
Abstract
Patients hospitalized after a moderate or severe traumatic brain injury (TBI) are at increased risk of nosocomial infections, including bacterial pneumonia and other upper respiratory tract infections. Infections represent a secondary immune challenge for vulnerable TBI patients that can lead to increased morbidity and poorer long-term prognosis. This review first describes the clinical significance of infections after TBI, delving into the known mechanisms by which a TBI can alter systemic immunological responses towards an immunosuppressive state, leading to promotion of increased vulnerability to infections. Pulmonary dysfunction resulting from respiratory tract infections is considered in the context of neurotrauma, including the bidirectional relationship between the brain and lungs. Turning to pre-clinical modeling, current laboratory approaches to study experimental TBI and lung infections are reviewed, to highlight findings from the limited key studies to date that have incorporated both insults. Then, practical decisions for the experimental design of animal studies of post-injury infections are discussed. Variables associated with the host animal, the infectious agent (e.g., species, strain, dose, and administration route), as well as the timing of the infection relative to the injury model are important considerations for model development. Together, the purpose of this review is to highlight the significant clinical need for increased pre-clinical research into the two-hit insult of a hospital-acquired infection after TBI to encourage further scientific enquiry in the field.
Introduction
Individuals with a traumatic brain injury (TBI) necessitating a hospital stay are at increased risk of infections, such as pneumonia. While it is well established that such infections typically worsen a patient's health, the mechanisms that underlie the interaction between a brain injury and infection remain poorly defined. Further, we are only just beginning to understand how a TBI drives immunological dysfunction to render an individual more vulnerable to infections. Animal models of experimental TBI combined with infections of bacterial or viral origin may provide important insights into these mechanisms, with increased understanding of the fundamental biology being an important step towards improved clinical management of post-TBI infections.
In this review, we first describe the scope of the clinical problem, highlighting evidence that infections contribute to increased mortality and/or morbidity after a TBI. We then provide an overview of the known mechanisms by which TBI alters the immune system, including inflammasome-mediated inflammation and immunosuppression. After introducing laboratory models of experimental TBI and lung infections as distinct entities, we then focus on the limited studies to date that have combined animal models of TBI with respiratory infections using live bacteria. Finally, drawing upon our own experiences in the field as well as the published literature, we consider practical issues relevant to the development and establishment of these two-hit models. The overall goal is to highlight both challenges and opportunities in the field in a “call-to-action” for further research in this space. While this review is focused on brain injuries sustained via trauma, it is noted that infections also influence outcomes after ischemic stroke, and the reader is directed to several recent articles on infections in the context of stroke. 1,2
Infectious Complications After TBI
TBI patients are uniquely susceptible to nosocomial infections in the intensive care unit (ICU), even compared with the general ICU population. 3,4 This is partially attributed to the high frequency of diagnostic and therapeutic procedures carried out in these patients, such as neurosurgical interventions, insertion of urinary and intravenous catheters, and endotracheal intubation for mechanical ventilation. 5 Such clinical procedures, although life-saving, increase the risk of patients acquiring surgical-site and catheter-related infections, including urinary tract infection (UTI), hospital-acquired pneumonia (HAP), and ventilator-associated pneumonia (VAP). Pneumonia is the most common hospital-acquired infection in TBI patients, affecting an estimated 40% of TBI patients. 6 –12 Other infections including UTIs, surgery-site infections, and catheter-related bloodstream infections, are the next most commonly reported, as noted by Hamada and colleagues 13 and others (Table 1). 8,12,14 –25
Key Clinical Studies Investigating Infection in TBI Patients
“Ventilation” refers to mechanical ventilation.
AIS, Abbreviated Injury Scale; CSF, cerebrospinal fluid; GCS, Glasgow Coma Scale; GOS-E, Glasgow Outcome Scale-Extended; HAP, hospital-acquired pneumonia; ICU, intensive care unit; ISS, Injury Severity Score; pts, patients; SSI, surgical site infection; TBI, traumatic brain injury; UTI, urinary tract infection; VAP, ventilator-associated pneumonia.
