Images were obtained as described above and permeability was assessed by analyzing colocalization of the TMR-dextran and CD31 as normalized to total dextran expression.
Statistical analysis
Sample sizes were formulated from power analyses based on the magnitude of differences detected in preliminary studies and from previous literature. Effect sizes were >0.45, with biochemical and histological changes being larger than behavioral effects. Thus, we powered the study to detect differences with a β > 0.8. Behavioral outcomes, infarct size, blood–brain barrier breakdown assays, and immunohistochemical results were assessed via a two-way analysis of variance (ANOVA; injury condition × ischemia condition, or injury condition × treatment). All significant overall results (p < 0.05) were followed up with a Tukey honestly significant difference post hoc analysis.
Results
Mild TBI increases susceptibility to MCAO in time- and sex-dependent manners
We measured infarct volume after mTBI and MCAO to determine whether there are sex differences or time-dependent responses in post-injury responses to ischemic stroke. These conditions resulted in eight total experimental groups, with four groups of mice for each sex (TBI-MCAO 7 days [males: n = 8, females: n = 8], TBI-MCAO 28 days [males: n = 14, females: n = 15], Sham-MCAO 7 days [males: n = 9, females: n = 8], and Sham-MCAO 28 days [males: n = 12, females: n = 12]). Infarct size was increased by TBI in both sexes at 7 days post-injury (F3,81 = 6.186, p = 0.011; Fig. 2). However, at 28 days post-injury, injured females have a persistent increase in infarct volume (p = 0.033), while the infarct volume of males did not differ between sham and injured animals at this time-point.
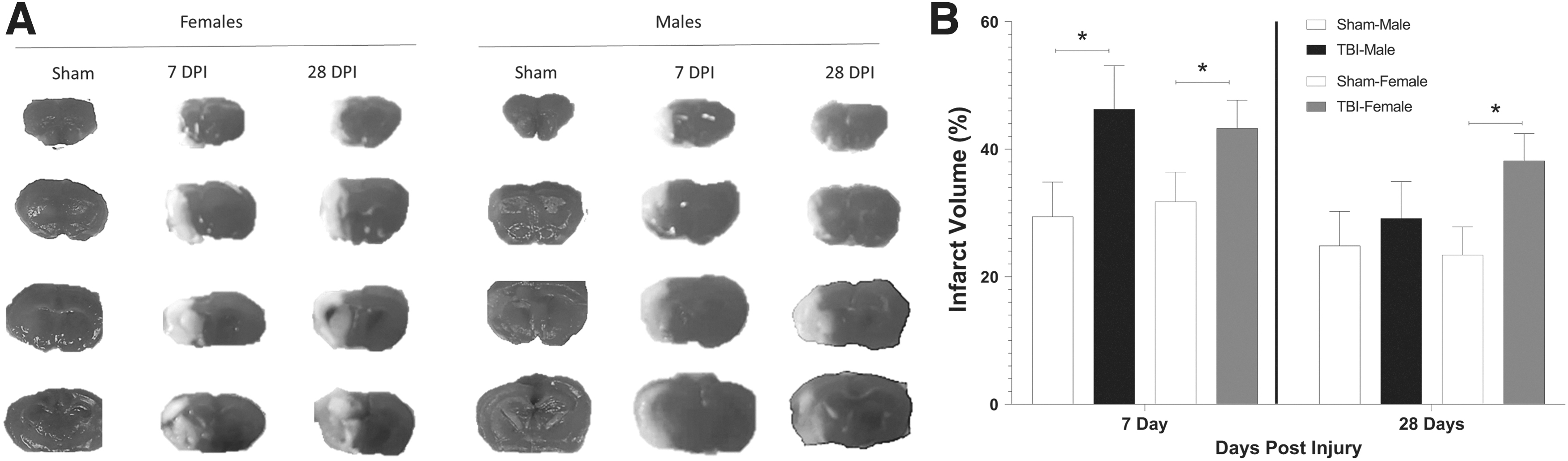
Mild traumatic brain injury (mTBI) exacerbates infarct volume in male and female mice.
