
Abstract
Repetitive mild traumatic brain injury (rmTBI, e.g., sports concussions) may be associated with both acute and chronic symptoms and neurological changes. Despite the common occurrence of these injuries, therapeutic strategies are limited. One potentially promising approach is N-methyl-D-aspartate receptor (NMDAR) blockade to alleviate the effects of post-injury glutamatergic excitotoxicity. Initial pre-clinical work using the NMDAR antagonist, memantine, suggests that immediate treatment following rmTBI improves a variety of acute outcomes. It remains unclear (1) whether acute memantine treatment has long-term benefits and (2) whether delayed treatment following rmTBI is beneficial, which are both clinically relevant concerns. To test this, animals were subjected to rmTBI via a weight drop model with rotational acceleration (five hits in 5 days) and randomized to memantine treatment immediately, 3 months, or 6 months post-injury, with a treatment duration of one month. Behavioral outcomes were assessed at 1, 4, and 7 months post-injury. Neuropathological outcomes were characterized at 7 months post-injury. We observed chronic changes in behavior (anxiety-like behavior, motor coordination, spatial learning, and memory), as well as neuroinflammation (microglia, astrocytes) and tau phosphorylation (T231). Memantine treatment, either immediately or 6 months post-injury, appears to confer greater rescue of neuroinflammatory changes (microglia) than vehicle or treatment at the 3-month time point. Although memantine is already being prescribed chronically to address persistent symptoms associated with rmTBI, this study represents the first evidence of which we are aware to suggest a small but durable effect of memantine treatment in mild, concussive injuries. This effect suggests that memantine, although potentially beneficial, is insufficient to treat all aspects of rmTBI alone and should be combined with other therapeutic agents in a multi-therapy approach, with attention given to the timing of treatment.
Introduction
As public health concerns surrounding traumatic brain injury (TBI) have increased, so has attention to the mechanisms underlying the acute symptoms, as well as long-term outcomes. Of particular interest has been repetitive mild TBI (rmTBI). Several lines of evidence suggest that rmTBI may be associated with long-term functional, structural, and molecular outcomes, including neurodegeneration. 1 –3 Pre-clinical work has elucidated acute changes that contribute to the post-injury signaling cascade, which includes glutamate-mediated excitotoxicity, 4,5 which appears to contribute to the clinical hallmarks of TBI. Although pre-clinical work and clinical evidence 5 suggest a pivotal role of excitotoxicity, clinical trials targeting this mechanism through the N-methyl-D-aspartate receptor (NMDAR) have not been successful. 6,7
Although several receptors for glutamate have been identified, the NMDAR is unique as its ion channels allow entry of calcium, in addition to sodium and potassium. 8,9 This calcium influx can increase intracellular Ca2+, activating the signaling cascade. 8,9 In TBI, this excess of intracellular Ca2+ indicates excitotoxicity. 4,5 It follows that NMDAR activation would be a therapeutic target.
Pre-clinical work has evaluated NMDAR activation in single, severe injuries, yielding promising data at acute time points. 4,10 –24 Yet, NMDAR antagonism has been used in the clinical setting for not only a single TBI 7,25 but also persistent symptoms following rmTBI, 7,26 –30 despite the paucity of data. Thus far, the NMDAR antagonist of choice is memantine, a drug used to mitigate chronic excitotoxicity and ameliorate symptoms in Alzheimer’s disease (AD). 31,32 Memantine is a particularly attractive option because, in addition to being approved by the Food and Drug Administration, it is noncompetitive and use-dependent, limiting side effects. 9,32,33
In consideration of this gap between pre-clinical research and clinical treatment, we conducted an initial study using a weight drop mouse model of rmTBI that results in behavioral deficits, as well accumulation of phosphorylated tau and chronic gliosis. 34 –39 In addition, in alignment with other pre-clinical work, we observed acute increases in cortical extracellular glutamate, with the ratio of extracellular glutamate/gamma-aminobutyric acid remaining higher 6 weeks post-rmTBI. 37 Our previous work revealed that early memantine treatment improved histopathological outcomes at 1 month. 38,39 We also observed reduced tau phosphorylation, glial activation, and long-term potentiation deficit, as well as normal NMDAR expression. 38,39 Despite these changes, no corresponding behavioral protection was observed. 38 Similar results were demonstrated in in vitro repetitive stretch injury. 40 Interestingly, recent work using a high-frequency head impact model demonstrated alterations in cognitive function as a result of glutamate-induced synaptic adaptation, with no gross histopathology. 41 In their model, memantine pretreatment prevented transcriptomic and electrophysiologic changes, as well as cognitive dysfunction up to 1 month after injury. 41
Although these acute effects of memantine treatment appear promising, from a clinical perspective, meaningful therapeutic intervention must improve long-term outcomes, especially when one considers the risk of progressive neurodegeneration. Similarly, although treatment immediately following rmTBI may be desirable, it is not necessarily feasible, especially in the case of former athletes with a history of head trauma and recent symptom onset. The potential for memantine treatment to improve outcomes when administered later remains an interesting, and relevant, question, with some support for chronic efficacy provided by work in AD. In this study, we aim to investigate (1) whether acute memantine treatment has long-term benefits and (2) whether delayed treatment following rmTBI is beneficial.
Materials and Methods
All experiments were approved by the Boston Children’s Hospital Institutional Animal Care and Use Committee and complied with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. Ninety-six (age 8 weeks) male C57BL/6 mice obtained from the Jackson Laboratories were used for these experiments and housed in a pathogen-free environment with inverse 12-h day–night cycles. Food and water were provided ad libitum.
Injury paradigm
Mice were randomized to either an rmTBI or a sham injury. 35,42 All mice were anesthetized for 45 sec using 4% isoflurane in oxygen. Anesthetized mice receiving an injury were placed prone on a Kimwipe and secured by the tail. The dorsal surface of the head was positioned under a hollow iron guide tube centered over bregma. A 54 g metal bolt was dropped through the tube from 42 inches, impacting the animal’s head and resulting in rotational acceleration through the Kimwipe. After the impact, mice were placed in a left lateral lying position to regain consciousness. Mice underwent a single injury daily for 5 consecutive days (rmTBI, n = 48; Fig. 1). A separate group underwent a sham injury (anesthesia exposures only) for 5 consecutive days (sham, n = 48). The duration of loss of consciousness was recorded daily and defined as the time from cessation of anesthesia until spontaneous righting occurred.
