
Abstract
Microglia-mediated neuroinflammatory responses play important roles in secondary neurological injury after traumatic brain injury (TBI). The TGF-β pathway participates in the regulation of M1/M2 phenotype transformation of microglia. TGF-β can activate the Smad pathway by binding to TGF-βRs, which is regulated by the cleavage function of A disintegrin and metalloproteinase 17 (ADAM17). However, the role of ADAM17 and the associated signaling pathways in the pathological process after TBI remain unclear. Herein, we assessed the transformation of microglia M1/M2 phenotype polarization and the neuroinflammatory response after the inhibition of ADAM17. The formation of TGF-βRs and TGF-β1/TGF-βRII complexes on microglia were detected to evaluate the effect of ADAM17 inhibition on the TGF-β1/Smad pathway. ADAM17 was highly expressed after TBI and mainly located in the microglia. the inhibition of ADAM17 improved neurological function after TBI. The neuroprotective effect of ADAM17 inhibition was related to a shift from the M1 microglial phenotype to the M2 microglial phenotype, thus reducing TBI-induced neuroinflammation. ADAM17 inhibition increased expression of TGF-βRs on the microglia membrane, promoted formation of TGF-β1/TGF-βRII complexes, and induced intranuclear translocation of Smads, which activated the TGF-β/Smad pathway. In conclusion, our study suggested that ADAM17 inhibition regulated microglia M1/M2 phenotype polarization through the TGF-β1/Smad pathway and influenced the neuroinflammatory response after TBI.
Introduction
Primary brain injury triggers a series of harmful inflammatory processes that aggravate the initial tissue damage and impair neural function, which are important pathological features after traumatic brain injury (TBI). 1 Microglia, the resident immune cells of the central nervous system, play significant roles in neuroinflammation. 2 –5 Microglia activated by traumatic stress exhibit two polarized phenotypes: the classic activated pro-inflammatory phenotype (M1) and the alternative activated anti-inflammatory phenotype (M2). 6 Our previous studies have confirmed that the M1/M2 polarization phenotype of microglia is closely related to the outcome of neuroinflammation after TBI. 7 Post-traumatic activation of pro-inflammatory phenotype microglia and the subsequent release of inflammatory mediators, such as tumor necrosis factor (TNF), interleukins (ILs), and interferons (IFN). 8,9 On the contrary, anti-inflammatory phenotype microglia secrete anti-inflammatory factors, such as transforming growth factor-β (TGF-β), IL-10, and IL-8. The recovery of neurological function after TBI can be improved by inhibiting the activation of microglia and regulating the M1/M2 ratio and neuroinflammation. 10 -12
At the same time, changes to neuroinflammation will also cause transformation to the polarization phenotype of microglia. 13,14 TGF-β secreted by anti-inflammatory phenotype microglia can promote the transformation of the M1 phenotype to the M2 phenotype, which is characterized by up-regulation of the anti-inflammatory cytokine IL-10 and inhibition of the synthesis of the proinflammatory cytokines TNF-α and IL-6. 7,15,16 After TGF-β binds to type I and II serine/threonine kinase receptors (TGF-βRI and TGF-βRII, respectively), activated TGF-βRII can phosphorylate TGF-βRI, thus activating the classic Smads pathway and non-Smad pathways, such as the PI3K/Akt and MPAK pathways, which convey a variety of biological effects and play important regulatory roles in inflammatory reactions. 17 -19
The shedding of the extracellular domain of membrane-anchored receptors mediated by the ectodomain shedding enzyme A disintegrin and metalloproteinase 17 (ADAM17) can cleave various substrates, including cytokine precursors (e.g., pro-TNF-α), cytokine receptors (e.g., IL-6R, TNF-R, and TGF-βRs), ErbB ligands (e.g., TGF-α and TGF-β), and adhesion proteins (e.g., L-selectin). 20,21 ADAM17 cleaves the extracellular domain of TGF-βRs, thereby down-regulating the downstream TGF-β/Smad pathways, and participates in the pathological processes of tumors and autoimmune diseases. 20,22 Relatively few studies have explored the role of ADAM17 in diseases of the central nervous system. Although studies have suggested that ADAM17 plays an important role in nerve repair, 23 -25 the role and underlying mechanism of ADAM17 in the regulation of inflammatory responses after TBI remain unclear. Considering that ADAM17 is an important regulatory factor of the TGF-β pathway, 26,27 it may be closely related to microglia polarization and the neuroinflammatory response. Therefore, the aim of the present study was to determine whether ADAM17 can influence the M1/M2 polarization of microglia and the neuroinflammatory response mediated by the TGF-β pathway in the pathological process after TBI.
Methods
Animals
Adult male Sprague-Dawley rats (weighing 230-260 g) were purchased from the Experimental Animal Center of Fujian Medical University (Fuzhou, Fujian, China) and housed in a clean, temperature-controlled environment (23°C ± 2°C) under a 12-h light/dark cycle with free access to food and water. The experimental protocols of the present study, including all surgical procedures and animal usages, were approved by the Experimental Animal Ethics Committee of Fujian Medical University and conducted in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (Bethesda, MD).
Cell culture and treatment
Murine microglial BV-2 cells were obtained from the Institute of Basic Medical Sciences of the Chinese Academy of Medical Sciences (Beijing, China) and maintained in Dulbecco's modified Eagle's medium (Nanjing KeyGen Biotech. Co., Ltd.) supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA) at 37°C under an atmosphere of 5% CO2/95% air. In vitro, BV2 cells were stimulated with 10 μg/mL of lipopolysaccharide (LPS; MB5198, Dalian Meilun Biotech Co., Ltd., Dalian, China) for 6 h and 20 ng/mL of exogenous IL-4 (Novoprotein_CD03, Novoprotein Scientific, Shanghai, China) overnight, respectively, to mimic the M1 and M2 polarization environments of the brain. Then, 50 nM LY2157299 (S2230, Selleck Chemicals, Houston, TX), a TGF-βRI kinase inhibitor, was applied to the BV2 cells for further study of the signaling pathways involved in polarization.