Blood–brain barrier changes after mTBI are dynamic over time
TBI has been shown to increase susceptibility to cerebrovascular dysfunction, even in the mildest of injuries. 41,42 One well-characterized mechanism of TBI-induced cerebrovascular dysfunction is acute opening of the BBB after injury. Here, we aimed to determine whether male and female mice that experienced an mTBI differ in susceptibility to long-term cerebrovascular leakage. In cohort 2, we had eight experimental groups: TBI-7 days (males: n = 6, females: n = 6), TBI-28 days (males: n = 6, females: n = 6), Sham-7 days (males: n = 6, females: n = 6), and Sham-28 days (males: n = 6, females: n = 6). Immunohistochemistry (IHC) on these frozen brains revealed an effect of time after injury on blood–brain barrier permeability (F2,46 = 42.511, p < 0.001; Fig. 3) and a sex by time after injury interaction (F2,46 = 4.892, p = 0.012). Male mice experienced an mTBI-induced increase in extravascular tracer at 7 and 28 days post-injury (7 days: p = 0.005; 28 days: p < 0.001), while female mice had significantly more leakage at 7 days post-injury compared with sham injured mice, which trended towards sham levels at 28 days post-injury (7 days: p < 0.001; 28 days: p = 0.059).
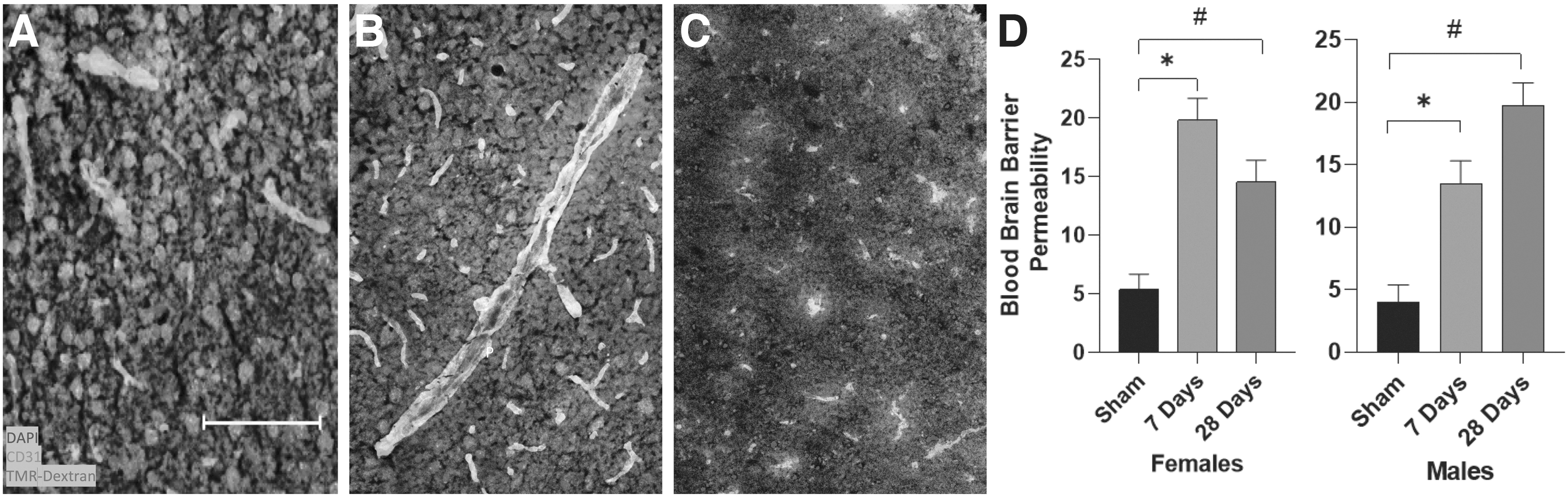
Mild traumatic brain injury (TBI) persistently increases blood–brain barrier (BBB) permeability to a 3kD dextran molecule coupled to tetramethylrhodamine.