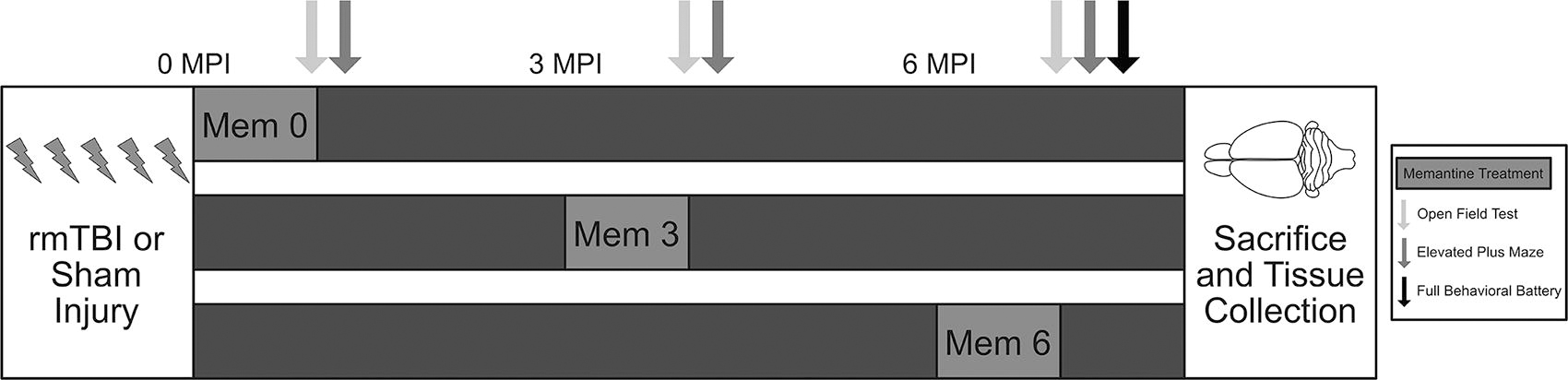
Experimental timeline. The experiment consisted of three cohorts (total: n = 96, per cohort: n = 32). To begin the experiment, animals received either sham injuries or repetitive mild TBI (five exposures/5 days; total: n = 48, per cohort: n = 16). From there, animals were randomized to receive either vehicle or memantine treatment for 1 month. Memantine 0 (Mem 0) received treatment or vehicle immediately following injury up until 1 month post-injury. Memantine 3 (Mem 3) received treatment or vehicle from 3 months post-injury until 4 months. Memantine 6 (Mem 6) received treatment or vehicle from 6 months post-injury until the 7-month time point. All cohorts were tested on the open field and elevated plus maze at 1 month, 4 months, and 7 months post-injury. Animals were also evaluated on rotarod and Morris water maze (full behavioral battery) at 7 months. Following completion of behavioral testing at 7 months, all cohorts were sacrificed and their brain tissue was collected for further analysis. Brain diagram has been adapted from an open-source drawing. 43
Memantine treatment
Mice were randomized to receive either memantine (20 mg/kg, in autoclaved water) or vehicle treatment (autoclaved water) via drinking water for 30 days. This dosing scheme is based on prior literature that demonstrated 20–30 mg/kg/day oral administration resulted in a clinically relevant plasma drug level of ∼1 μm. 44,45 Furthermore, oral administration was selected to match the clinical route of memantine administration. Treatment began at one of three time points post-injury (Fig. 1). One cohort received treatment immediately following the last injury (mem 0, n = 32), another received treatment at 3 months (mem 3, n = 32), and the final group received treatment at 6 months (mem 6, n = 32). Within each cohort, mice were randomized to four groups: sham and vehicle (n = 8, sham vehicle), rmTBI and vehicle (n = 8, rmTBI vehicle), sham and memantine (n = 8, sham mem), and rmTBI and memantine (n = 8, rmTBI mem).
Behavioral testing
All mice completed a battery of behavioral assessments at 1, 4, and 7 months post-injury (Fig. 1). Mice were assessed on the open field and elevated plus maze at all three time points and on the rotarod and Morris water maze (MWM) at 7 months post-injury. All behavioral testing was conducted by investigators blinded to experimental status.
Anxiety-like behavior and locomotion were assessed using the Open Field (OF) test. 46,47 The arena consisted of a plastic transparent box (40 × 40 × 30 cm) with infrared beams crossing the x- and y-axes to plot movements. The arena was split into two concentric square regions: the peripheral region (outer ring of 10 cm width) and center region (inner 20 × 20 cm). Each mouse was placed in the center of the arena facing the back wall and allowed to roam for 10 min while movements were recorded using the MotorMonitor II tracking software (Kinder Scientific). The time spent in each zone and distance traveled were measured. Time spent in the center region constituted more explorative behavior and less anxiety. Locomotor activity was defined as the total distance traveled. The arena was cleaned between each individual trial using Peroxigard wipes (Virox Technologies Inc.).
Anxiety-like behavior was assessed using the elevated plus maze (EPM). 35,36,47 The EPM apparatus (Lafayette Instruments) was an elevated (85 cm) plus-shaped platform that consisted of two open and two closed arms (30 × 5 cm) extending out opposite of each other, with a square-shaped center (decision zone). Each mouse was placed in the decision zone of the maze facing a closed arm and allowed to roam for 5 min. The Noldus Ethovision XT 11.5 video tracking system recorded the total time spent in the three zones and entries into the decision zone. The percent time spent in the open versus closed arms was used as a metric of anxiety-like behavior, with more time spent in the closed arms signifying greater anxiety. The maze was cleaned between trials using Peroxigard wipes.
Motor function was assessed using the rotarod. 36 The device, a ROTO-ROD Series 8 (IITC), consists of several lanes with rotating drums (3.18 cm diameter). Testing was conducted over 3 days. Habituation/training was conducted on day 1. Each animal was placed on the drum and performed the task for 5 min (4 Revolutions per minute (RPM), no acceleration). If the animal fell off, it was gently placed back on to the drum to continue. Testing was conducted on days 2 and 3 (four trials/day). For each test, the animal was placed on a rotating drum (3 RPM, acceleration of 1 RPM/sec), and the latency to fall was recorded.
Hippocampal-dependent spatial learning and memory were assessed using the MWM paradigm. 36,48,49 MWM testing was conducted in a white pool (83 cm diameter, 60 cm depth) that was filled with water (29 cm depth). Water temperature was maintained at ∼24°C, and a round, plastic target platform (10 cm diameter) was positioned ∼1 cm below the water surface. Several highly visible maze cues were located on the pool walls. During hidden and visible platform trials, mice were randomized to one of four starting quadrants. At the start of each trial, each mouse was placed in the tank facing the wall, given 90 sec to locate and mount the platform and remain on it for 5 sec. The latency for the animal to mount the platform was recorded and used as a measure of learning/memory, with a shorter latency indicating better performance. Mice that failed to find and mount the platform were guided to it and given 10 sec to remain on it. Each mouse was subjected to one trial a day (two runs per trial) for 4 days. On the fourth day, following the hidden trial, a probe to test recall for the platform location was performed. For the probe, the platform was removed and the animal was given 60 sec to explore. Noldus Ethovision XT 11.5 software was used to track the time spent in the quadrant of the pool where the platform was previously (target quadrant). The time spent in the target quadrant was used as a measure of spatial memory; mice with deficits spend less time in the target quadrant. On the fifth day, the animal performed two visible trials followed by a final probe trial. For visible platform trials, a red reflector marked the top of the platform, and each mouse was again given 90 sec to locate it. Latency to the platform on the visible trials was recorded to account for the effect of non-learning-based factors.
Histopathological analyses
All mice were euthanized at 7 months post-injury via cardiac perfusion (Fig. 1). 38,50 Before the perfusion, animals were anesthetized with ketamine (100–120 mg/kg) and xylazine (100 mg/kg) in sterile saline via an IP injection. Mice were placed abdomen up on a surgical pad. A vertical incision was made from the abdomen to thorax, and the chest flap was secured to allow access to the heart. A needle was inserted into the left ventricle to draw blood. A butterfly needle was then placed into the left ventricle and the right atrium was cut, followed by perfusion with phosphate saline buffer. The brain was removed, and the left hemisphere was post-fixed in 4% paraformaldehyde solution, and the right hemisphere was dissected and stored at −80°C. The blood was centrifuged at 5000 RPM for 5 min, and then the serum was collected and stored at −80°C.
Immunohistochemistry
Following post-fixation, brains were moved to 30% sucrose. They were flash frozen and sliced at 50 μm using a Leica CM1950 Cryostat (Leica Biosystems). Several frontal sections and several caudal sections were stained using anti-IBA1 Rabbit (1:200, WAKO), anti-GFAP Rabbit (1:600, Cell Signaling), and anti-Tau phospho-T231 Rabbit (1:400, AbCam). For tau T231, antigen retrieval was performed in advance of staining, via incubation in antigen unmasking solution (Vector Laboratories) in an 80°C water bath. Briefly, sections were treated with hydrogen peroxide and incubated in blocking solution (3% normal goat serum) before being incubated in primary antibody. The following day, sections were washed and incubated sequentially with appropriate secondary antibody, Vectastain Elite ABC Kit (Vector), and diaminobenzadine and mounted with Permount (Thermo-Fisher Scientific). Slides were imaged at 40× using the MoticEasyScan (Motic Microscopes).