Cell transfection
BV2 cells were transfected with small interfering RNA (siRNA) against ADAM17 and the plasmid pcDNA3.1-Flag-ADAM17, respectively. Mouse ADAM17 siRNA and a negative control were chemically synthesized by Guangzhou RiboBio Co., Ltd. (Guangzhou, China). Mouse pcDNA3.1-Flag-ADAM17 (open reading frame sequence MR210844) was purchased from PPL (Nanjing KeyGen Biotech. Co. Ltd.). The BV2 cells were transiently transfected with ADAM17 siRNA and the plasmid with a corresponding negative control using Lipofectamine 2000 reagent (Thermo Fisher Scientific Waltham, MA) for 24 h at 37°C in accordance with the manufacturer's instructions. Afterward, transfected BV2 cells were treated with 50 nM LY2157299 for 24 h at 37°C for signaling pathway assessment. The target sequences of ADAM17 siRNA and RealTime-PCR primers of ADAM17 are showed in Supplementary Table S1 and Supplementary Table S2.
Animal model and drug delivery
Rats were randomly assigned to one of four groups: sham, TBI, TBI+Vehicle, or TBI+ TNF-alpha protease inhibitor I (TAPI-1, an ADAM17 inhibitor, 28 579051; Sigma-Aldrich Corporation, St. Louis, MO; n = 48 each), each of which were further divided into four time-based subgroups (1, 3, 7, and 14 days). The rat TBI model was established as previously described under sodium pentobarbital anesthesia (50 mg/kg by intraperitoneal injection). An incision was made to the scalp and a 5 mm-diameter hole was made to the right side of the coronal suture (anterior-posterior, -2 mm; lateral-anterior, 2 mm). A 40 g hammer was dropped on the brain from a height of 20 cm to simulate TBI. The hole in the posterior bone was then sealed closed with wax. In sham rats, the surgery was performed but the impact was omitted. At 0.5 h after surgery, these rats were implemented with an osmotic pump (Model 2006, Alzet Osmotic Pump, 1μL/h) for a 3-day intraventricular infusion of the TAPI-1 (0.5μg/g/day) or vehicle (20% dimethyl sulfoxide) as a negative control. The injection site was determined to be 2 mm from the anterior fontanel to the posterior fontanel and 1.5 mm lateral to the right of the sagittal suture.
Neurological impairment score
Rats were subjected to exercise (muscular state and abnormal action), sensation (visual, tactile, and balance), and reflex examinations and assigned a modified neurological severity score (mNSS) that was recorded when the rat failed to complete the task or there was no corresponding reflex. 29 The mNSS ranged from 0 to 18 points, where a total score of 18 points indicated severe neurological deficits and a score of 0 indicated normal performance (13-18 points indicated severe injury, 7-12 indicated moderate injury, and 1-6 indicated mild injury). Neurological function was measured on days 1, 3, 7, and 14 after TBI by two investigators who were blinded to group information.
Measurement of brain water content and blood–brain barrier permeability
Brain water content was calculated using the wet weight-dry weight method. Animals were sacrificed after neurological assessment and the brain tissue was removed from the injured side. Filter paper was used to remove excess blood and cerebrospinal fluid. The wet weight was measured and the brains were dried in an oven at 100°C for 24 h until a constant weight was achieved, at which point the dry weight was measured. The % brain water content was calculated as: (wet weight − dry weight)/wet weight × 100%.
Blood–brain barrier (BBB) permeability was investigated by measuring the extravasation of Evans blue dye (2% in saline; 4 mL/kg; E2129, Sigma-Aldrich Corporation), which was intravenously injected 2 h prior to sacrifice on post-injury Day 3. Following sacrifice, the mice were transcardially perfused with phosphate-buffered saline and the brains were removed. Each tissue sample (whole brain) was immediately weighed, homogenized in 1 mL of 50% trichloroacetic acid, and centrifuged. Then, the absorption of the supernatant was measured with a spectrophotometer (UV-1800 ENG 240V; Shimadzu Corporation, Kyoto, Japan) at a wavelength of 620 nm. The quantity of Evans blue dye was calculated using a standard curve and expressed as μg/g of brain tissue.
Nissl staining
Formaldehyde-fixed specimens were embedded in paraffin and cut into 4 μm-thick sections that were deparaffinized with xylene and rehydrated in a graded series of alcohol. Samples were treated with Nissl staining solution for 5 min. Damaged neurons were shrunken or contained vacuoles, whereas normal neurons had a relatively large and full soma and round, large nuclei. Quantitative analysis of Nissl staining results was performed by the proportion (%) of damaged neurons. 30 Five regions of interest (ROIs) around the injured area were randomly selected by investigators who were blinded to group information, and the damaged neurons were counted using ImageJ software. Five ROIs were averaged for statistical analysis.
Immunohistochemical analysis
Formaldehyde-fixed specimens were embedded in paraffin and cut into 4 μm thick sections that were deparaffinized with xylene and rehydrated in a graded series of alcohol. Antigen retrieval was carried out by microwaving in citric acid buffer. Sections were incubated with an antibody (Ab) against ADAM17 (1:200; ab57484, Abcam plc, Cambridge, UK) or ionized calcium-binding adapter molecule (Iba)-1 (1:200; sc-32725, Santa Cruz Biotechnology, Inc., Dallas, TX), and then incubated with a secondary Ab for 1 h at room temperature. Finally, the cell nucleus was stained with DAB and re-stained with hematoxylin. The negative control was prepared without adding the anti-ADAM17 or Iba-1 Ab. Quantitative analysis of immunohistochemical results was performed by integration system. Stained slides were reviewed and scored by two pathologists with more than 10 years' experience who had not participated in this experiment. Five fields were randomly selected for analysis by pathologists: 0, no positive cells; 1, 1-25% positive cells; 2, 26-50% positive cells; 3, 51-75% positive cells; and 4, >75% positive cells. Two pathologists independently assessed the slides. Discrepancies were resolved by discussion between pathologists.