Mild TBI promotes intravascular coagulation
Next, we performed immunohistochemistry on collected tissue to determine if there were changes in intravascular coagulation following mTBI. Cohort 3 had eight experimental groups: TBI-7 days (males: n = 7, females: n = 7), TBI-28 days (males: n = 6, females: n = 6), Sham-7 days (males: n = 6, females: n = 6), and Sham-28 days (males: n = 7, females: n = 5). The IHC revealed that there was a main time after injury effect of IgG accumulation within the vessels (F2,48 = 4.783, p = 0.013; Fig. 4). We found that among females, mTBI produced significantly less IgG buildup in the vasculature at 7 days post-injury compared with shams but returned to sham levels by 28 days post-injury (7 days: p = 0.043). Injured male mice exhibited a significant time-dependent increase in IgG accumulation at 7 and 28 days post-injury compared with sham-injured animals (7 days: p = 0.037; 28 days: p = 0.010)
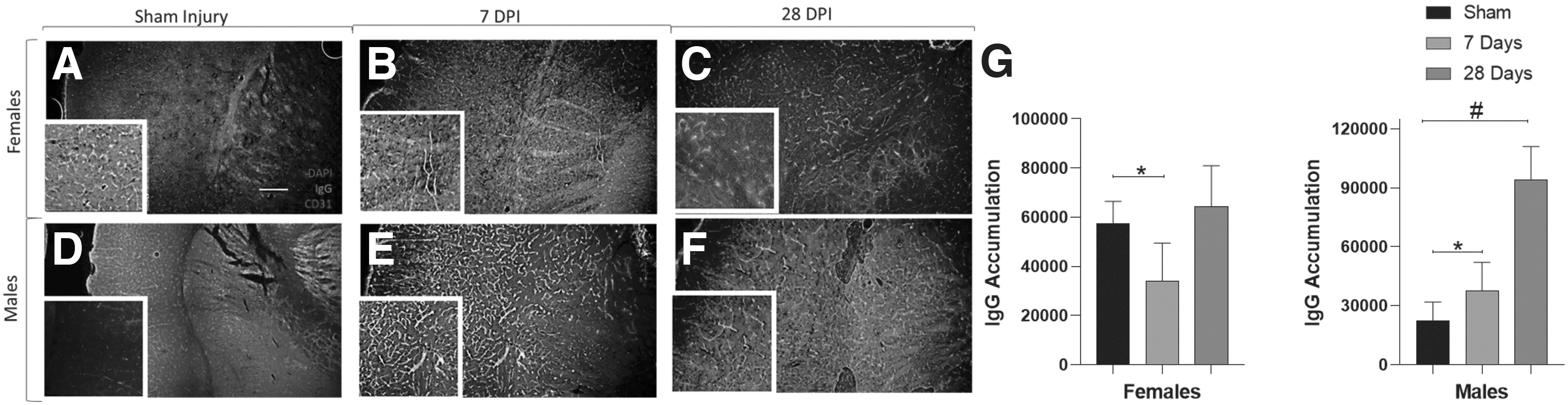
Time-dependent cerebrovascular immunoglobulin G (IgG) protein accumulation increases in male but not female mice following mild traumatic brain injury. Representative immunohistochemistry images in females
To determine whether mTBI altered stroke-induced intravascular coagulation, we performed IHC on collected tissue from cohort 4. In cohort 4, there were eight experimental groups: TBI-7 days (males: n = 6, females: n = 6), TBI-28 days (males: n = 6, females: n = 6), Sham-7 days (males: n = 6, females: n = 7), and Sham-28 days (males: n = 7, females: n = 6). The IHC measured fibrin(ogen) deposits within the cerebrovasculature in both the stroke and the contralateral hemispheres. The hemisphere ipsilateral to the stroke showed a main effect of time on fibrin(ogen) buildup in the cerebrovasculature (F2,48 = 5.297, p = 0.010; Fig. 5G), indicating a significant mTBI-induced increase in both males and females compared with sham injured animals at both the 7- and 28-day time-points (male 7 days: p = 0.039; male 28 days: p = 0.041; female 7 days: p = 0.029; female 28 days: p = 0.033). Within the contralateral hemisphere, there was an interaction of time and sex on intravascular fibrin(ogen) buildup (F2,48 = 4.907, p = 0.014; Fig. 5N). This interaction was largely driven by the injured female mice, which had a large spike in fibrin(ogen) accumulation at 7 days post-injury (DPI).

Fibrin(ogen) deposits increase significantly in mice that experienced a middle cerebral artery occlusion (MCAO) after mild traumatic brain injury. MCAO promotes time-dependent increased levels of fibrin(ogen) in the ipsilateral
Vascular morphology and glial responses dynamically change over time after injury
Glial cells regulate inflammatory cascades following mTBI to limit damage and begin to promote repair. We performed immunohistochemistry to determine the changes in astrocyte responses and aquaporin expression after ischemia. IHC analysis determined that there are main effects of both sex and time on AQP4 expression (Sex: F1,49 = 4.336, p = 0.043; Time after injury: F2,48 = 7.347, p = 0.02; Fig. 6). Female mice showed an increase in AQP4 expression surrounding the infarct in mice that had an MCAO 7 days after mTBI compared with those with no prior injury (sham; p = 0.039). Male mice showed a significant injury effect at both 7- and 28- days post-injury compared with sham injured animals (7 days: p = 0.037; 28 days: p = 0.004).