Tissue lysis and enzyme-linked immunosorbent assays
Both cortical and hippocampal tissues were lysed in RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM sodium chloride, 1 mM ethylenediaminetetraacetic acid, 1% NP-40, 1% sodium deoxycholic acid, 0.1% sodium dodecylsulfate, 1 mM phenylmethylsulfonyl fluoride, protease inhibitor cocktail) with phosphatase inhibitor cocktail (Santa Cruz Biotechnology).
Tauopathy, the formation of insoluble tau aggregates, is a hallmark of several neurodegenerative disorders 51,52 and has been reported following rmTBI. Not only is tau phosphorylation an early event in tauopathy, 53 –55 but neurotoxic effects of soluble tau have also recently gained attention. 56 –59 Soluble phospho–tau T231 and total tau were measured in the hippocampus using a MULTI-SPOT enzyme-linked immunosorbent assay (ELISA; Meso Scale Diagnostics). The assay was run according to the manufacturer’s guidelines. Briefly, the plate was incubated with Blocker A solution, followed by incubation of calibrators and samples. Next, detection antibody solution was incubated. Read Buffer T was added to the plate, and it was analyzed using an Meso Scale Discovery instrument. A calibration curve was generated to calculate sample concentrations.
Single molecule array quantification
To investigate chronic inflammation, 60,61 IL-6 in the hippocampus was quantified using the mouse IL-6 Discovery kit for Simoa HD-X (Quanterix Corporation). 62 IL-6, while capable of serving both pro- and anti-inflammatory roles, was selected owing to evidence suggesting chronically elevated levels following TBI are correlated with worse outcomes. 63 –65 Before the assay, lysates were diluted with Tris-buffered saline to reach a final total protein concentration of 0.6 mg/mL. All reagents were prepared according to the manufacturer’s guidelines. Samples were thawed, reconstituted, and centrifuged at 10,000 g for 5 min. Calibrators were run neat, and controls and samples were run using a 4× dilution. The output data were converted from pg/mL to pg of IL-6/mg of total protein. The assay had a dynamic range of 0–700 pg/mL, with all samples falling above the lower limit of quantification (0.120 pg/ml).
Immunohistochemistry analysis
IBA1 was quantified using a cell counting technique in ImageJ (NIH). Briefly, regions of interest (ROIs) were drawn (cortex, corpus callosum, hippocampus, dentate gyrus (DG), cornu ammonis 3 (CA3), and CA1). The thresholder was then used to highlight only the IBA1+ cells. The smallest cell size was determined, and particle analysis was used to count the number of cells. To better understand morphological changes, in addition to cell density, QuPath was used as previously reported. 50,66,67 QuPath is a semi-automated image analysis software that allows for high-throughput measurement of immunohistochemistry. Although it relies on similar principles and generates output comparable with that of ImageJ and other more traditional methods, it leverages algorithms to increase the accuracy and efficiency of the analyses. Activated microglia feature both a larger cell body and an increased number of processes 68 and, in turn, cover a larger area, constituting a larger number of positive pixels. To train the pixel classifier, regions of the image were identified as positive or negative. Performance was confirmed using a classification map, and retraining of the classifier was performed as appropriate. To quantify potential changes in morphology, the ratio of positive pixels to cell density was calculated.
Glial fibrillary acidic protein (GFAP) and tau T231 were also analyzed using the QuPath pixel classifier. Similar ROIs were used for GFAP and T231, with the addition of the thalamus for T231.
Tissue atrophy, a measure of neurodegeneration, was investigated in the cortex, corpus callosum, and hippocampus using the Motic DSAssistant software (Motic Microscopes). Cortical thickness was measured in the motor cortex and two locations within the sensory cortex, with these values being averaged. A similar procedure was conducted for the corpus callosum and the hippocampus (using area instead of thickness).
All ROIs were drawn using the Allen Mouse Brain Atlas (2004). 69
Statistical analysis
Data are presented as mean ± standard error of the mean.All histological, protein quantification, and atrophy measures reflect fold change values normalized using the sham vehicle group. To capture subtle changes across measures and better characterize improvements on aggregate, composite z-scores were calculated for IBA1, GFAP, and T231 expression, as well as atrophy and behavior. Composite scoring has several advantages. In addition to decreasing type I error by reducing the number of measures, it also improves sensitivity to changes in outcomes. Furthermore, composite scores tend to be more highly correlated with biomarkers and are better at predicting disease progression in conditions such as AD. Values were normalized using sham vehicle performance and then weighted equally. For the composites, a higher score indicated worse condition. Continuous variables were compared looking at the effects of injury (rmTBI vs. sham), treatment timing (vehicle, mem 0, mem 3, mem 6), and the interaction between injury and treatment timing using analysis of variance (ANOVA) and Tukey post-hoc tests with the rmTBI vehicle animals as the referent group. For behavior, analyses were performed both within a single time point and across time points using a repeated measures ANOVA. A cutoff of p < 0.05 was used for statistical significance. A cutoff of p ≤ 0.1 was used to detect potential trends in the data. All statistical analysis was performed in R (R Core Team, 2020).
Results
Composite scoring
Composite scores were calculated to gain an understanding of the global effects of memantine. The composite score for behavior at 7 months revealed an effect of injury (Fig. 2A; p < 0.00001), with rmTBI animals exhibiting greater deficits than the shams. The microglial composite score revealed an effect of injury and an interaction between injury and treatment but no effect of treatment alone (Fig. 2B; ps < 0.01). Composite IBA1 expression was higher in rmTBI animals. Post-hoc analysis of the interaction indicates a benefit of treatment immediately and at 6 months post-injury (vs. rmTBI vehicle, p < 0.001). The astrocytic composite score demonstrated a trend toward an effect of injury, with rmTBI animals scoring higher than their sham counterparts (Fig. 2C; p = 0.05294). The composite score for T231 also trended toward an increase in injured animals (Fig. 2D; p = 0.09797). The composite scores for the atrophy measure revealed no effects (Fig. 2E).
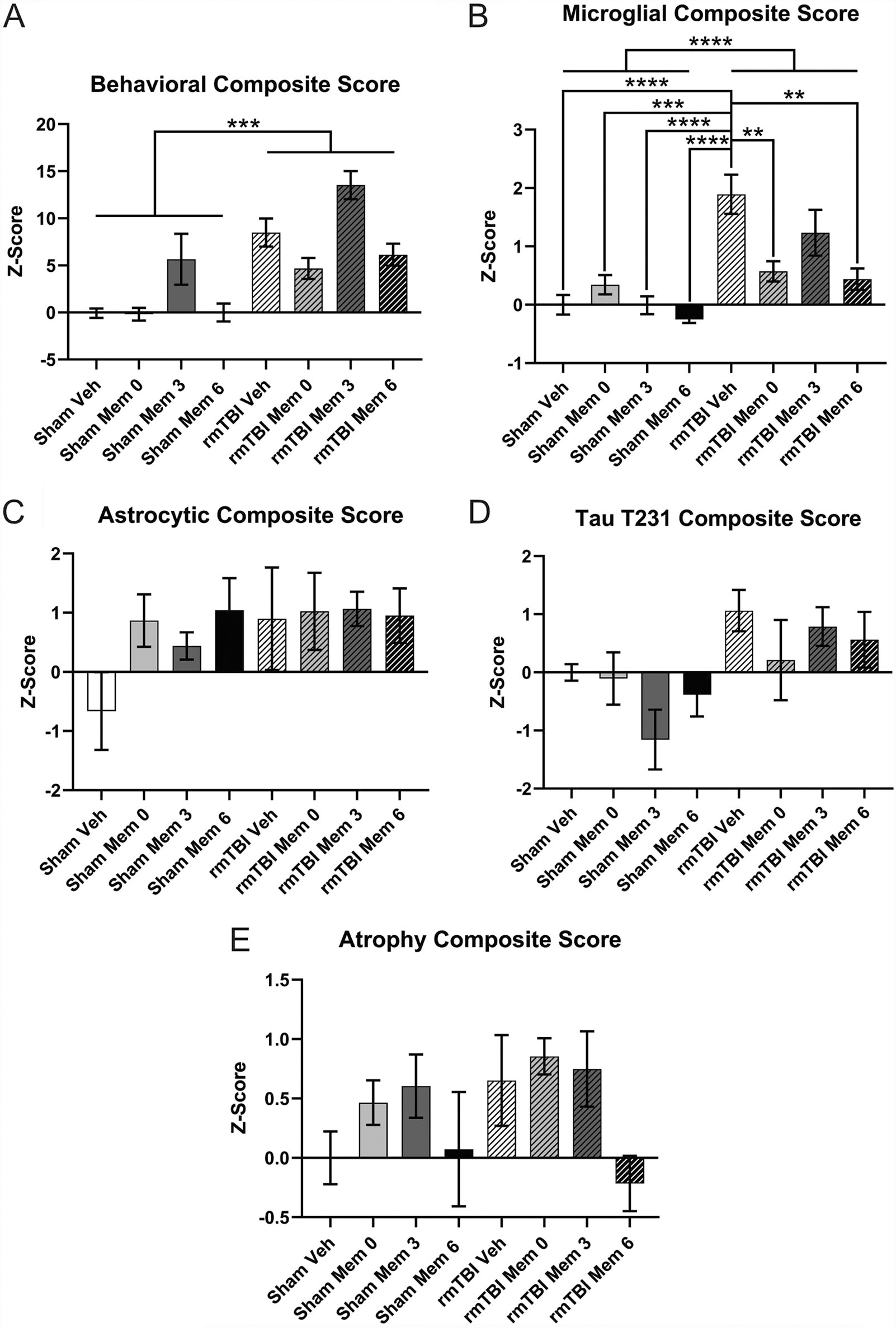
Composite scoring.