Immunofluorescence analysis
Formaldehyde-fixed specimens were embedded in paraffin and cut into 4-μm thick sections that were deparaffinized with xylene and rehydrated in a graded series of alcohol, followed by antigen retrieval. Sections were incubated overnight at 4°C with Abs against neuronal nuclei (1:100; A11954-1, Wuhan Boster Biological Technology, Ltd., Wuhan, China), Iba-1 (1:200; sc-32725, Santa Cruz Biotechnology, Inc., Dallas, TX), glial fibrillary acidic protein (GFAP) (1:200; sc-33673, Santa Cruz Biotechnology, Inc.), cluster of differentiation CD16 (1:100; M01408-1, Wuhan Boster Biological Technology, Ltd.), CD206(1:100; sc-70586, Santa Cruz Biotechnology, Inc., Dallas, TX), TGF-βRII (1:100; bs-0117R, Bioss, Beijing, China), and p-Smads (1:100; A00090T220, Wuhan Boster Biological Technology, Ltd.). After washing, the sections were incubated with secondary Abs for 1 h at room temperature. The cell nuclei were stained with 4′,6-diamidino-2-phenylindole. Immunopositive cells in five randomly selected fields were counted under a microscope (Leica, Wetzlar, Germany) at 400 × magnification by investigators who were blinded to the experimental group.
Enzyme-linked immunosorbent assay
Inflammatory factors (i.e., TNF-α, IL-1 β, IL-6, and IFN-γ) of brain tissues and BV2 cell culture supernatants were detected using mouse enzyme-linked immunosorbent assay (ELISA) kits (Nanjing KeyGen Biotech. Co., Ltd.) at an optical density of 450 nm using a microplate reader (SpectraMax M3; Molecular Devices, Inc., San Jose, CA).
Western blot analysis
The samples, including brain tissues and BV2 cells, were prepared using nuclear and cytoplasmic protein purification assays (Nanjing KeyGen Biotech. Co., Ltd.) with modified radioimmunoprecipitation lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate). The p-SMAD2 protein concentrations of the cell nuclear and cytosolic lysates were determined separately with a bicinchoninic acid assay (Beyotime Institute of Biotechnology, Shanghai, China). Other proteins were not separated into nuclear and cytosolic fractions. Approximately 25 μg of protein were loaded to each well of a polyacrylamide gel, separated by electrophoresis, and then transferred to polyvinylidene difluoride (PVDF) membranes, which were incubated with primary Abs against B cell lymphoma (Bcl)-2 (1:2000; ab196495), Bcl-2-associated X factor (Bax) (1:5000; ab32503), cleaved caspase-3 (1:3000; ab13585), Iba-1 (1:1000; ab178846), CD16 (1:1000; ab223200), CD206 (1:1000; ab300621), IL-1β(1:1000; ab254360), Arg-1 (1:2000, ab96183), TGF-β1(1:1000; ab187085), TGF-βRI (1:1000; ab235578), TGF-βRII (1:1000; ab259360), TGF-βRIII (1:1000; ab166705), p-Smads (1:1000; ab280888), and Smads (1:1000; ab202445) (all purchased from Abcam, Shanghai, China), followed by incubation with appropriate secondary Abs. Immunoreactivity was visualized with the ECL Western Blotting Detection System (EMD Millipore Corporation, Billerica, MA). Gray value analysis was conducted with UN-Scan-It 6.1 software (Silk Scientific Inc., Orem, UT). Expression levels were normalized against β-actin (1:5000; M01263-3, Wuhan Boster Biological Technology, Ltd.)
Co-immunoprecipitation analysis
BV2 cells were homogenized in IP lysis Buffer (Nanjing KeyGen Biotech. Co., Ltd.) and then incubated with 1 μg of TGF-β1 (1:30; ab187085, Abcam plc) or TGF-βR II (1/30; ab259360, Abcam plc) Abs or immunoglobulin G (ab6789; Abcam plc) for 1 h at 4℃. A 10-μL volume of protein A agarose beads (Roche Diagnostics Deutschland GmbH, Mannheim, Germany) was added to the sample lysate mixture and incubated overnight with primary Abs at 4℃. After immunoprecipitation and centrifugation, the agarose beads were washed three times with lysis buffer and used for immunoblotting to detect expression of the TGF-βRII and TGF-β1 proteins in order to estimate the expression level of the TGF-β1/TGF-βRII complex.
Statistical analysis
Data were analyzed using PASW Statistics for Windows, version 18.0. (SPSS Inc., Chicago, IL). All experiments were performed in triplicate unless otherwise noted and the results are expressed as the mean ± standard deviation. Comparisons between groups were made with the unpaired Student's t-test. Multiple-group comparisons were assessed by one-way analysis of variance and post hoc multiple comparisons were performed using the Student-Newman-Keuls test. A probability (p) value of <0.05 was considered statistically significant.
Results
ADAM17 expression increased after TBI and was specific to the microglia of the injured cerebral cortex
In the rat TBI model, a molecular biology study was performed on post-injury Days 1, 3, 7, and 14 (Fig. 1A). The western blot results showed that ADAM17 levels were significantly increased on post-injury Days 1, 3, and 7, with the most significant increase on post-injury Day 3 (p < 0.01), which then gradually decreased to almost normal at post-injury Day 14 (Fig. 1B). Immunohistochemistry also indicated that ADAM17 was highly expressed in the cortical injury area of the TBI group on post-injury Day 3 (Fig. 1C).

The expression of a disintegrin and metalloproteinase 17 (ADAM17) increased after traumatic brain injury (TBI), and it is specifically located in the microglia at the injured cerebral cortex.
To clarify the location of ADAM17 in cortical cells after brain injury, immunofluorescence double staining was performed to detect ADAM17 as well as markers specific to neurons (NeuN), microglia (Iba-1), and astrocytes (GFAP). The results showed that ADAM17 was mainly expressed in microglia, with no obvious expression in neuron and astrocytes (Fig. 1D).