Time-dependent aquaporin4 (AQP4) expression changes in mice that experienced an MCAO after mild traumatic brain injury. MCAO expression of AQP4 changes in a time- and sex-dependent manner in mice that experienced a prior mTBI compared with those that did not have a prior injury (sham). Representative immunohistochemistry images of AQP4 staining in females (A-C) and males (D-F) following sham injury
Additionally, we assessed IBA1+ microglia and reported that total microglial area in both male and female mice changed over time in tissue collected from cohort 3 (F2,48 = 5.648, p = 0.007; Fig. 7D–E), with a significant increase by 28 days post-injury (p = 0.014). There was also a time-dependent effect on the number of Vascular Associated Microglia (VAMs) (F2,48 = 7.518, p = 0.002; Fig. 7F–G), with an increase in microglia that are closely associated with the vasculature by 28 days post-injury (p = 0.005). Given that these microglial changes are consistent between the sexes but vary across the time after injury, these data indicate a lasting vascular-associated inflammatory cascade following mTBI. Importantly, the increase in microglial staining is proportional to mTBI-induced increase in total vascular area. Indeed, we report that the total amount of tomato lectin+ cerebrovasculature increases significantly over time (F2,48 = 6.665, p = 0.003; Fig. 7H–I), with an increase in total area of tomato lectin+ vasculature by 28 days post-injury compared with sham injured animals (p = 0.003). These data are consistent with previous reports that mTBI leads to angiogenesis and long-term changes in vascular structure. 43
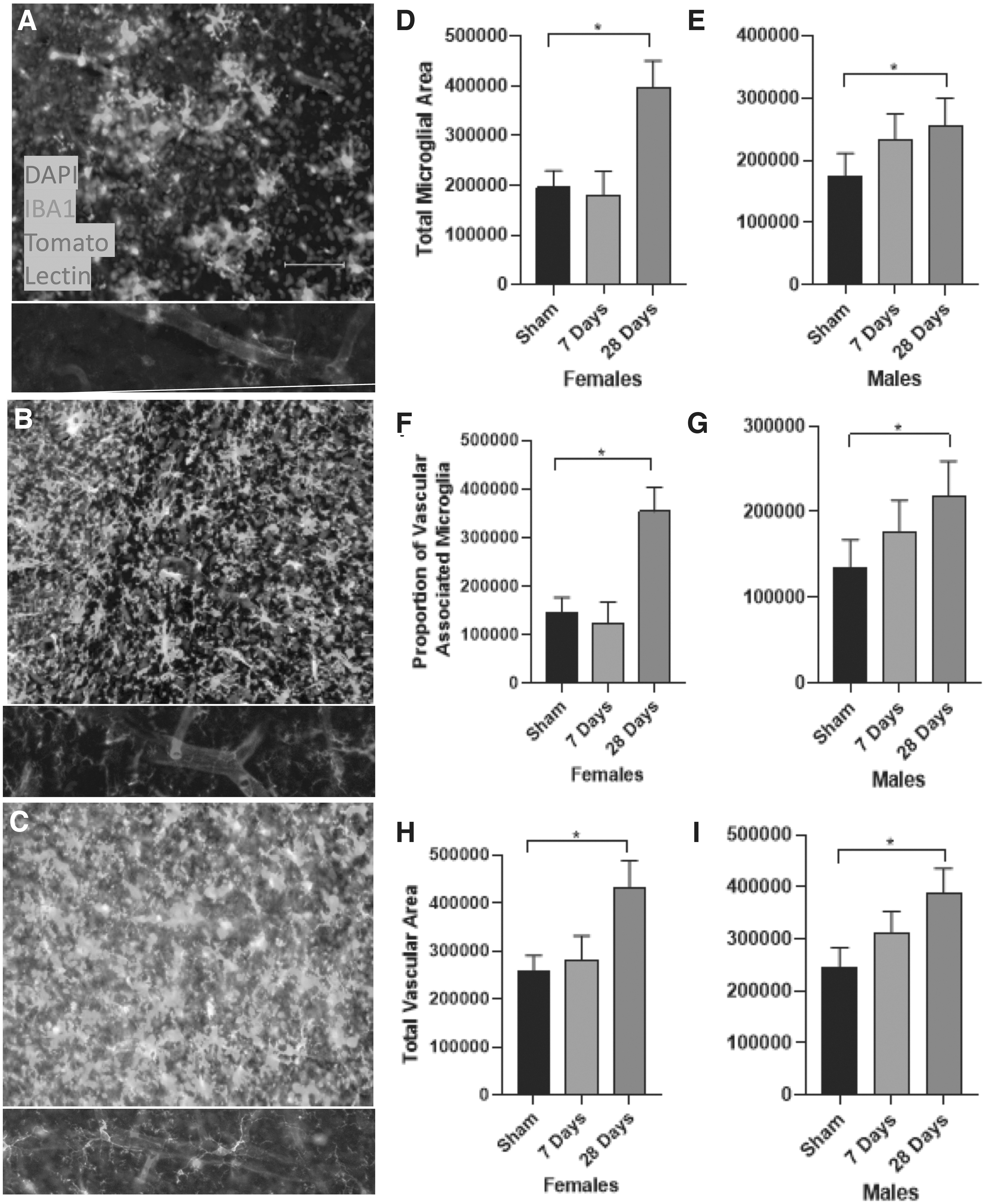
Middle cerebral artery occlusion (MCAO) increases overall and vascular-associated microglial expression in mice that had a prior mild traumatic brain injury. Representative immunohistochemistry images of IBA1 and tomato lectin staining in MCAO mice following sham-injury
This change in vascular area led us to investigate whether growth factor expression associated with angiogenesis is altered over time in male and female mice after mTBI. In cohort 3, we measured total VEGF levels in both gray matter (corticostriatal [CS]) and white matter (corpus callosum [CC]) regions of the brain, we subdivided the VEGF expression into both intravascular and extravascular components based on colocalization with tomato lectin. IHC results showed main effects of both sex and time on total VEGF in CS regions (Time after injury: F2,48 = 6.860, p = 0.003; Sex: F1,49 = 6.455, p = 0.016; Fig. 8M), and main effects of sex and time after injury in the CC regions (Time after injury: F2,48 = 7.391, p = 0.002; Sex: F1,49 = 6.018, p = 0.019; Fig. 8M). Total VEGF significantly increased