Loss of consciousness
For loss of consciousness, there was an effect of injury and an interaction between injury and day but no effect of day alone (Fig. 3A; ps < 0.0001). rmTBI animals took significantly longer to right themselves.
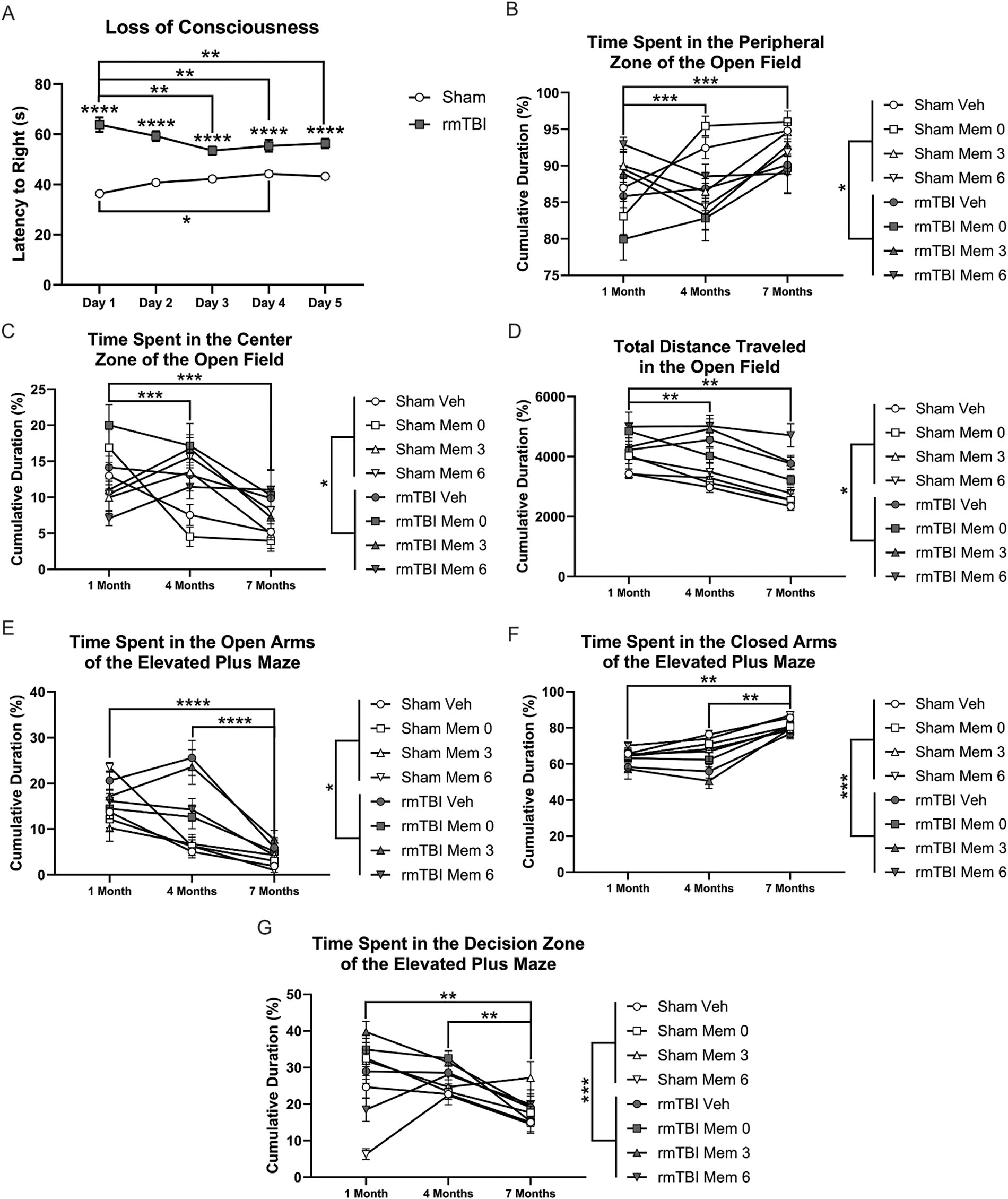
Behavioral outcomes—loss of consciousness, open field, and elevated plus maze.
Behavioral outcomes
Analysis of the time spent in the peripheral zone of the OF revealed an effect of injury and of time point, as well as injury–treatment, injury–time point, and treatment–time point interactions (Fig. 3B; ps < 0.05). rmTBI animals spent less time in the periphery, which is indicative of decreased anxiety-like behavior. Post-hoc tests demonstrated a significant difference between performance at 1 month and either 4 or 7 months (Fig. 3B; ps < 0.001), regardless of injury or treatment, with all groups spending less time in the periphery at the earlier time point. This likely represents a repeat testing effect. In terms of time spent in the center zone, there was also an effect of injury and of time point, as well as injury–treatment, injury–time point, and treatment–time point interactions (Fig. 3C; ps < 0.05). Injured animals spent significantly more time in the center than sham animals, demonstrating decreased anxiety-like behavior. Similarly, time spent in the center zone decreased across testing time points (Fig. 3C; vs. 1 month: ps < 0.001), with all animals spending greater time there at 1 month. Analysis of locomotor activity demonstrated main effects of injury and time point, in addition to injury–time point and treatment–time point interactions (Fig. 3D; ps < 0.05). Injured animals traveled significantly further, suggesting increased exploration or hyperactivity. Similarly to what was observed in terms of time spent in the zones, animals traveled significantly further at 1 month (Fig. 3D; vs. 4 and 7 months: ps < 0.01).