Inhibition of ADAM17 can improve neurological function after TBI
TAPI-1 was used to inhibit the activity of ADAM17 and the mNSS was used to evaluate post-TBI neurological function after the inhibition of ADAM17. The results showed that as compared with the TBI group, the mNSS of the TBI+TAPI-1 group was improved at post-injury Day 3 (10.51 ± 0.38 vs. 12.21 ± 0.52, respectively, p < 0.05), and this beneficial effect persisted until 14 days post-injury (7.26 ± 0.32 vs. 8.04 ± 0.24). This suggests that inhibition of ADAM17 contributes to improved neurological function after TBI (Fig. 2A).

Specific inhibition of a disintegrin and metalloproteinase 17 (ADAM17) can improve neurological function and reduce neuronal apoptosis after traumatic brain injury (TBI).
The contents of Evans blue dye and brain water can be used to quantitatively evaluate the degree of BBB destruction and brain edema. The results showed that on post-injury Day 3, the brain water content of the TBI+TAPI-1 group was appreciably reduced as compared with the TBI group (Fig. 2B). Concurrently, the corresponding penetration of Evans blue dye was significantly reduced in the damaged cortex (Fig. 2C, 2D).
Inhibition of ADAM17 can reduce neuronal apoptosis after TBI
Nissl staining and western blot analysis were used to detect apoptosis-related factors as well as neuronal apoptosis. The results of Nissl staining of the cerebral cortex showed that the apoptosis rate of neurons in the Sham group was low, at about 4 to 6%. In contrast to the Sham group, the apoptosis rate of neurons in the TBI group was significantly increased after injury at each time period (p = 0.00). After TAPI-1-induced inhibition of ADAM17, as compared with the TBI group, the neuronal apoptosis rate in the TBI+TAPI-1 group on post-injury Day 3 was significantly reduced (53.00 ± 6.22% vs. 89.05 ± 8.04%, respectively, p < 0.05; Fig. 2E, 2F).
The Western blot results showed that as compared with the TBI group, the expression levels of apoptotic factors (i.e., cleaved capase-3 and Bax) in the cerebral cortex of the TBI+ TAPI-1 group were significantly reduced on post-injury Day 3, while expression of the anti-apoptotic factors Bcl-2 was increased (p < 0.05; Fig. 2G). These results suggest that the inhibition of ADAM17 can reduce neuronal apoptosis after TBI.
Inhibition of ADAM17 promoted M1/M2 phenotype transformation and inhibited inflammatory reactions
Immunohistochemistry staining and Western blot analysis were performed to detect changes in the expression levels of markers of the microglia (positive Iba-1 staining). The results showed that on post-injury Day 3, as compared with the sham group, positive staining of Iba-1 was notably increased (p < 0.05) in the TBI group. As compared with the TBI group, Iba-1 expression was decreased in the TBI+TAPI-1 group (p < 0.05; Fig. 3A, 3B).
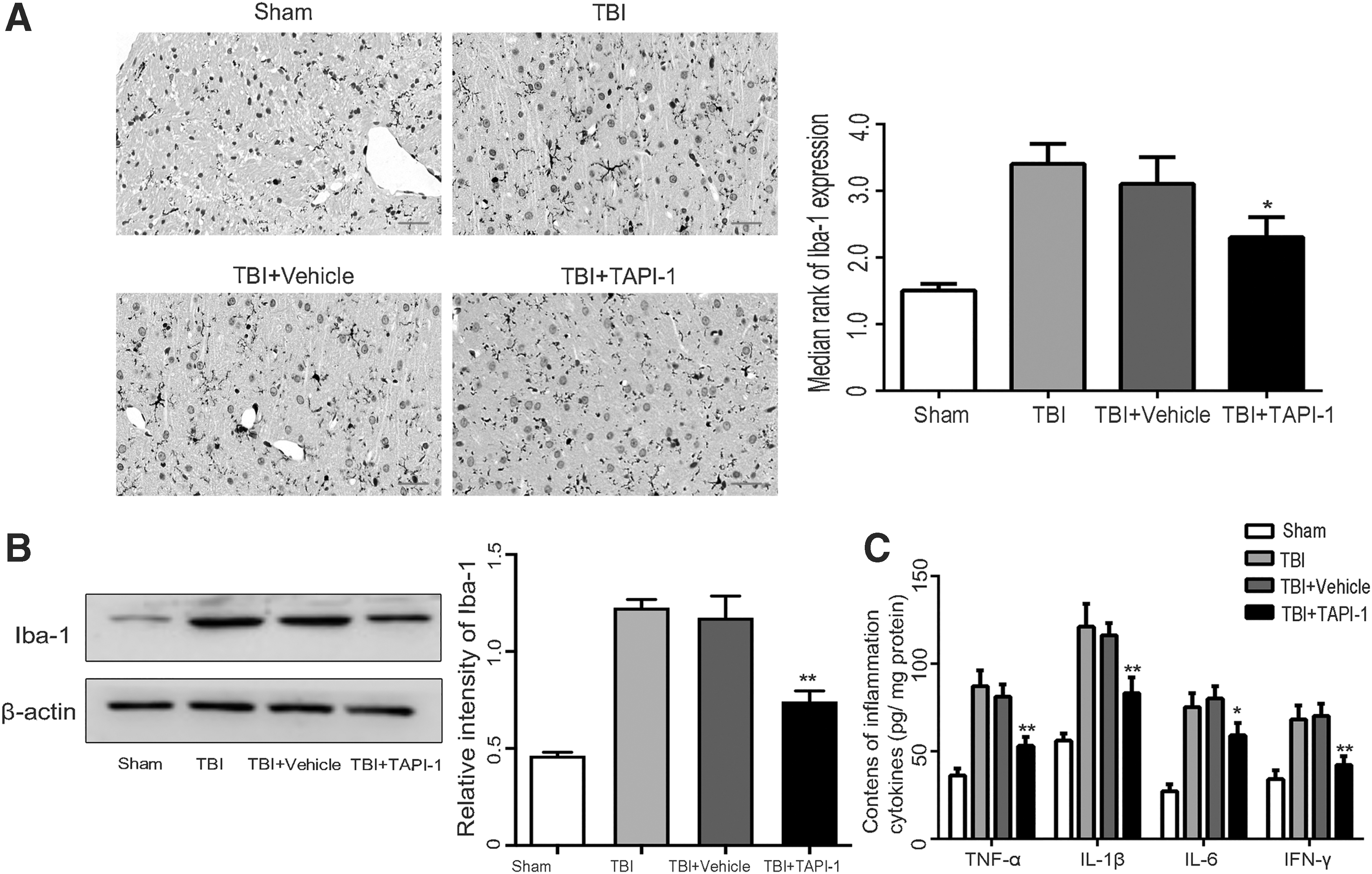
Specific inhibition of a disintegrin and metalloproteinase 17 (ADAM17) inhibits microglial activation and inflammatory reaction in lesion cortex after traumatic brain injury (TBI).