Clinical evidence demonstrates that TBI patients who also sustain infections have increased morbidity and a poorer long-term prognosis (Table 1). For those with a moderate or severe TBI, an infection increases the length of ICU and in-hospital stay, frequency of hospital readmissions, and duration of mechanical ventilation, in addition to a lower Glasgow Outcome Scale-Extended (GOS-E) score indicating poorer functional outcomes. 18,22,24,25 The evidence linking post-injury infections with changes in mortality is more contentious, with some studies reporting an increase in in-hospital mortality, 26 -28 while others found no effect. 12,18,21 This variability may be attributed to several factors, such as differences between hospitals in terms of their mortality rates, incidence of antimicrobial resistance, study sample size, and the geographical region in which the study was conducted. 28 Further, the definition of hospital-acquired infections differ widely between studies, with some researchers using established criteria (e.g., from the American Thoracic Society), while others made diagnoses based upon clinical, radiological, and/or microbiological assessments. 12
Hospital-acquired infections in TBI patients are most often bacterial in origin. The most common microorganisms that cause nosocomial infections include Gram-negative bacteria such as Hemophilus influenza, Klebsiella pneumoniae, Pseudomonas aeruginosa, and Acinetobacter baumannii, in addition to the Gram-positive bacterium Staphylococcus aureus (Table 1). 14,16,29 –32 Multi–drug-resistant bacteria as a cause of respiratory infections are increasingly detected in critically-ill patients. 33
Although viral infections are less common, they are also notable causes of respiratory infections in hospitals, where the incidence of viral infections tends to mirror the level of viral activity in the community. 34 –37 However, few studies have investigated the incidence or effects of viral infections in neurotrauma patients to date. Yet, it is increasingly appreciated that viral infections such as SARS-CoV-2 (COVID-19) can induce neuroinflammation and contribute to injurious neurological sequelae. 38 –41 With at least one study reporting that COVID-19 increased the mortality rate in elderly patients with moderate-to-severe TBI by over 5-fold, 42 further investigation into both pre- and post-injury viral infections in the context of neurotrauma is clearly warranted. More broadly, further research into the risk factors associated with infections, both bacterial and viral in origin, is needed in order to prevent and better treat infections in TBI patients. 12
Lung-Brain Interactions in TBI, Lung Injury, and Pneumonia
A TBI rarely occurs as an isolated injury. Rather, TBI most often occurs concurrently in the context of polytrauma alongside extracranial injuries or complications, which contribute to worsened neuropathological outcomes. 43 Pulmonary dysfunction, especially due to pneumonia, is one of the most common and life-threatening complications in severe TBI patients. 21,30,44,45 Therefore, it is prudent to focus on the salience of lung pathologies with regard to TBI. Increasing evidence indicates that TBI itself may be an independent contributor to these complications, 46 highlighting the need to further investigate the bidirectional relationship between the brain and the lungs.