Analysis of the time spent in the open arms of the EPM indicated an effect of injury and of time point and injury–time point and treatment–time point interactions (Fig. 3E; ps < 0.05). Injured animals spent significantly more time in the open arms than sham animals, indicating decreased anxiety-like behavior. Post-hoc tests also revealed significantly greater time spent in the open arms of the EPM across all groups at 1 month (Fig. 3E; vs. 4 and 7 months: ps < 0.0001), which likely represents an artifact of repeated testing. Analysis of the time spent in the closed arms also demonstrated an effect of injury and time point as well as an injury–time point interaction (Fig. 3F; ps < 0.001). Regardless of treatment, animals that were injured spent significantly less time in the closed arms. In regard to time point, all animals spent significantly greater time in the closed arms at 7 months (Fig. 3F; vs. 1 and 4 months: ps < 0.01). The number of entries into the decision zone was affected by injury, treatment, and time point, in addition to a treatment–time point interaction (Fig. 3G; ps < 0.001). rmTBI animals entered the decision zone more frequently. Post-hoc tests demonstrated that animals had significantly fewer entries into the decision zone at 7 months (Fig. 3G; vs. 1 and 4 months: ps < 0.01).
For rotarod testing, there was an effect of injury and of trial, as well as a treatment–trial interaction (Fig. 4A; ps < 0.001). Injured animals had a significantly shorter latency to fall, indicating worse performance. Although the overall latency to fall was longer on day 2, this was likely driven by the improvement of the rmTBI animals. Sham animals performed comparably on both trials.

Behavioral outcomes—rotarod and Morris water maze.
Analysis of the hidden trials of the MWM revealed effects of injury, treatment, and trial, as well as an injury–trial and injury–treatment–trial interaction (Fig. 4B; ps < 0.001). rmTBI animals had a significantly longer latency to the platform, indicating deficits in learning or memory. The effect of trial demonstrates a significantly longer latency to the platform on hidden one (Fig. 4B; vs. two, three, four: ps < 0.001) and two (Fig. 4B; vs. four: p = 0.01). Analysis of the probes revealed effects of treatment and trial and a treatment–trial interaction (Fig. 4C; ps < 0.01). Animals spent significantly more time in the target quadrant during the first probe trial. On the visible trials, there was an effect of injury (Fig. 4D; p < 0.0001). Injured animals took significantly longer to reach the visible platform. This difference indicates that the observed deficits may not be solely due to memory and may reflect visual dysfunction. 70,71
Immunohistochemistry
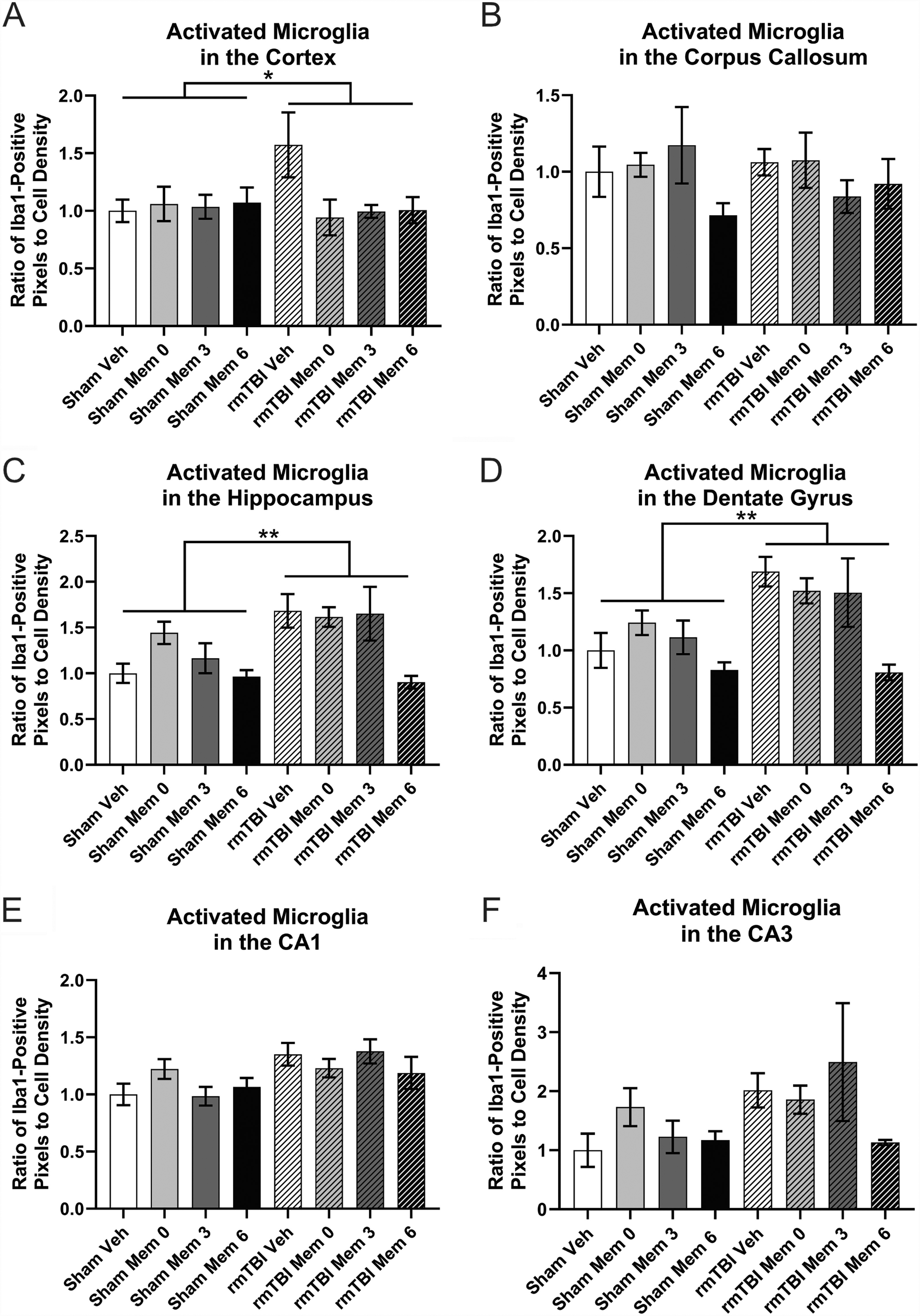
Both the density and morphology of microglia, measured by IBA positivity, were quantified. Chronic neuroinflammation following TBI is associated with worse outcomes, making attenuation of this effect beneficial. 63,72,73 Microglial density in the cortex was significantly increased as a result of injury (Fig. 5A; p < 0.0001), with no treatment-related effects. Within the corpus callosum, there was a significant increase in microglia in response to injury and an injury–treatment interaction (Fig. 5B; ps < 0.01), as well as a trend toward an effect of treatment (p = 0.078). There were more IBA1+ cells in the corpus callosum of animals who received treatment at 6 months regardless of injury (Fig. 5B; vs. vehicle: p = 0.0648028; vs. mem 0: p < 0.01). The density of microglia in the hippocampus also increased post-injury (Fig. 5C; p < 0.01), but there was no effect of treatment. Similarly, in the dentate gyrus of the hippocampus, there was a trend toward an injury-induced increase in microglia (Fig. 5D; p = 0.05176). In the CA1, another subregion, there was also an effect of injury (Fig. 5E; p < 0.01), with greater IBA1 expression in the rmTBI group, and a trend toward an injury–treatment interaction (p = 0.087131). This trend might indicate benefits of immediate memantine treatment (vs. rmTBI vehicle: p < 0.05) or at 6 months post-injury (vs. rmTBI vehicle: p = 0.10). Finally, in the CA3, there was a trend toward an effect of treatment (Fig. 5F; p = 0.09147), with a decrease in IBA1+ cells in response to treatment either immediately (vs. vehicle: p < 0.05) or at 6 months post-injury (vs. vehicle: p = 0.06). Although rmTBI leads to an increase in microglia, there is also a repeated trend toward benefits of memantine treatment, particularly when administered immediately, as well as at 6 months.

Immunohistochemistry for IBA1+ microglia count.