The ELISA results showed that as compared with the sham group, the serum expression levels of the inflammatory factor TNF-α, IL-1 β, IL-6, and IFN-γ were significantly increased in the TBI group on post-injury Day 3 (all p < 0.05). As compared with the TBI group, the expression levels of the serum inflammatory factors were significantly decreased in the TBI + TAPI-1 group (p < 0.05; Fig. 3C).
To further explore the mechanism of ADAM17 Promoted M1/M2 phenotype transformation. Immunofluorescence double staining was performed to detect changes in the expression levels of markers of the microglia (positive Iba-1 staining, red), M1 phenotype (positive CD16 staining, green), and M2 phenotype (positive CD206 staining, green) at the cellular level. Exposure of BV2 microglia to LPS induced a switch to the M1 phenotype and positive expression of CD16 was notably increased. As compared with the siCtrl+LPS group, CD16 expression was decreased in the siADAM17+LPS group, while CD206 expression was increased, indicating M2 phenotype polarization (p < 0.05). After overexpression of ADAM17, the BV2 microglia in the ADAM17 group were switched to the M1 phenotype. As compared with the ADAM17 group, CD16 expression was decreased and CD206 expression was increased in the ADAM17+TAPI-1 group, indicating M2 phenotype polarization (p < 0.05; Fig. 4A, 4B).

A disintegrin and metalloproteinase 17 (ADAM17) inhibition promoted M1/M2 phenotype transformation and inhibited inflammatory reaction in vitro.
The results of Western blot analysis also showed that as compared with the siCrl+LPS group, the expression levels of CD16 and IL-1β were significantly inhibited, while those of CD206 and Arg-1 were increased in the siADAM17+LPS group, indicating a switch from the M1 phenotype to the M2 phenotype (p < 0.05; Fig. 4C). As compared with the ADAM17 group, the expression levels of CD16 and IL-1β were significantly decreased, while those of CD206 and Arg-1 were increased in the ADAM17+TAPI-1 group. In the ADAM17+TAPI-1 group, the expression levels of CD16 and IL-1β were decreased, while those of CD206 and Arg-1 were observably increased, indicating M2 phenotype polarization (p < 0.05; Fig. 4C).
The ELISA results showed that as compared with the siCtrl+LPS group, the expression levels of inducible nitric oxide synthase (iNOS) and IL-1β were significantly decreased, while those of Arg-1 and TGF-β1 were increased in the siADAM17+LPS group (p < 0.05). Overexpression of ADAM17 promoted the expression of iNOS and IL-1 β, but decreased that of Arg-1 and TGF-β1. TAPI-1 obviously inhibited the expression of ADAM17, which counteracted the M1 phenotype polarization of microglia induced by ADAM17. Meanwhile, cells in the ADAM17+TAPI-1 group had the M2 phenotype (p < 0.0; Fig. 4D). As compared with the siCtrl+LPS group, the expression levels of TNF-α, IL-6, and IFN-γ were significantly decreased in the supernatant of the siADAM17+LPS group (all p < 0.05). Similarly, the expression levels of inflammatory factors were considerably lower in the ADAM17+TAPI-1 group than in the ADAM17 group (p < 0.05; Fig. 4E).
Inhibition of ADAM17 activate genes related to the TGF-β 1 pathway
TGF-β1 and its receptors (TGF-βRI and, TGF-βRII), affected by the cleavage function of ADAM17, are important factors in regulating M1/M2 polarization of microglia. 15 -17 As compared with the siCtrl+LPS group, the expression levels of TGF-β1, TGF-βRI and TGF-βRII were appreciably increased in the siADAM17+LPS group. Overexpression of ADAM17 inhibited expression of TGF-β1, TGF-βRI, and TGF-βRII. The TGF-β1 pathway was activated after inhibition of ADAM17 expression in the ADAM17+TAPI-1 group (p < 0.05; Fig. 5A).