in all groups at 7 days post-injury compared with sham levels (CS males: p = 0.016; CS females: p = 0.048; CC males: p < 0.001; CC females: p = 0.012); however, VEGF expression in the CC of males was significantly decreased by 28 days post-injury compared with 7 days post-injury (p = 0.006). There was a main effect of time on intravascular VEGF expression (F2,48 = 5.665, p = 0.007; Fig. 8N), which increased in the CS of male mice at 7 days post-injury compared with sham injuries (p = 0.004) and decreased at 28 days compared with 7 days (p = 0.031). Similarly, extravascular VEGF expression followed a similar pattern, with an effect that changed over time (F2,48 = 6.666, p = 0.003; Fig. 8O), indicating an increase at 7 days post-injury in male mice compared with sham injuries (p = 0.005) and a decrease at 28 days post-injury compared with 7 days post-injury (p = 0.012).
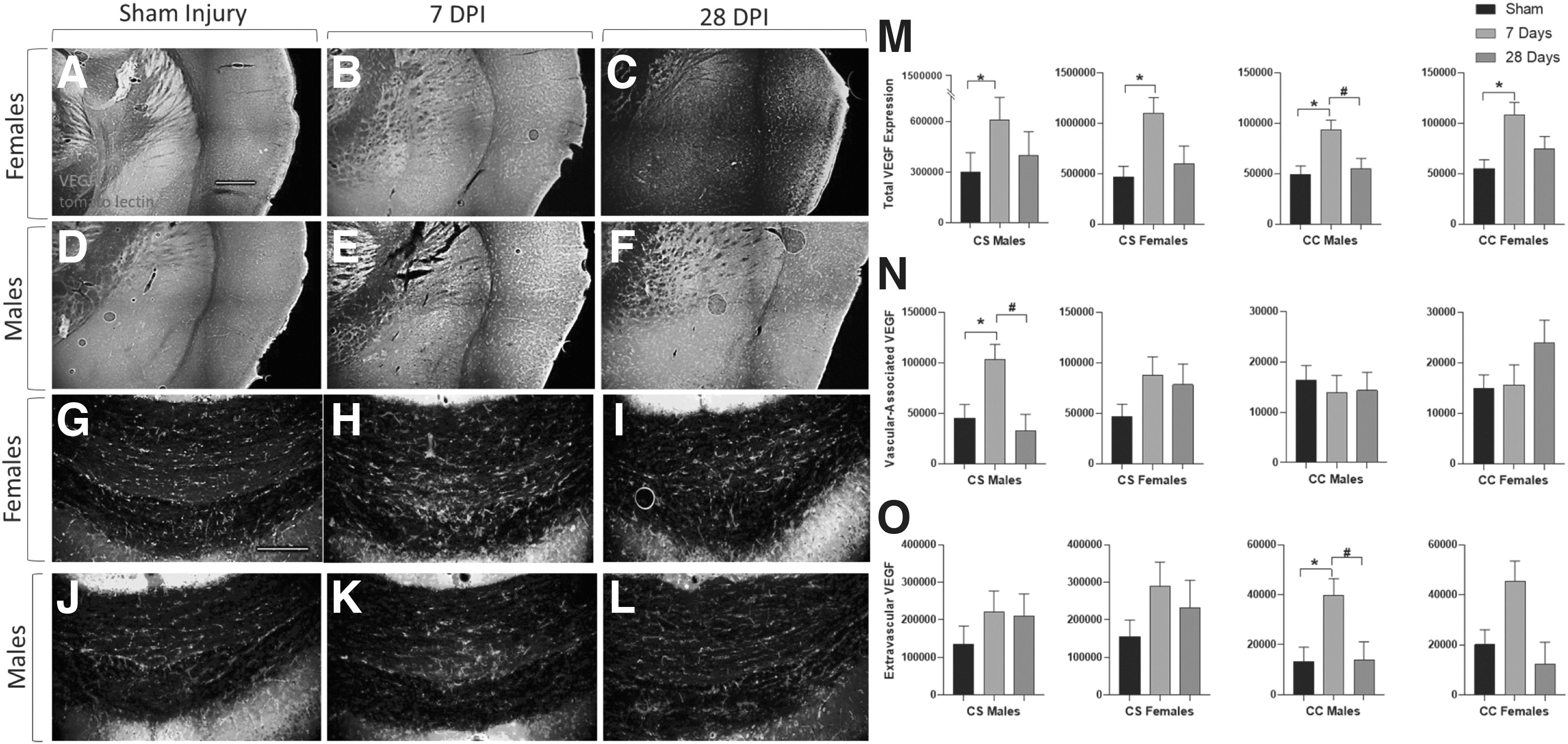
Mild traumatic brain injury (TBI)–induced vascular endothelial growth factor (VEGF) expression increases differentially in gray and white matter. Representative immunohistochemistry images of VEGF expression in gray matter (corticostriatal-CS) sections
Discussion
TBI is linked to an increased incidence rate of – and mortality after – ischemic stroke, 1 –5 and one of the potential mechanisms that we have identified is cerebrovascular dysfunction. The cerebral vasculature and the neurovascular unit more broadly are central to TBI pathophysiology. 34,44 A large percentage of individuals with a history of TBI exhibit postmortem evidence of damage to both large and small vessels. 45,46 Additionally, the vasculature plays critical roles in the BBB, which can promote ischemia when damaged. 47 Here, we hypothesized that experimental mTBI would induce lasting cerebrovascular deficits that would in turn result in persistent susceptibility to poorer ischemic stroke outcomes. We previously explored these disruptions at 1 week post-injury; however, to better understand the relationship among TBI, vascular dysfunction, and poor ischemic outcomes, here we also included a longer time after injury and also included both male and female mice. We selected 28 DPI specifically, as our prior publication 38 shows persistent secondary metabolic effects at least 3 weeks after mTBI. The present study demonstrates that the vulnerability to larger infarcts post-injury exists in both male and female mice at 7 DPI, while only the female mice maintain this increased susceptibility to 28 DPI. This lasting ischemic susceptibility is associated with chronic cerebrovascular dysfunction and BBB permeability.