Acute lung injury (ALI) can occur in the immediate aftermath of TBI. Similarly, a TBI alone can also cause major pathologic changes in the lung in acute respiratory distress syndrome (ARDS), a more severe form of ALI. 47 ALI and ARDS are defined as clinical hypoxemia, hypercapnia, and diffuse filling of alveolar spaces with proteinaceous material and blood. 48 From a pathological perspective, ARDS is characterized by edema and diffuse alveolar damage featuring distinctive hyaline membranes, necrotic endothelial cells, and alveolar epithelial type I cells. 49 Mechanical ventilation is required to correct imbalances in blood oxygen and carbon dioxide levels in ALI/ARDS patients. 48 Indeed, approximately 20% of severely-injured TBI patients are reported to develop ALI or ARDS symptoms, 50,51 although the molecular and cellular mechanisms for post-TBI lung injury are not well defined.
First, it is important to consider how a TBI can affect the lungs (Fig. 1). 46,47,52 –82 Mechanical trauma to the head can cause primary brain damage, including axonal injury, neuronal death, and vascular damage. This leads to the release of damage-associated molecular patterns (DAMP) that stimulate secondary injury. 52 “Secondary injury” refers to an extensive series of metabolic, biochemical, and inflammatory processes including microglial activation, oxidative stress, glutamate excitotoxicity, and astrogliosis. 53 These cellular and molecular cascades act in concert to disrupt the blood–brain barrier (BBB), driving the infiltration of peripheral immune cells into the brain, and leakage of inflammatory cytokines and chemokines into the systemic circulation. 54 The latter causes a state of systemic inflammation which, in turn, can lead to the development of ALI and neurogenic pulmonary edema. 46,60,61
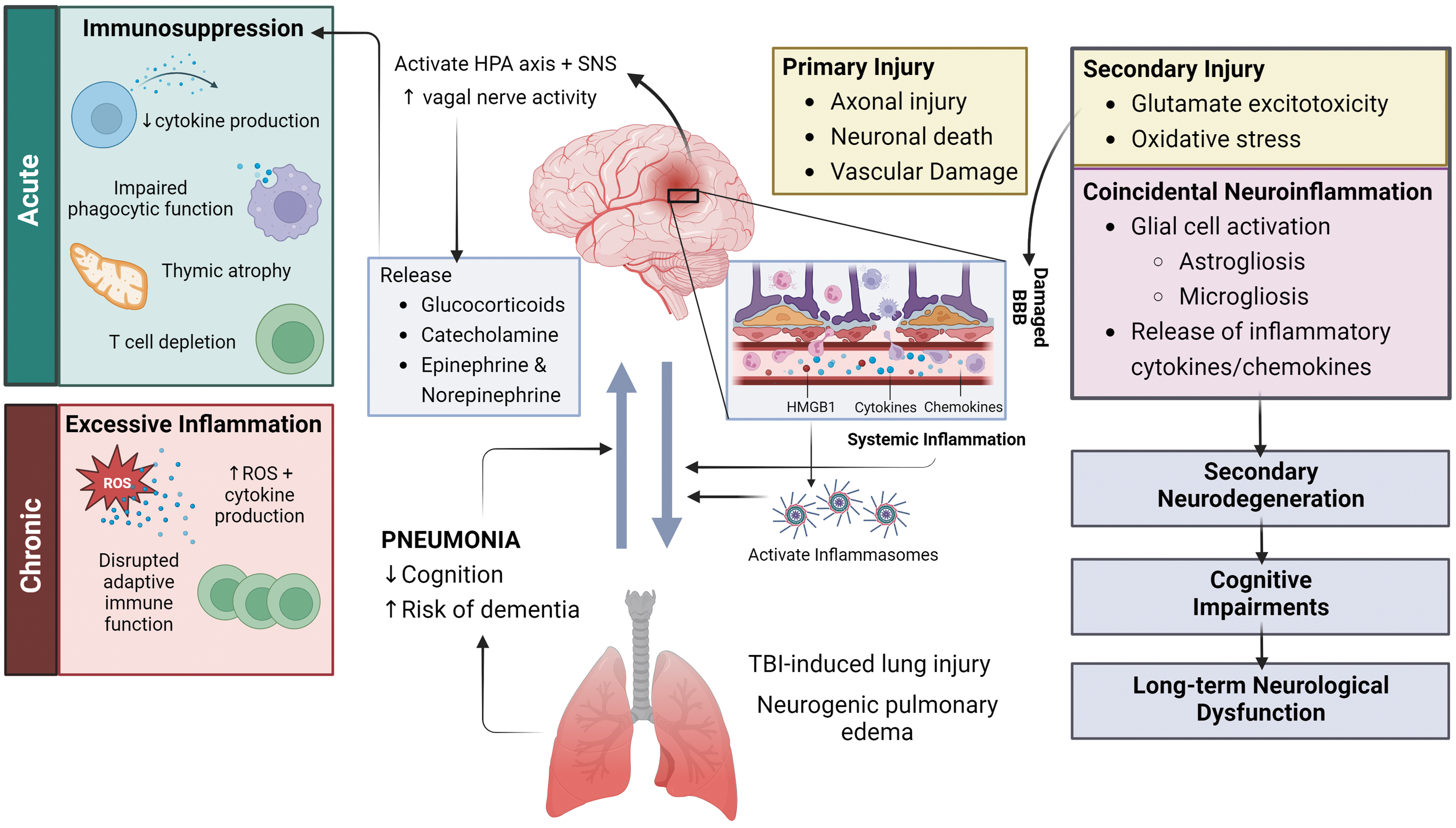
Brain-lung interaction during TBI/pneumonia. Mechanical trauma to the head causes two phases of injury—primary and secondary injury. 52 -54 The release of damage-associated molecular patterns, such as high-mobility group box protein-1 (HMGB1), during primary injury 55 stimulates a cascade of events that drives secondary injury. 56 This includes glial cell activation and inflammatory cytokine/chemokine release. 57 These pathological changes progressively lead to secondary neurodegeneration and, ultimately, long-term neurological dysfunction. 58,59 Further, endothelial dysfunction and exuberant neuroinflammation also result in the disruption of the blood–brain barrier (BBB). 54 This then permits the infiltration of peripheral innate immune cells into the brain parenchyma and leakage of inflammatory cytokines and chemokines into the systemic circulation, consequently resulting in a state of systemic inflammation that may harm the lungs and instigate the development of acute lung injury (ALI) and neurogenic pulmonary edema. 46,60,61 Additionally, the presence of HMGB1 in systemic circulation triggers the activation of Toll-like receptor 4 (TLR4) and inflammasomes 47 that contribute to the development of lung pathology. 56,62,63 Conversely, patients with pneumonia were found to suffer from cognitive dysfunction and increased risk of dementia, independent of neurotrauma. 64 –72 These findings highlight the bidirectional relationship between the brain and the lungs. TBI is also known to stimulate the hypothalamic-pituitary-adrenal (HPA) axis and sympathetic nervous system (SNS), 73 –76 as well as increase vagal nerve activity, 77 which stimulates the release of hormones such as glucocorticoids, catecholamines, epinephrine and norepinephrine, leading to peripheral immune dysfunction. 78,79 Although the immune response appears to become acutely suppressed initially—which then leads to an increased vulnerability to secondary infections, the immune response then shifts towards a state of excessive inflammation which can persist chronically. 80 -82 ROS, reactive oxygen species.