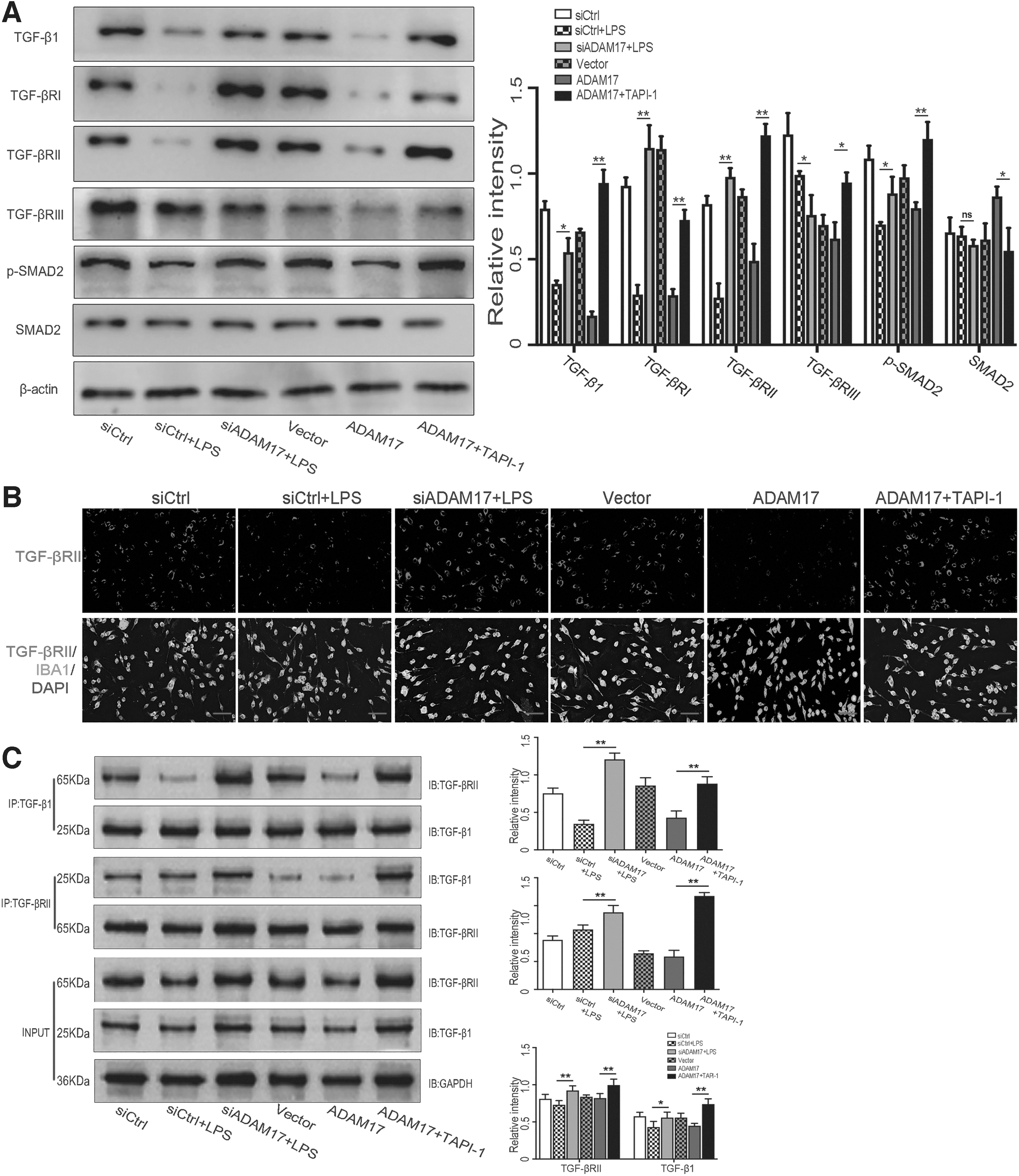
Specific inhibition of a disintegrin and metalloproteinase 17 (ADAM17) activated the transforming growth factor (TGF)-β1 pathway in vitro.
As compared with the siCtrl+LPS group, TGF-βRII (red) expression by microglia (Iba-1 positive, green) increased in the siADAM17+LPS group, while inhibited in the ADAM17 group. As compared with the ADAM17 group, TGF-βRII expression was significantly increased in the ADAM17+TAPI-1 group (Fig. 5B).
Co-immunoprecipitation (Co-IP) analysis showed that as compared with the siCtrl+LPS group, TGF-β1/TGF-βRII complex formation was notably increased in the siADAM17+LPS group. ADAM17 inhibited TGF-β1/TGF-βRII complex formation. As compared with the ADAM17 group, TGF-β1/TGF-βRII complex formation was promoted in the ADAM17+TAPI-1 group (Fig. 5C).
TGF-β1/TGF-β RII complex formation can activate the downstream classic Smads pathway. Western blot analysis and immunofluorescence staining further showed that as compared with the siCtrl+LPS group, siADAM17 significantly increased translocation of p-Smads from the cytosol to the nucleus and increased p-Smads expression, while ADAM17 inhibited intranuclear translocation of p-Smads. On the contrary, the ADAM17 inhibitor TAPI-1 counteracted the inhibitory effect of ADAM17 on translocation of p-Smads (Fig. 6A, 6B).