Obviously, the central nervous system (CNS) vasculature is central to ischemic pathophysiology. From a severity perspective, the critical predictors of ischemic outcomes are the presence of sufficient collateral blood flow and whether the infarcted territory can be reperfused. Moreover, the primary strategy for the minimization of ischemic damage is to restore blood flow to the ischemic territory; clinically, this can be accomplished with pharmacological or mechanical clot removers. However, it is becoming increasingly clear that reperfusion of the infarcted tissue and not recanalization of the blocked vessel per se is the primary predictor of ischemic outcomes. 48 Immediate and or delayed failure to reperfuse the ischemic tissue is a common feature of stroke in both experimental animals and patients. The causes of incomplete reperfusion are not fully understood but involve vascular inflammation (with associated accumulation of serum proteins and immune cells), intravascular coagulation, and endothelial dysfunction. 49,50
TBI pathophysiology represents the interactions between direct immediate physical damage to cells and tissues and the secondary responses induced by the trauma, which can evolve over much longer time scales. These secondary responses, including BBB breakdown, edema, hemorrhage, and coagulopathies, can all contribute to the overall neurological dysfunction. 51,52 Moreover, there is extensive evidence that after an injury, the brain enters an inflammatory state and often increases the permeability of the BBB. While initially the permeability increases in response to the mechanical or biochemical stress of an injury, 25,26,30 there is additional evidence that this permeability has an adaptive capacity to assist the necessary immune response and limit longer-term damage. 30,53 BBB function is critical to the regulation of cerebral vascular coagulation. 54 When the barrier is relatively diffuse, peripheral immune cells can traffic into the areas of injury, which can amplify inflammatory responses. 55 Moreover, serum proteins such as albumin and fibrinogen and other potential triggers of coagulation (and inflammation) cascades, which are normally excluded from the CNS, are now able to enter. Consistent with existing studies, 34,53,56 our data indicate that the BBB remains leaky after a mild injury up to 28 DPI. This persistent permeability has been implicated in stroke recovery, with permeability detected long after injury in humans that suffer TBIs and strokes. 57 -59 The integrity of the blood–brain barrier is critical for sustained recovery after an ischemic stroke. 60,61 The long-lasting dysfunction could be indicative of poorer post-stroke recovery and suggests that mediating this barrier dysfunction may be a potential therapeutic route to reduce the post-injury vulnerability to ischemic stroke.