It is also important to understand the crosstalk between the brain and the lungs during a lung infection, and how an initial localized pulmonary inflammatory response can lead to neurological sequelae. Upon detecting a microbial presence in the alveoli, alveolar macrophages—resident sentinels of the alveolar space—secrete cytokines and chemokines to swiftly recruit neutrophils into the lung parenchyma, from where they usually reside within the pulmonary capillaries. 83,84 Neutrophils are often the first line of defense against an invading pathogen. They have several microbicidal capabilities, such as phagocytosis, production of reactive oxygen species (ROS), and extrusion of neutrophil extracellular traps. 85 Together with macrophages, the presence of neutrophils in the lung parenchyma is a hallmark of pulmonary inflammation. While this immune response is necessary to clear the infection, excessive inflammation can damage the lung epithelia and impair gas exchange, which can then lead to hypoxia and in severe cases, death of the host. 36
Crosstalk between the lung and the brain, the “lung-brain axis,” is mediated by several complex pathways including the neuroendocrine, neural, and inflammatory systems. 86 During a lung infection, pathogens can exert a neuropathological effect either directly or indirectly via these pathways. For instance, previous studies have shown that some pathogens are capable of direct translocation into the central nervous system via the nervous and/or hematogenous route. 87 -89 More commonly, however, respiratory pathogens stimulate the release of systemic inflammatory mediators that leak from the lungs into the bloodstream, which can then access the brain parenchyma via a disrupted BBB to provoke a neuroinflammatory response.
Intriguingly, several studies have uncovered an association between cognitive function and pneumonia. In the absence of a brain injury, an increased risk of delirium, 90,91 cognitive impairment, 64 and dementia 65 has been reported in patients hospitalized with pneumonia or ARDS, particularly in elderly patients. 66 –70 Although the underlying pathophysiological mechanisms are not well understood, it has been suggested that neuroinflammation and hypoxia, due to perturbations in gas exchange indirectly triggered by the infection, may drive these neurocognitive deficits. 66,71,72
Mechanisms of Vulnerability to Infection
Several mechanisms have been proposed that may explain why individuals are particularly vulnerable to sustaining respiratory infections after a TBI. Further, the mechanisms by which a TBI and infection may interact to facilitate worse outcomes for the patient are beginning to be elucidated. This section will describe some of these mechanisms, with a particular focus on systemic inflammation, immunosuppression, and inflammasome activation.
TBI-induced systemic immunosuppression
TBI initiates a systemic stress response associated with activation of the HPA axis and the sympathetic branch of the autonomic nervous system, resulting in the release of glucocorticoids and catecholamines, respectively. 73 –76 Catecholamine release into the circulation remained elevated for 14 days post-injury and was found to be proportional to the severity of injury in TBI patients. 92 While this response may acutely compensate for TBI effects, a prolonged stress response has deleterious effects and can lead to multi-organ dysfunction. 43,73,93
In addition to the increased release of corticoids and catecholamines, TBI induces a systemic immune response that can progress to systemic immune response syndrome, releasing immune mediators of inflammation (e.g., cytokines, chemokines) into the circulation. 58,94 -96 Systemic inflammation can persist for months in patients suffering from mild to severe TBI. 94,95,97,98 In prospective cohort studies, TBI patients had elevated serum interleukin (IL)-1β, IL-6, IL-8, IL-10, and tumor necrosis factor (TNF)-α levels over the first year post-injury when compared with age-matched healthy controls. 97 A subacute systemic cytokine load score identified individuals at risk for unfavorable outcomes following TBI, and was predictive of global outcomes at 6 and 12 months. 98 In particular, a higher IL-6/IL-10 ratio was associated with increased risk of poor outcomes at 6 months. Moreover, the acute phase cytokine load score predicted cognitive performance in moderate-to-severe TBI patients, 99 such that IL-1β, TNF-α, soluble IL6 receptor (sIL-6R), regulated upon activation, normal T cell expressed and secreted (RANTES), and macrophage inflammatory protein (MIP)-1β were negatively associated with cognition. Thus, there is a significant correlation between circulating cytokines and chemokines and worsened patient outcomes. Once in circulation, these immune mediators exert deleterious effects on other peripheral organ systems. 58,100
Animal studies indicate that TBI results in acute and chronic changes in peripheral immune function, prominently skewed towards widespread immunosuppression in the innate and adaptive arms of systemic immunity acutely after TBI, and then transforming to a hyper-inflammatory and dysfunctional state during chronic recovery periods. 80 -82 In the acute phase, systemic immune changes include suppressed pro-inflammatory cytokine production and impairments in critical respiratory burst and phagocytosis functions in myeloid cells (neutrophils and monocytes), as well as marked thymic atrophy and circulating T lymphopenia. 80 In contrast, during the chronic phase, excessive inflammation ensues with increased ROS and pro-inflammatory cytokine production in myeloid cells, and disrupted adaptive immune function as demonstrated by chronic T lymphopenia, CD4:CD8 inversion, fewer naïve T cells, and greater memory T cell conversion. 80 Such changes in systemic immunity parallel robust activation of neuroinflammatory pathways in the injured brain, which have been shown to drive secondary neurodegeneration at distant sites (e.g., hippocampus and thalamus), leading to cognitive impairments and long-term neurological dysfunction in TBI mice. 59