Analysis of the microglial morphology, as measured by the average positive pixels per cell, demonstrated a significant effect of injury (Fig. 6A; p < 0.05) and a trend toward an interaction between injury and treatment (p = 0.08685) in the cortex. Post-hoc tests revealed an increase in the average pixels as an effect of injury, as well as a trend toward significant improvement with immediate treatment following injury (vs. rmTBI vehicle, p = 0.0842253). Within the corpus callosum, no significant differences were observed (Fig. 6B). There was both an effect of injury (Fig. 6C; p < 0.01) and a trend toward an injury–treatment interaction (p = 0.09905) in the hippocampus. The ratio significantly increased as a result of injury, with a potential further increase resulting from treatment at 6 months (p < 0.05). Similar injury effects were observed in the dentate gyrus (Fig. 6D; p < 0.01) and CA3 (Fig. 6F, p = 0.09593), but no changes were observed in the CA1 (Fig. 6E).
Immunohistochemistry for IBA+ microglia morphology.
GFAP staining for astrocytes revealed a significant increase in cortical astrocytes due to injury (Fig. 7A; p < 0.05) but no effect of treatment. No differences between groups were observed in the corpus callosum (Fig. 7B). Analysis of the hippocampus revealed an effect of injury (Fig. 7C; p < 0.05), as well as a trend toward an injury–treatment interaction (p = 0.05231). Greater GFAP expression was found in the hippocampus following rmTBI. A similar pattern was observed in the hippocampal subregions, with trends toward an effect of injury (Fig. 7D, Dentate gyrus (DG): p = 0.07404; Fig. 7E, CA1: p = 0.05532; Fig. 7F, CA3: p = 0.09396) and no treatment-related changes.
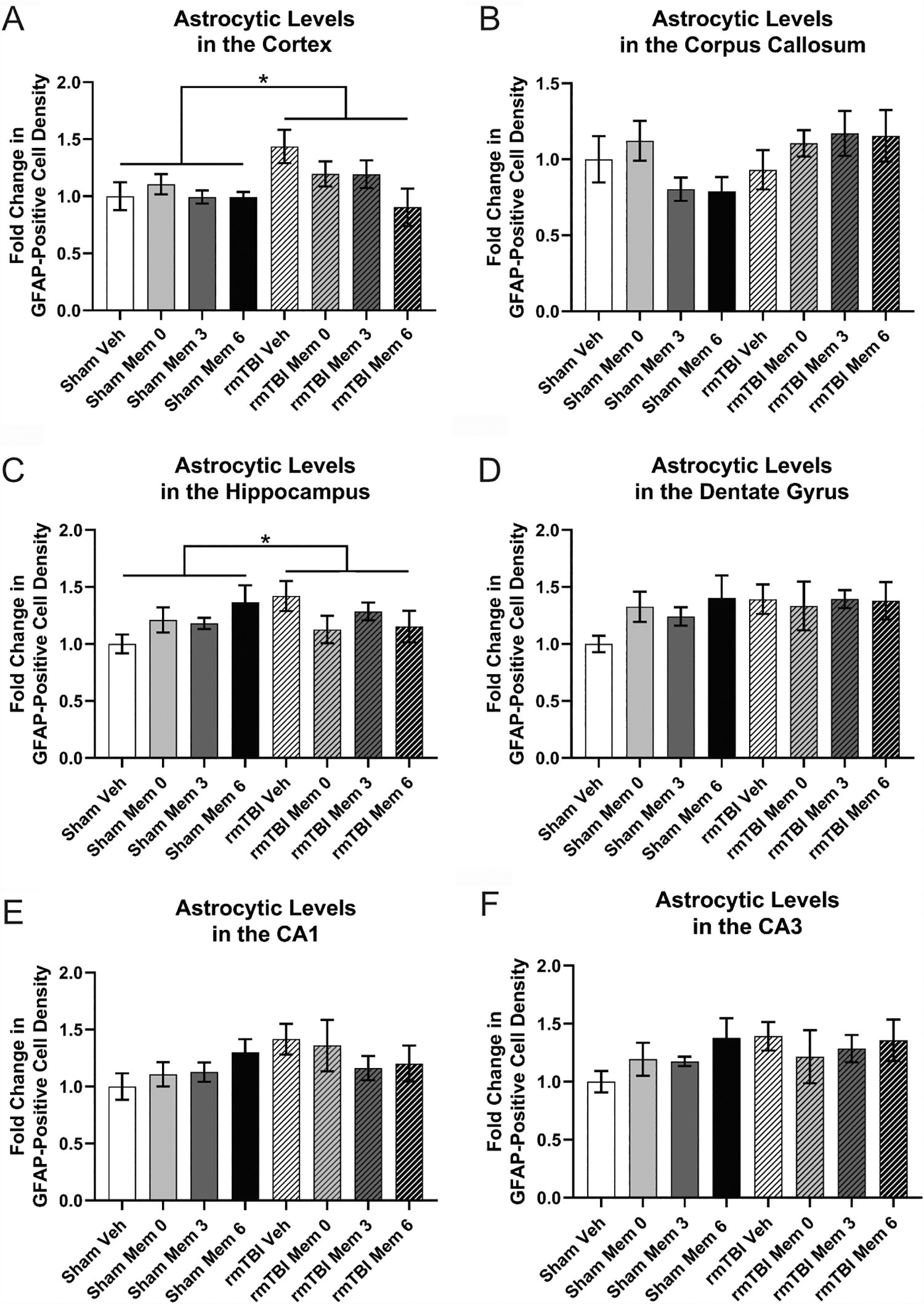
Immunohistochemistry for GFAP+ astrocytes.
Tau phosphorylation, as measured by positive staining for the T231 isoform, revealed no effects of injury, treatment, or interaction in the cortex (Fig. 8A), corpus callosum (Fig. 8B), or the hippocampus (Fig. 8C). T231 abundance did differ as a function of the hippocampal subregion examined. Although there were no changes in phosphorylated tau in the DG (Fig. 8D) or CA1 (Fig. 8E), T231 did increase owing to injury in the CA3 (Fig. 8F; p < 0.05). There was also an effect of injury in the thalamus (Fig. 8G; p < 0.01), with elevated T231 in the rmTBI groups. There were no clear benefits of treatment regarding injury-induced phospho–tau.

Immunohistochemistry for T231 tau phosphorylation.
Tau ELISAs
Both total tau (Fig. 9A) and T231 (Fig. 9B) levels in the hippocampus, as measured by ELISA, are unaffected by injury but do change as a function of treatment (ps < 0.05) and an interaction between injury and treatment (ps < 0.05). Similarly, the ratio of T231–total tau in the hippocampus, which serves as a measure of the relative abundance of phospho–tau, revealed no injury effect but did demonstrate an effect of treatment and injury–treatment interaction (Fig. 9C; ps < 0.05).

ELISA measures of total tau and phosphorylated tau in the hippocampus.
Inflammatory analyses
IL-6 levels were investigated in the hippocampus, which revealed no changes, although there was a trend toward an effect of treatment (p = 0.06823).
Atrophy measures
No atrophy was observed in the cortex (Fig. 10A), corpus callosum (Fig. 10B), or hippocampus (Fig. 10C; p = 0.09588), although there was a trend toward decreased hippocampal volume in response to rmTBI.