Pathological thrombosis is a very common finding around sites of trauma. 62 Tissue damage is often associated with the need to protect against microbial infection and restore hemostasis. 63 Our findings signify that thrombus formation persists out at least 28 DPI and could be implicated in wide ranges of downstream inflammatory signaling that occurs following TBI. Injured or chemically activated endothelial cells and platelets release von Willebrand factor (VWF) which can be immobilized at sites of vascular inflammation. 64 Platelets can then interact with VWF through their glycoprotein Ib-IX-V receptor complexes, which can begin the process of platelet adhesion and thrombus formation. 64,65 Other processes including deposition of fibrinogen and cleavage into insoluble fibrin can help stabilize thrombus formation. Damage-associated molecular patterns can activate platelets, promoting thromboinflammation. 66 Microthrombi form acutely after TBI and can negatively impact both cerebral blood flow and functional recovery. 54 The traumatized brain (and potential clinical interventions) thus faces a significant challenge as TBI induces at least a temporary state of hypercoagulation and induces inflammatory signaling, while simultaneously increasing the risk of intracranial hemorrhage which means that anticoagulants must be used carefully.
Various glial cell types respond after injury by changing both their number and morphology. Microglia, for one, are highly regulated and activated after TBI and even mild injuries can cause marked increases in microglial numbers over a short period of time. 13,14 Moreover, microglia are major players in limiting vascular damage by promoting inflammatory responses and phagocytosing damaged or shredded vasculature and surrounding cells. 67,68 In our injured animals, both male and female mice had significant increases in total microglial area and vascular-associated microglia by 28 DPI. This suggests that there is a chronic activation and response of microglia on the cerebral blood vessels that help to contain the widespread dysfunction and damage of the vasculature.
This chronic activation of microglia has been shown previously to exist beyond 3 months in injured mice, and out to years after injury in humans. 69,70 VAMs have been implicated previously in inflammatory and apoptotic responses on damaged endothelial cells or abnormally formed vessels after rampant angiogenesis. 71,72 After ischemia, large numbers of microglia migrate and adhere to cerebral blood vessels near the penumbra, which can result in disintegration of unstable or damaged vessels. 68,73 In TBI, this propensity of microglia to respond to the vasculature can be attributed to the shear forces that release factors like tumor necrosis factor-α, lipopolysaccharide, and CCL2, which result in activation of inflammatory cascades that lead the microglia to phagocytose damaged vessels. 74,75 As such, it may be a potential future target to attempt to limit VAM responses after the acute inflammatory responses of an injury to prevent lasting chronic impairments that may predispose some to ischemia. Notably, further information is needed on transcriptional activation and secretory pathways of microglia to fully capture the role of this cell type in mediating chronic immune activation and the observed sex differences in vulnerability to ischemia.
In addition to microglia, astrocytes are also activated in response to mTBI. 13,14,16,76 We therefore investigated the impact of mTBI and subsequent ischemic stroke on astrocytes expressing aquaporin (AQP4), a water channel that is necessary for maintaining the water to solute ratios in the brain. 21 AQP4 increases after experimental TBI, though this has only been shown previously in moderate or severe injury models. 77,78 Our data suggest a persistent change after mild injury in male mice, with an acute change in female mice that resolves by 28 DPI. This time course of AQP4 expression changes has been underexplored, with only a few studies suggesting a chronic decrease in AQP4 after more severe blast injuries. 79 In ischemic stroke, AQP4 expression increases have been linked to increases in cytotoxic edema and infarct volume via impaired water travel and leakage of other soluble factors into the brain's extracellular spaces, with notable expression changes in both male and female mice. 80 -82
An additional prominent response to TBI is a marked remodeling of the vasculature that can include loss of microvasculature, and significant new vessel growth. 56 These processes are largely induced by tissue hypoxia and can be associated with tissue and functional recovery. 30,83 Our data suggesting increases in VEGF and subsequent angiogenic growth following a mild injury is consistent with existing literature that shows acute increases in VEGF and angiogenic properties of cerebrovasculature after brain injuries. 84 -86 This increase in VEGF has previously been explored acutely by 1 week after injury and while VEGF increases drive angiogenesis, which improves neurological outcomes, 72,85,87 it also promotes BBB dysfunction. This VEGF-mediated dysfunction has been suggested to occur via formation of abnormal vessels and prolonged leakiness of the BBB. 88 Additionally, while in TBI there are clear roles for neurological recovery, after ischemia there is evidence that increases in VEGF expression results in worse outcomes like cerebral edema, infarct volume, and cognitive impairments. 89 -91 Therefore, it seems there is a “sweet-spot” in expression levels of angiogenesis that promotes neurological recovery and microvascular repairs after injury but will not induce further inflammatory cascades and dysfunctional vasculature that result in worsened ischemic strokes.