TBI-induced immunosuppression is hypothesized to result from three major mechanisms: 1) HPA axis activation leading to excess glucocorticoid release from the adrenal cortex; 2) excess sympathetic activation followed by catecholamine release from the adrenal medulla and sympathetic terminals; and 3) increased vagal nerve activity. 46,78,81,101 These mechanisms have been implicated in driving injury-induced central nervous system (CNS) immunosuppression in ischemic stroke, spinal cord injury, and TBI. 78,79
Clinical studies investigating cortisol and the HPA axis demonstrate high circulating cortisol levels following acute brain injury. 102 In an experimental mild TBI study in mice, decreased numbers of circulating T cells was inversely correlated with a transient and robust increase in plasma cortisol levels. 103 Cortisol reduces pro-inflammatory cytokine production and boosts levels of anti-inflammatory cytokines, such as IL-10 and transforming growth factor (TGF)-β. 104 TBI-induced transient increase in cortisol impaired circulation of lymphocytes and sequestered T cells in lymphoid tissue. 103 High cortisol levels have been shown to suppress phagocytosis, cytokine production (e.g., TNF-α, IL-1β, IL-6), and downregulate toll-like receptor 4 (TLR4) signaling in macrophages throughout the body, resulting in an anti-inflammatory state, 105,106 which may contribute to increased susceptibility to infection.
Acutely following TBI, there are increased circulating catecholamines in patients. 107 Sympathetic storm or increased norepinephrine and epinephrine have also been shown to induce immunosuppression in experimental models of spinal cord injury, 101 and both spinal cord injury and stroke-induced immunosuppression is sufficient to cause spontaneous pneumonia, typically attributed to the translocation of gut-derived bacteria. 101,108 In contrast, spontaneous pneumonia has not been demonstrated following TBI in experimental models. 109 Pre-clinical studies on the role of epinephrine in TBI have shown that there is a profound effect of sympathetic activation on the activation of T cells. 110 However, the effects of the sympathetic nervous system on monocytes and macrophage function in the context of TBI have not been elucidated.
The vagus nerve, a key bidirectional mechanism for neuro-immune communication, is activated following acute brain injury to regulate peripheral inflammatory responses. Vagal parasympathetic innervation of the celiac ganglion activates the splenic nerve to control the release of norepinephrine and epinephrine in the spleen. 77,111 This activates T cells in the spleen via β2-adrenergic receptors, resulting in acetylcholine release. 112 Acetylcholine binds α7 nicotinic acetylcholine receptors on macrophages, which inhibits TLR signaling resulting in decreased TNF-α production. 112 In studies of experimental stroke with secondary P. aeruginosa lung infection, there is a strong association between immunosuppression and the activation of α7 nicotinic acetylcholine receptors. 55 Interestingly, antagonism of this receptor abrogated stroke-induced immunosuppression, increasing phagocytosis and protecting the lung from Pseudomonas infection. 55 However, whether this occurs in TBI as the overriding mechanism of immunosuppression, and whether potential targeting of this mechanism can modulate the risk of infection after acute CNS injury, is unknown.
HMGB1 and inflammasome activation
High mobility group box protein-1 (HMGB1) is a DAMP that has been implicated as pivotal to the brain-lung axis after injury. HMGB1 is released from necrotic neurons, 56 enters the bloodstream, and travels to the lungs, where it acts as an agonist for TLR4 and RAGE receptors and activates NF-κB signaling. 113 In trauma patients without brain injury, circulating HMGB1 has been shown to mediate ALI, and higher levels of HMGB1 are associated with poorer outcomes in ALI patients. 114 TBI patients also exhibit enhanced HMGB1 levels that correlate with poorer outcomes. 115,116
In mouse experiments, increased expression of inflammasome components (e.g., AIM2 and ASC) have been reported in the lung at 4 and 24 h following moderate-severe TBI, and have been associated with lung pathology. 47 Extracellular vesicles containing HMGB1 and inflammasome components are released from damaged endothelial cells in the brain, enter circulation and traffic to the lung where they induce local cell death and pathology. 47 The NLRP3 inflammasome also plays a prominent role in the pathophysiology of ALI and TBI. 62,63 Following acute brain injury, the NLRP3 and AIM2 inflammasomes have been implicated in cell death and alveolar collapse in the lung. 47 In TBI patients, lung cell pyroptosis has been reported, 117 and animal TBI studies have demonstrated that NLRP3/AIM2 inflammasome activation in the lung results in increased IL-1β and pyroptosis. 47 Thus, post-TBI lung injury due to circulating HMGB1-induced inflammasome activation may be perpetuated by increased TLR4/RAGE signaling and increased NLRP3 and AIM2 inflammasome ligands in the lung. 113
Experimental Models of TBI and Infections
In this section, we delve into the currently available models of experimental TBI and infections, both separately and in combination, which have utility for exploring the complex interplay between neurotrauma, infections, and immunity.