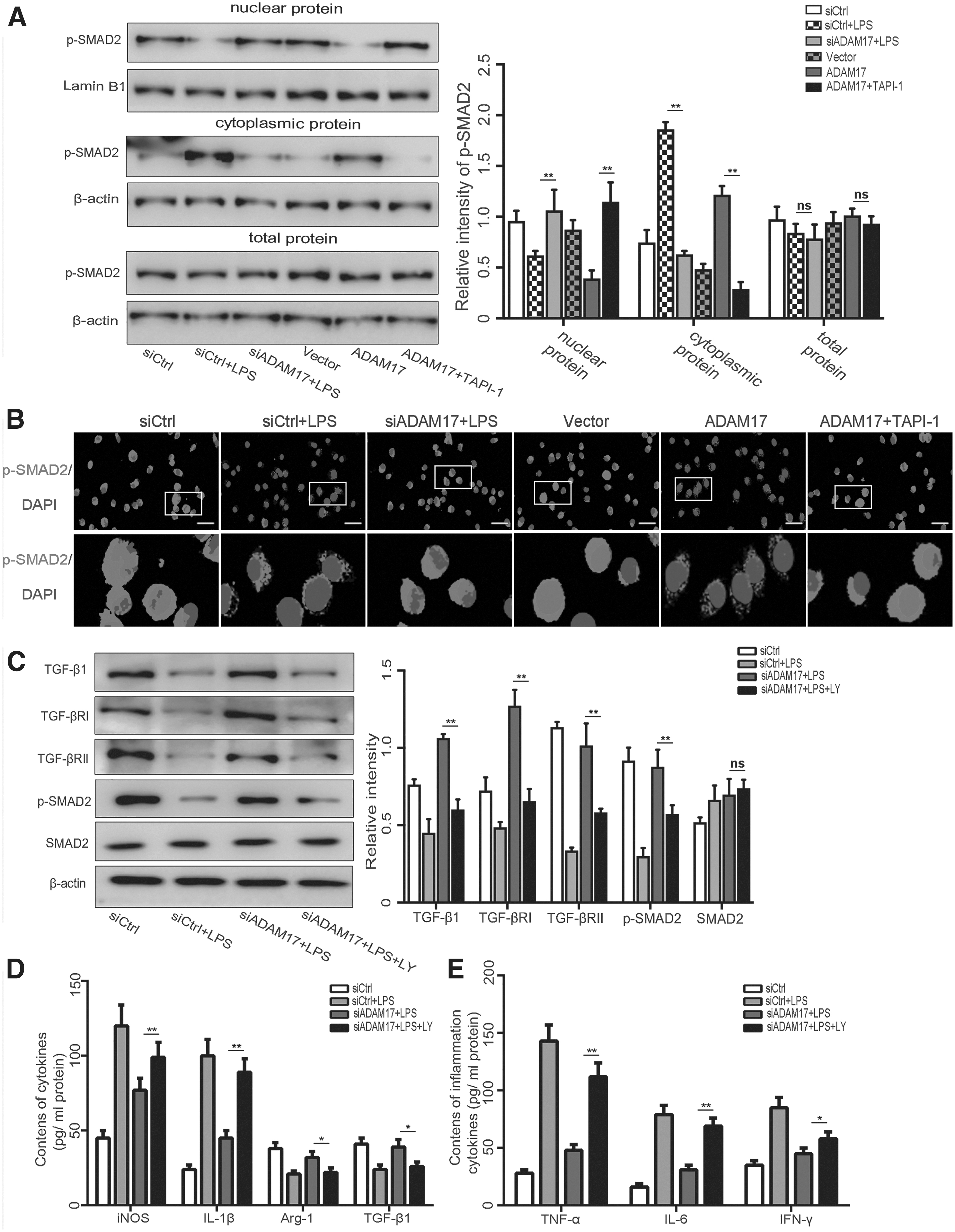
Inhibition of TGF-β1/Smad pathway hinders regulation of M1/M2 phenotype transformation of microglia by a disintegrin and metalloproteinase 17 (ADAM17) in vitro.
Inhibition of the TGF-β1 pathway hindered ADAM17 regulation of the M1/M2 phenotype transformation of microglia
To further clarify whether the TGF-β1 pathway participates in the regulation of microglia polarization by ADAM17, TGF-β receptor inhibitor (LY2157299) was used to specifically inhibit these pathways. As compared with the siADAM17+LPS group, LY2157299 notably inhibited the expression of TGF-βRI, TGF-βRII, and downstream p-Smads (Fig. 6C). As compared with the siADAM17+LPS group, the expression levels of iNOS and IL-1β were increased, while those of Arg-1 and TGF-β were decreased in the siADAM17+LPS+LY group, indicating counteraction of the effect of ADAM17 on M2 phenotype polarization of microglia and a switch from the M1 phenotype in the siADAM17+LPS+LY group (p < 0.05; Fig. 6D). After LY2157299 specifically inhibited TGF-βRs, the expression levels of related inflammatory factors were considerably increased in the siADAM17+LPS+LY group as compared with the siADAM17+LPS group, which offset the anti-inflammatory effect of siADAM17 (p < 0.05) (Fig. 6E).
Discussion
ADAM17 is an important factor in the process of central nervous system tumors, spinal cord injury, ischemic brain injury, and brain degenerative diseases, 22,24,31 -33 but current research in the field of TBI is still scarce. Our study focused on the ADAM17-derived neuroinflammation for pathophysiology of TBI. Our findings provide more new information on ADAM17-mediated microglial polarization for the secondary brain injury after TBI and also provide a rationale for targeting ADAM17 to modulate neuroinflammation in the treatment of TBI.
Shedding of the extracellular domain of the membrane anchored receptor mediated by the shedding enzyme ADAM17 can affect the expression level and biological effects of the substrate, which plays a significant role in the regulation of inflammatory responses. 22,24,31 After TBI, the expression levels of metalloproteinases, including ADAM17, are increased. 25,34 However, the underlying pathological mechanism remains unclear. The results of the present study revealed high expression of ADAM17 in the damaged area of the cerebral cortex after TBI injury and was mainly located in microglia, which are closely related to the inflammatory response. In TBI rats, inhibition of ADAM17 lowered permeability of the BBB, reduced the degree of brain edema, and inhibited apoptosis of nerve cells, thereby improving nerve function. Inhibition of ADAM17 suppressed microglia M1 phenotype polarization and induced M2 phenotype polarization, which can alleviate neuroinflammation. Further studies indicated that inhibition of ADAM17 can increase the expression of TGF-βRs on the microglia membrane and promote the formation of TGF-β1/TGF-βRII complexes in addition to inducing nuclear transfer of Smads, then activating the TGF-β1/Smad pathway and finally regulating M1/M2 polarization of microglia (Fig. 7). Collectively, these results demonstrate that the ADAM17 pathway can accelerate polarization of the M1/M2 phenotypes and repress activation of microglia as well as the neuroinflammatory response, thereby facilitating neural repairment after TBI. ADAM17 is relatively specific to the microglia specificity and, thus, should be considered as a therapeutic target for neuroinflammation after TBI.