Existing literature broadly describes sex differences after TBI, but several gaps and contradictory evidence remains. Female rats perform better after a single injury in locomotor assays like rotarod and beam walk, but in repetitive injury models, females perform worse than males. 92,93 Sex differences have also been noted in TBI-related pathophysiology, with males displaying persistent edema and permeability that resolves in females by 5 DPI. 94,95 This persistent BBB permeability in males is consistent with our data that shows a chronic increase in permeability out to 28 DPI, while permeability begins to reduce at this timepoint in females. Moreover, TBI produces a greater constriction of cerebral blood flow in male piglets compared with females. 96 This post-injury vasoconstriction is likely related to our observation of increased intravascular coagulation in males and may drive the pathophysiological differences in vascular dysfunction between injured male and female mice.
Finally, extensive evidence exists to demonstrate sex differences in ischemic stroke prevalence and outcomes, 97 and while females have a lower incidence of stroke across the lifespan, when accounting for age, older females actually have a higher incidence compared with older males. Additionally, the quality of life and functional post-stroke outcomes of females are significantly worse than males. 39,97 Our data suggest that female mice may respond to TBI in ways that promote post-injury ischemic vulnerability.
There are well-documented sex differences in the cerebrovascular responses to brain injuries. 92,94 Therefore, we hypothesized that the recovery trajectory after mTBI would differ between male and female mice. Moreover, sex differences in the vulnerability to post-injury ischemia would also vary across time and sex. It is likely that by observing these changes over time we could uncover sexually differentiated responses in several of the outcomes, including coagulopathy, BBB permeability, and angiogenesis. Additionally, it is likely that at our acute timepoint of 7 DPI, the impact of the female sex steroid hormones is not fully involved in the recovery after mTBI, but by a chronic 28 DPI, the estrus cycle has completed numerous times and could be partially responsible for the persistent vulnerability to ischemia and worsened ischemic outcomes, including the prolonged BBB permeability.
Conclusions
The present study demonstrates that following a mild traumatic brain injury, male and female mice experience differentially expressed cerebrovascular dysfunction and stroke vulnerability over time. These findings suggest that the sex differences that underlie stroke vulnerability could be in part due to previous brain injuries that result in lasting changes in intravascular coagulation, blood-brain barrier permeability, and angiogenesis changes. This work opens up a potential field of study comparing injury-related dysfunctions in male and females.
Footnotes
Acknowledgments
We appreciate the assistance of R. Oliverio, B. White, J. Ivey, and all members of our laboratory and institution who have contributed to the completion of this project.
Transparency,Rigor,and Reproducibility Statement
The sample size for each group was 6 mice per group, based on power analysis of previous infarct volume data which indicated >80% power to detect overall significant effects of ANOVAs as primary analyses, with alpha <0.05 for post-hoc difference detections. A total of 272 mice were subjected to experimental TBI or sham injury, 19 died, and the rest were tested to completion of studies, with complete data sets obtained for all remaining mice. Investigators were blinded to experimental conditions when performing IHC analyses and infarct volume measurements. IHC antibodies came from the same laboratories over the course of the stains and pilot studies were conducted to validate antibody specificity and accuracy. Normal distribution of primary histological data was verified using a Kolmogorov-Smirnoff test. Data from this study are available upon request.
Authors' Contributions
BW, KK, and ZW contributed to the conception and design of the study. BW, RV-C, AA, and NZ performed the experiments. BW, KK, and ZW analyzed the data, and BW wrote the first draft of the manuscript, with KK and ZW contributing to figure design. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding Information
This study was supported by the National Institutes of Neurological Disorders and Stroke (P30-NS045758), as well as the West Virginia University Stroke and Alzheimer's Related Dementias Training Program (T32 AG052375), and the National Institute of General Medicine Sciences (P20 GM109098).
Author Disclosure Statement
No competing financial interests exist.