Animal models of experimental TBI
Various animal models have been developed to better understand the pathophysiology of TBI as well as to explore potential therapeutics. The choice of animal used must take into account theoretical and ethical considerations. This includes focusing on certain aspects of TBI; namely, the nature, severity, and location of injury. It is important to note that each model chiefly focuses on a certain aspect of TBI, yet none of these models can perfectly mimic all aspects of TBI in humans.
Rodents are most commonly used in TBI research due to their small size, modest cost, and standardized outcome measurements. Among the many different models that have been developed, the three most prominent in the field include the fluid percussion injury (FPI), controlled cortical impact (CCI) injury, and the weight-drop (WD) model. 118 Other less common, and more recently developed, models include blast-induced TBI, 119 the Closed-Head Impact Model of Engineered Rotational Acceleration (CHIMERA), 120,121 and the awake closed-head injury (ACHI) model. 122 Readers are referred to several excellent reviews that describe these models in depth. 118,123,124
Despite the frequent use of these animal models to explore injury mechanisms and potential therapeutics, it has been difficult to translate pre-clinical findings to the clinic to date. Certain clinical and pathological features have been successfully modeled in animals, such as the neuroinflammatory response; yet other aspects, such as the severity of the injury, are rarely recapitulated to the same extent. For example, inconsistent with the human condition, rodents with a “moderate or severe TBI” are ambulant and resume normal behavior shortly after the injury. 125 Functional deficits seen may range from sensorimotor to cognitive and social behavior changes, depending upon several variables including the age at the time of injury, injury location, and severity. Of note, most studies use healthy young adult male animals, which does not necessarily reflect the human population who sustain TBI (i.e., presence of pre-existing comorbidities, alcohol/drug abuse, obesity, young or old age, different sexes, and post-TBI complications such as infection). Therefore, further studies are needed to address such translational gaps.
Animal models of lung infection
Various animals, from rodents to non-human primates, have been used to study the pathophysiology of lung infections. 126 Mouse is the most common species, particularly for models of pneumonia, due to practical considerations. 127 There are several routes to administer microorganisms into the lungs to model a respiratory infection. 128 Aerosol infection involves the inhalation of aerosolized microorganisms into whole–body or nose-only chambers: the aerosol is generated by one or more nebulizers and sprayed into an enclosed chamber (“whole–body” model), or directly onto the restrained animal's snout (“nose-only” model). This method usually results in the symmetrical and uniform deposition of microorganisms in both lungs, 129 and is relatively simple and noninvasive. However, it is not suitable for all pathogens, as some pathogens survive poorly in aerosols, and the extent to which the bacteria reach the lower respiratory tract is variable due to multiple factors (e.g., the performance of the device and size of the droplet or particle).
An alternative method is intranasal inoculation, which better mimics natural infection. Droplets of the inoculum are administered onto the animal's nares, while the animal is held in a vertical position, and then aspirated by the animal upon inhalation. Anesthesia is often required to prevent the inoculum from being swallowed or sneezed out. 130 While this technique is simple, it can also result in a highly variable, non-uniform and asymmetric deposition of microorganisms in the lungs, dependent upon the technician's skill. A more reproducible method is instead intratracheal inoculation via endotracheal or oropharyngeal injection. 131 This involves directly administering the pathogen into the trachea, with or without a tracheotomy, thereby bypassing the upper airways. While this technique requires no surgery and causes the animal minimal pain and discomfort, it is technically more difficult to perform. It has the advantage of being highly consistent and allows for greater control over the dose of the pathogen entering the lungs. 132
Following pulmonary infection, animals may demonstrate behavioral changes such as lethargy and lack of response to environmental stimulus, as well as physical changes such as piloerection, weight loss, fever, and nasal discharge. Severe infection often results in signs of respiratory distress such as gasping or wheezing. Some animals may succumb to the infection, with mortality rate depending on many factors such as the infectious agent, isolate and dose, and host defense. 133
Experimental studies investigating infection in TBI models
To date, only limited studies have considered the experimental combination of a TBI and infection. Infection is most commonly induced by administering isolated or synthetic endotoxins such as lipopolysaccharide (LPS), heat-killed bacteria, or viable bacteria. Although these mechanisms all serve to mimic infection in vivo, the use of live bacteria most closely emulates human disease in animal models, is thereby most clinically-relevant, and as such is the focus of this review. Relevant animal studies investigating infection in TBI models are summarized in Table 2. 134 –140 It should be noted that animal models of TBI and infection has so far been established in the three most common models of TBI: FPI, CCI and WD, but not in blast injury, CHIMERA, or ACHI models.