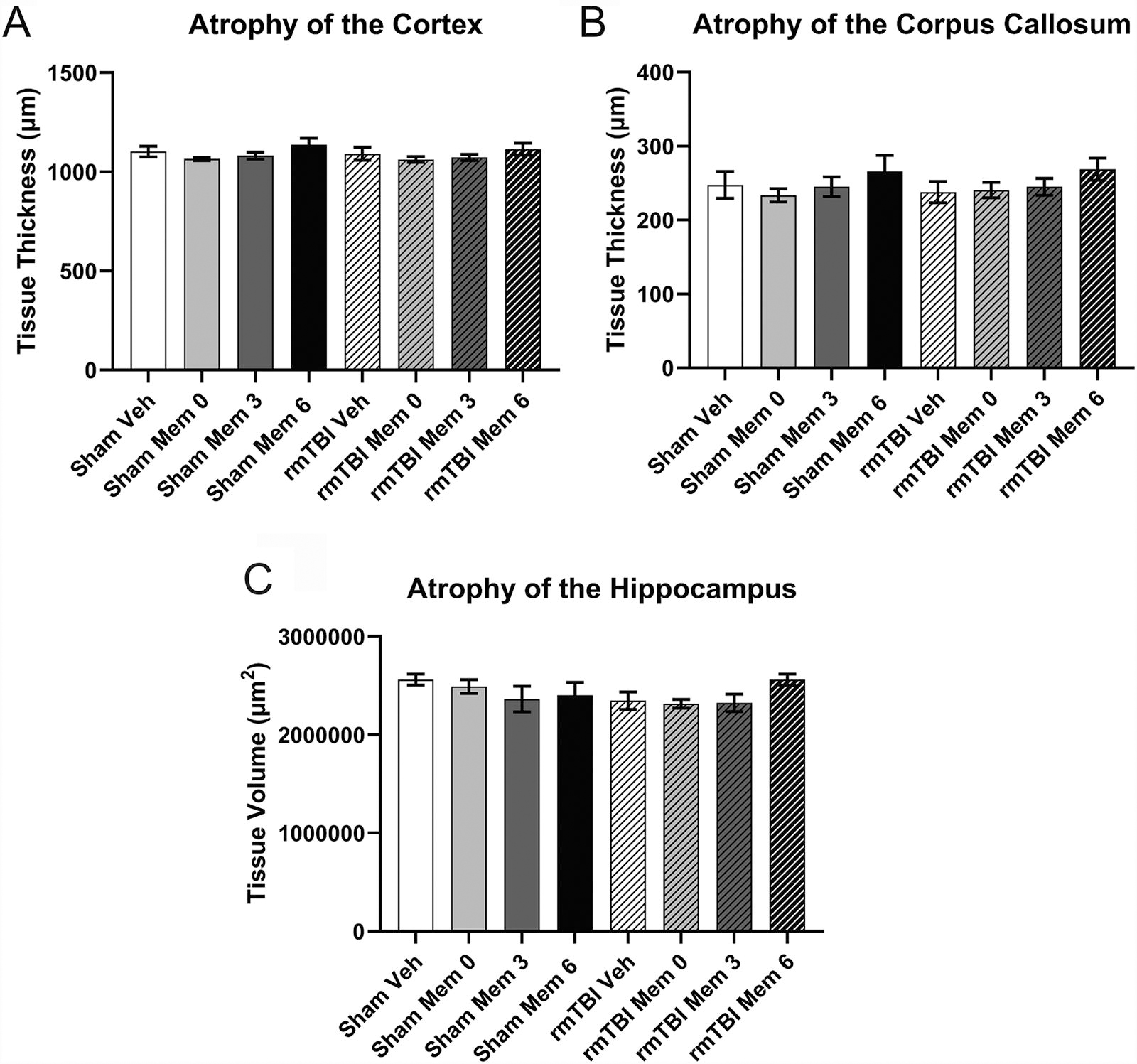
Atrophy.
Discussion
We found that an acute course of memantine treatment following rmTBI offers a modest improvement in histopathology, particularly microglia, but this effect depends on the timing of treatment. Our weight drop rmTBI results in chronic changes in behavior, as well as neuroinflammation (microglia in cortex, corpus callosum, and hippocampus; astrocytes in cortex and hippocampus) and tau phosphorylation (T231 in CA3 and thalamus). Memantine treatment, either immediately or 6 months post-injury, appears to confer greater rescue of neuroinflammatory changes than vehicle or treatment at 3 months. Although memantine is already being prescribed chronically to address persistent symptoms associated with rmTBI, 7,26 –30 this study represents the first evidence we are aware of to suggest a small but durable effect of treatment after mTBIs. This suggests that memantine, although potentially beneficial, is insufficient to treat all aspects of rmTBI alone and should be combined with other interventions in a multi-therapy approach.
Microglial levels in the cortex, corpus callosum, and hippocampus remained elevated 7 months after injury. Furthermore, analysis of microglial morphology suggests that injury also results in more chronically activated microglia. Recurrent trends across brain regions suggest benefits of memantine treatment, particularly when administered immediately or at 6 months post-injury, which was supported by a significant improvement in the composite score. These results suggest that memantine, especially when provided during glutamatergic excitotoxicity, is capable of modulating microglial behavior. Both neuronal death and excess glutamate alone induce neuroinflammation. 74 Although astrocytes are viewed as the primary regulators of glutamate homeostasis, 75 microglia are known to express glutamate receptors 75 –77 and transporters upon entering their reactive state. 78 In fact, microglia release several cytokines in response to glutamate receptor activation, further exacerbating neurotoxicity and negatively affecting myelination. 79 –81 It is believed that the NMDAR plays a pivotal role in this response. 82 Therefore, the benefits observed in this study could represent direct action on the microglia or indirect action via protection of the neurons. There is evidence, in LPS-induced models, that memantine can act directly on microglia to confer neuroprotection. 83,84 It is likely that both direct and indirect effects of memantine are responsible for the observed outcomes.
A trend toward a significant effect of injury on the astrocytic composite score at 7 months was observed as well as subtle differences within individual brain regions. GFAP expression is significantly higher in the injured cortex and hippocampus, but no changes were observed in the corpus callosum or hippocampal subregions. As there was not a robust injury effect on chronic GFAP expression, it is difficult to determine the effect of treatment. Although astrocytes may accumulate chronically as an astroglial scar following more severe, focal injury, generally, astrocytosis tends to resolve, 85,86 whereas microglial activity can be persistent and prolonged. 85,87 This might explain the differences between the cell types observed here. There is also variability among shams, further complicating our interpretations. There is evidence that memantine may act through a novel astrocyte-mediated neurotrophic effect, 83 increasing the expression of glial-derived neurotrophic factor (GDNF), 83 which might influence astrocyte behavior in the absence of injury. 88 Future studies investigating memantine treatment should examine GNDF levels as a potential astrocyte-derived mechanism.
We observed injury-induced increases in T231 phosphorylation. This was evident within the CA1 and thalamus specifically, with further support offered by the composite score. Memantine treatment did not affect these changes. An effect of treatment and injury–treatment interaction was observed in ELISA-based measurements of both soluble total and phospho–tau, although the potential relevance of this pattern is difficult to determine. The discrepancy between the immunohistochemical and ELISA results may be due to the measurement of soluble tau alone via ELISA. Tau phosphorylation is observed following TBI and, in some cases, accumulates and aggregates creating the insoluble tangles associated with chronic traumatic encephalopathy and other tauopathies. 53 –55,89 The exact mechanism underlying TBI-induced tau pathology is unclear, although it is known that tau phosphorylation negatively affects microtubule stability and axonal transport, 90,91 which is also disrupted by axonal injury, 92,93 and there is a bidirectional relationship between tau phosphorylation and neuroinflammation. 94 –98 As such, memantine may influence tau accumulation through modulation of neuroinflammation and microglia. In addition, tau phosphorylation is associated with synaptic dysfunction, excitotoxicity, and increased NMDAR transmission. 99,100 In models of AD, evidence suggests that memantine treatment can protect against beta-amyloid toxicity by attenuating tau phosphorylation. 101,102 This supports a role for chronic memantine treatment to mitigate the effects of toxic tau.
We did not observe improvements in behavior following memantine treatment, mirroring the results of our acute study. 38 The data must be interpreted with substantial caution given recent reports, including our own, 70,71 that reveal deficits in vision resulting from various rodent models. 103 –106 It is, therefore, difficult to determine what contribution vision loss has in the behavioral deficits observed, as opposed to TBI-related cognitive dysfunction. Ultimately, investigation either in a TBI model that preserves vision or via a vision-independent behavioral test will be important in understanding potential functional improvements.