Schematic of the possible neuroprotective mechanisms of a disintegrin and metalloproteinase 17 (ADAM17) inhibition after traumatic brain injury (TBI). As illustrated, ADAM17 mediates the shedding of the extracellular domain of transforming growth factor (TGF)-βRs on the cell membrane and decrease of its quantity. In addition, the ADAM17 negatively regulates the TGF-β1/Smad pathway, so that it can promote the polarization of M1 phenotype of microglia and inhibit M2 phenotype of microglia. Specific inhibition of ADAM17 can increase the expression of TGF-βRs on microglia membrane and promote the formation of TGF-β1/TGF-βRII complex. TGF-β1/TGF-βRII complex then induces intranuclear translocation of Smads, activates TGF-β 1/Smad pathway, regulates M1/M2 polarization of microglia, and finally affects the neuroinflammatory response as well as play a neuroprotective role after TBI.
ADAM17, as an important inflammatory regulatory mediator, has been demonstrated to be regulated by a variety of factors and plays an important role in the inflammatory response of the central nervous system. 35 Zou and colleagues 34 found that the activation of S100B/RAGE signal pathway could regulate the expression, translocation, and activation of ADAM17 after TBI. In addition, glycinergic neurotransmission can also modulate ADAM17 expression, which in turn affects neuroinflammation. 36 Our study demonstrated that the TGF-β1/Smad pathway plays a crucial role in the regulatory role of ADAM17 in the M1/M2 phenotype polarization of microglia as well as the neuroinflammatory response. However, the underlying mechanism of how ADAM17 regulates the TGF-β pathway after TBI still needs to be further explored. The activity of TGF-β1/Smad pathway is closely associated with the number of TGF-βRs. 23,24 ADAM17 cleaves the extracellular domain of TGF-βRs, thereby reducing expression, and down-regulates signal transduction of the downstream pathways. 22 The results of this study suggest that the expression and activity of ADAM17 were increased in the cortical microglia after TBI. At the same time, the expression levels of TGF-βRI and TGF-βRII were notably reduced on the cell membrane, which also affects downstream intranuclear translocation and phosphorylation of Smads. In addition, it has been shown that ADAM17 can not only produce soluble transmembrane protein vasorin, 22 but also promote the overproduction of epidermal growth factor receptor ligands, 37 both of which can inhibit the TGF-β pathway.
Interestingly, controversy currently exists regarding the role of TGF-β in TBI. TGF-β can mediate neuroinflammation and neurodegeneration by activating proinflammatory cytokines and through the Smad protein pathway or transforming growth factor-beta-activated kinase-1 pathway. 38,39 However, our findings support a beneficial effect of TGF-β after TBI, which is consistent with the findings of Li and colleagues. 40 The results of this study suggest that inhibition of ADAM17 reduces the cleavage and detachment of TGF-βR on the cell membrane to increase expression levels. Consequently, TGF-β1 and TGF-βRII form a complex that induces intranuclear translocation of Smads and then activates the TGF-β1/Smad pathway. The activated pathway can regulate M1/M2 phenotype polarization of microglia and the neuroinflammatory response, which play significant roles in neuroprotection. Induction of M2 phenotype polarization in microglia can improve BBB permeability, reduces the degree of brain edema, inhibits neuronal apoptosis, and thus improves neurological function, 41 which is consistent with our conclusions. In addition, the improvement of BBB function can also limit immune cell exudation to a certain extent, inhibit the release of inflammatory factors, and improve the inflammatory microenvironment. 42 In short, inhibition of ADAM17 up-regulated the expression levels of TGF-βRs on the microglia membrane and boosted the formation of TGF-β1/TGF-βR complexes, while inducing intranuclear translocation of Smads, activating the TGF-β1/Smad pathway, and promoting switching to the M2 phenotype of microglia, ultimately exerting a neuroprotective effect.
However, there are some limitations in this study. Although our study has confirmed that the M1/M2 polarization phenotype of microglia is closely related to the outcome of neuroinflammation after TBI, the simple dichotomy of microglia into M1 and M2 phenotypes is currently considered to not fully account for the complex post-injury environment. Therefore, future studies should investigate different microglial subtypes in more depth after TBI. In addition, future studies involving ADAM17 knockout mice are warranted to explore the mechanism underlying the effect of ADAM17 on inflammation after TBI. Meanwhile, primary microglia cells are also needed to confirm the direct effects of ADAM17 on microglial activation.
Conclusions
In summary, M1 phenotype polarization of microglia along with neuroinflammation plays a significant role in neural damage after TBI injury. ADAM17, which is highly expressed on the membrane of microglia, mediates shedding of the extracellular domain of TGF-βRs on the cell membrane, thereby decreasing the quantity of receptors. In addition, ADAM17 negatively regulates the TGF-β1/Smad pathway to promote polarization of the M1 phenotype and inhibit the M2 phenotype of microglia, resulting in the aggravation of LPS-induced neuroinflammation and neuronal damage. Inhibition of ADAM17 can up-regulate the expression of TGF-βRs, which are located on the membrane of microglia, and then activate the TGF-β1/Smad pathway, promote the polarization of the M2 phenotype and suppress LPS-induced neuroinflammation in vitro. In addition, inhibition of ADAM17-induced changes in microglial polarization ultimately effectively improves neurological function in TBI mice.
Footnotes
Acknowledgments
We thank International Science Editing for editing this manuscript. Our article was previously submitted to Research Square as a preprint (DOI: 10.21203/rs.3.rs-171893/v1).
Authors' Contributions
X.C.: Conception, design, and manuscript preparation.
J.Y.: Experimental studies, data analysis, and manuscript editing.
J.L.: Experimental studies, and manuscript preparation.
L.L.: Experimental studies, data acquisition, and data analysis.
Y.C.: Experimental studies and data acquisition.
Y.L.: Statistical analysis and manuscript review.
W.F.: Literature search and data analysis.
C.D.: Statistical analysis and manuscript editing.
D.K.: Definition of intellectual content, design, and manuscript preparation.
Funding Information
This work was supported by grants from the funds for the Quanzhou City Science and Technology Program of China (2022C030R, the Young and middle-aged backbone talent foundation of Fujian Provincial Commission of Health Construction (2020GGA058) and the Joint Funds for the innovation of science and Technology Fujian province (Grant number: 2020Y9033) from Dr. Xiangrong Chen.
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.