Key Animal Studies Investigating Infection in TBI Models
CCI, controlled cortical impact; IL, interleukin; i.v., intravenous; LCMV, lymphocytic choriomeningitis virus; LPS, lipopolysaccharide; mTBI, mild traumatic brain injury; polyI:C, polyinosinic:polycytidylic acid; TBI, traumatic brain injury; VSV, vesicular stomatitis virus; WD, weight drop.
Most experimental studies modeling hospital-acquired infections after TBI have examined pulmonary injury and respiratory infections. For example, Vermeij and colleagues 135 used the Marmarou WD model to induce mild TBI in male rats, and induced pneumonia via intratracheal inoculation of heat-killed S. aureus (1 × 109 CFU) at 24 h after TBI. Rats with TBI alone had increased plasma levels of phosphorylated neurofilament heavy chain (pNF-H), a marker of neuronal damage; alongside indices of lung damage such as increased relative lung weight (reflecting pulmonary edema), vascular permeability, and increased levels of interleukin (IL)-1 and IL-6 cytokines in the bronchoalveolar lavage fluid. TBI rats that sustained S. aureus infection showed a marked reduction in TNF-α release, indicative of systemic immune suppression. Additionally, extensive histological pulmonary inflammation, pulmonary edema, and lung damage was observed at 24 h post-infection. However, TBI prior to lung infection was not found to affect the severity of subsequent pneumonia, perhaps due to the mild nature of the injury model. 135
A recent study by Pittet and colleagues 136 sought to investigate sex differences in the response to pneumonia post-TBI, revealing a beneficial effect of estrogen. In this study, C57BL/6 male and female mice underwent CCI-induced severe TBI, followed by intratracheal instillation of P. aeruginosa (5 × 107 CFU K-strain) 24 h post-TBI. Increased mortality and decreased lung bacterial clearance (i.e., increased bacterial burden) were observed in TBI male mice challenged with secondary bacterial pneumonia, compared with similarly infected sham-operated (i.e., craniotomy only) mice. Male mice were significantly more vulnerable than females to mortality resulting from bacterial pneumonia after TBI (25% vs. 100% survival), while oophorectomized female mice had a similar mortality rate to male mice. The lung bacterial load was also reduced in female mice, alongside increased production of alveolar macrophages and higher levels of TNF-α secretion compared with male mice. Administration of estradiol to male and oophorectomized female mice were found to rescue these deficits, i.e., reduced mortality and increased lung bacterial clearance. 136 Together, these findings demonstrate greater mortality and decreased lung bacterial clearance in infected mice post-TBI compared with similarly infected sham-operated mice. These outcomes were found to be sex-dependent, with estrogen playing a salutary role.
Paradoxically, contrasting results were reported by Vaickus and colleagues, 137 using P. aeruginosa at the same concentration (5 × 107 CFU), but from a different source (Boston 41501 strain; ATCC), when administered at 48 h post-TBI. Mild TBI was induced using a closed-head WD model, with tail trauma as a control group, in female ICR132 (CD-1) mice. The authors reported that mild TBI prior to infection improved survival at least in part due to release of the neuropeptide substance P, which acts as a chemokine and increased neutrophil recruitment within the lungs, thereby facilitating increased bacterial clearance. 137 Similar findings were reported by Ruan and colleagues, 138 when they induced a mild TBI using the WD model in FVB/N mice of both sexes, followed 30 min later by an intratracheal injection of P. aeruginosa following a tracheotomy to model a lung infection concurrent to a TBI. In this study, AN engineered strain of P. aeruginosa was used (Xen5 at 2 × 104 CFU) which exhibits bioluminescence, allowing for in vivo imaging of the infection. 138 Despite this difference, they reported similar findings to those of Vaickus and colleagues in terms of the mild TBI reducing bacterial burden and pulmonary microvascular permeability; fewer immune cells, and lower levels of the pro-inflammatory cytokines IL-1β, IL-6, and TNF-α in the bronchoalveolar lavage fluid and lung tissue of mice that sustained the dual insult compared with infected mice without a TBI. Additionally, alveolar macrophages isolated from mild TBI mice demonstrated enhanced bactericidal capacity.
Altogether, these results suggest that a mild TBI attenuate, rather than exacerbate, inflammation and ALI following a subsequent pulmonary infection. 138 Such discrepancies in findings may be attributed to differences in experimental model and study design, 136 including baseline differences in the immunological profiles of rodent strains. 141 Together, this highlights the need for further research in animal models of TBI and infection to gain a more comprehensive understanding of interacting pathophysiological mechanisms in this context.