It is notable that the protection provided by memantine treatment appears to be both pathology-specific and time-dependent. It should be acknowledged that neuropathological outcomes were investigated only at a single chronic time point, which was selected owing to our initial studies demonstrating efficacy at 1-month post-injury, 38,39 but these measures do not account for the complex onset and progression of pathology. Although immediate and 6-month post-injury treatments appear to confer benefits, treatment at 3 months did not. The strongest effect was observed in the immediate treatment group. Excitotoxicity is believed to occur acutely and persist subacutely following injury. 4,5,37 Mechanistically, it would follow that memantine treatment initiated immediately, and sustained for 1 month, would align with this window of vulnerability, partially blocking Ca2+ influx and limiting activation of the secondary injury cascade. It is interesting that we also observe benefits of treatment at six months, but not at three months. Although this may indicate a window in which NMDAR blockade is disadvantageous, it may also represent an artifact of experimental design. Although both groups received treatment for a month, the cohort that received memantine at 6 months post-injury received treatment just before the 7-month behavioral testing and investigation of histological outcomes, as opposed to the 3-month cohort that was further removed from the cessation of treatment. This might suggest that treatment begun later is effective, but not durable, in an acute course and that, if treatment is initiated chronically, it must be sustained to confer lasting benefits. Although we are limited in our ability to distinguish between these alternatives, it provides strong evidence emphasizing the importance of timing in treatment for rmTBI. Future work is needed to determine if sustained treatment is necessary chronically.
Although the post-traumatic neurometabolic cascade is not fully understood, glutamatergic excitotoxicity is believed to represent a significant driver of secondary injury, 4,5,107 –109 which may be the impetus of the clinical manifestations of mTBI. Despite the perceived importance of excitotoxicity, the effect of memantine treatment reported here is modest at best. We suggest several reasons for this. First, notable variability can be seen in certain outcomes, which may indicate a lack of statistical power. As previously reported, our injury paradigm results in a highly replicable rmTBI. 34 –39,42,50,110 The variability may represent the aging process magnifying the inherent variability associated with animal studies. Second, memantine is a noncompetitive and use-dependent NMDAR antagonist, 9,32,33 which is why it is safe, well-tolerated, and has few side effects. Although these features are advantageous, its noncompetitive nature means it is not particularly potent and, in cases of high glutamate flux, such as TBI, 4,5,107,108 may not completely block NMDAR activity. 111 As a consequence, Ca2+ influx, and some level of excitotoxicity, may still occur. Our results, demonstrating partial rescue, support this potential mechanism. Further investigation using a more potent NMDAR antagonist would help determine the validity of this partial blockade hypothesis. It should also be recognized that although memantine is generally well tolerated, NMDAR antagonism is not without potential side effects. 112,113 Other NMDAR antagonists have been associated with increased arousal, as well as deficits in learning and long-term potentiation, 114 because of the effects that antagonism can have on the NDMAR itself and the synthesis of other neurotransmitters, including dopamine. 115 Furthermore, more potent NMDAR antagonists, such as ketamine, have been associated with psychotic, manic, and dissociative symptoms. 116 In addition, although excitotoxicity is an important aspect of TBI pathophysiology, it is accompanied by many other changes. These include cytoskeletal damage, axonal dysfunction, inflammation, and even cell death. 107 –109 Although glutamatergic excitotoxicity may contribute to, or exacerbate, some of these other changes, aspects of these pathologies are independent. With such a heterogeneous set of pathological effects, a multi-therapy approach that targets multiple mechanisms might prove more efficacious. For example, branched-chain amino acids, 117 –121 omega-3 fatty acids, 122,123 tau antibodies, 34,124,125 or anti-inflammatory agents, 126 among other prospective treatments, may be combined with memantine. Furthermore, a temporal component should be considered. Although either immediate acute or delayed continuous memantine treatment appears most effective, there are likely optimal treatment windows for different aspects of injury. Not only the combination of interventions but also the sequence of administration will likely prove important to maximize recovery. Future work is necessary to investigate these multi-therapy approaches and how memantine can be leveraged within such a regimen.
This study suggests that NMDAR antagonism via memantine has a small but detectable beneficial effect as a monotherapy following rmTBI, which is time-dependent. Particularly, immediate treatment, or treatment initiated at 6 months post-injury, demonstrates a greater benefit than vehicle or treatment at 3 months. This effect was the most evident in microglial accumulation. To our knowledge, this is the first study to investigate whether an acute course of treatment with memantine has durable benefits and whether the timing of treatment influences efficacy. Together, these results suggest that memantine alone is insufficient to treat the myriad effects of rmTBI but may be an effective component of a multi-therapy approach and that treatment timing is an essential consideration in the development of such a therapeutic strategy.
Transparency, Rigor, and Reproducibility Summary
ANOVA (histological outcomes) and repeated measures ANOVA (repeated behavioral testing) were used as appropriate. The statistical tests used were based on the assumptions of normality, homoscedasticity, and no multicollinearity, which were tested in advance of statistical interrogation to ensure the correct methods were being applied. Outliers were determined by calculating Cook’s distance to identify data points of high leverage, which would have a significant effect on the outcome of the analysis. Multiple comparisons were corrected for using Tukey’s method. The formal analysis was performed by authors with graduate training in statistics. The required sample size for this study was determined based on prior investigation of both injury and treatment effects. 35,38,42 Both the injury model and the outcomes investigated (behavioral and histological) are standard within the field. Furthermore, owing to the complexity of the study—two injury conditions (sham, rmTBI), two treatment groups (vehicle, memantine), and three treatment time points (immediate, 3 months, 6 months)—a total sample of 96 animals was used, with 48 per injury group, 48 per treatment, 32 per time point, and, within each time point, 8 per injury–treatment group. Animals were randomized to each condition. Statistical methods and sample sizes were reported within the article. With the exception of the administration of treatment (via in cage water bottles) and measurement of the latency to right, which could not be blinded, all outcomes (behavioral and histological) were quantified by investigators blinded to the animal’s experimental status. Two controls were in place for this study—sham procedure and vehicle treatment. Sham animals received five isoflurane exposures to align with the rmTBI group and control for anesthetic effects. A vehicle treatment of autoclaved water alone was used to match the intervention that consisted of memantine in autoclaved water. Replication studies are not currently planned. Owing to the effect observed, as explained in the article, future investigation, using memantine, alongside other therapeutic interventions in a multi-therapy approach, would be a likely next step to interrogate the therapeutic potential of memantine following rmTBI. The data and analytic code can be made available to other investigators upon request. The authors have agreed to provide the full content of the article upon request by contacting the corresponding author (W.P.M.).
Footnotes
Acknowledgments
Part of the work was performed in the Animal Behavioral and Physiology Core at Boston Children’s Hospital (CHB IDDRC, 1U54HD090255).
Authors’ Contributions
M.L.B. was responsible for investigation, formal analysis, data interpretation, and writing—original draft preparation. G.C. and N.J.M. were responsible for investigation and data interpretation. S.O.-M. was responsible for investigation. J.Q. was response for conceptualization, methodology, supervision, data interpretation, and revision. R.M. was responsible for conceptualization, methodology, supervision, investigation, formal analysis, data interpretation, writing, revision, and funding acquisition. W.P.M. was responsible for conceptualization, methodology, supervision, investigation, data interpretation, writing, revision, and funding acquisition. All authors approved the final draft.
Funding Information
This work is part of the NFL-LONG study, which is funded by a grant from the National Football League.
Author Disclosure Statement
W.P.M. receives royalties from (1) ABC-Clio publishing for the sale of his book, Kids, Sports, and Concussion: A Guide for Coaches and Parents and Concussions; (2) Springer International for the book, Head and Neck Injuries in Young Athletes; and (3) Wolters Kluwer for working as an author for UpToDate. His research is funded, in part, by philanthropic support from the National Hockey League Alumni Association through the Corey C. Griffin Pro-Am Tournament and a grant from the National Football League. R.M. is funded by grants from the National Institutes of Health, the Department of Defense, and the National Football League. R.M. received research support from Abbott Pharmaceuticals.
For the other authors, no competing financial interests exist.