Inducing pneumonia caused by a different bacterium, Streptococcus pneumoniae, Doran and colleagues 139 first investigated the consequences of TBI combined with infection across a more prolonged time course. Twelve-week-old male mice with a moderate CCI injury were administered intranasal S. pneumoniae (1500 CFU) at 3- or 60-days post-injury, then euthanized at 3- or 7-days post-infection. Mice that received the dual insult suffered higher mortality rates (60% vs. 5% when infected at 60-days post-injury, for TBI + S. pneumoniae vs. Sham + S. pneumoniae), poorer motor function recovery, and enhanced neuroinflammation. Further, infiltrating lung monocytes were found to be acutely immunosuppressed, as they were not able to produce IL-1ß, TNF-α or ROS. Notably, delayed infection resulted in the opposite outcome, with lung monocytes becoming hyper-reactive and producing increased levels of pro-inflammatory cytokines at chronic time points. Further, there was increased expression of NLRP3, AIM2, cleaved caspase-1 and ASC, indicative of chronic inflammasome activation in the lungs in the delayed infection model. In both experimental paradigms, mice with both TBI and S. pneumoniae were found to have increased bacterial burden in the lung, worsened lung pathology (infiltrating neutrophils, and alveolitis), and increased circulating HMGB1 levels. This study highlights the importance of the timing of a post-injury infection and identified differential responses dependent upon the pre-existing immune state. 139
To the best of our knowledge, only one study to date has considered the consequences of viral pathogens on TBI outcomes. Mastorakos and colleagues 140 challenged adult (8-12 weeks old) male and female C57Bl/6J mice with mild TBI, induced by compression of the meningeal space by thinning the skull bone and applying a downward pressure. 142 They combined this with one of several pathogens/mimics, including: 1) the lymphocytic choriomeningitis virus Armstrong strain, a non-cytopathic arenavirus; 2) vesicular stomatitis virus Indiana strain; 3) polyinosinic:polycytidylic acid, a synthetic analogue of double-stranded RNA that activates antiviral pathogen-associated molecular pattern (PAMP) receptors; 4) the pathogenic yeast Candida albicans; or 5) LPS derived from Escherichia coli. They found that these immune challenges, regardless of their etiology, interfered with meningeal vascular repair and prevented neurological recovery after mild brain injury. Viral infection specifically resulted in altered angiogenic programming and reparative macrophage distribution; alongside a marked and prolonged increase in Type I interferon (IFN) expression, which then acts on Type I IFN receptors on myelomonocytic cells, preventing meningeal repair. Failed revascularization then led to neuronal loss and sustained BBB breakdown. Therefore, this study revealed the deleterious effects of single or recurrent infections of various infectious agents in impeding neurological recovery post-TBI. 140
Although we have focused here on reviewing models of respiratory infections post-TBI, it is pertinent to note that the gut-brain axis has also been implicated in TBI, which may influence how other types of infection interact with a brain injury. 143 Many studies have now shown that TBI alters the gut microbiota, 144 with microbial dysbiosis associated with gastrointestinal dysfunction and intestinal inflammation, 145 as well as exacerbated neuroinflammation. 146
Two key experimental studies to date have examined intestinal infections after TBI. Firstly, Ma and colleagues 134 examined the effects of the enteric pathogen Citrobacter rodentium (5 × 1010 CFU administered via oral gavage at 28 days post-injury) on gut inflammation after moderate TBI, in young adult (8-10 weeks old) male mice. Infection with C rodentium after TBI worsened brain injury and neuroinflammation, in addition to changing colon morphology and mucosal barrier function at 12 days post-infection (but to a similar extent in sham and TBI mice). 134 Another study by the same group sought to understand the effects of colonic inflammation induced by dextran sodium sulfate (administered via drinking water, from 28 days post-TBI for 7 days), on neurobehavior in young adult (9 weeks old) male mice after a moderately-severe CCI injury. 147 Persistent neurological deficits (e.g., impaired cognition and increased anxiety-like behavior), exacerbated TBI-associated hippocampal neurodegeneration, and altered autonomic balance, were observed in chronic TBI mice with acute colonic inflammation, 147 which may have been due to how injured mice handled the prolonged systemic immune response following delayed administration with dextran sodium sulfate. Together, these studies demonstrate that a post-TBI gastrointestinal inflammatory challenge adversely impacts outcome in TBI mice.
Practical Considerations for Experimental Modeling of Post-Injury Infections
As detailed above, despite the high rate of hospital-acquired infections sustained by patients after a TBI, 3,14 and increasing evidence that infections worsen outcomes after a TBI, 24,148,149 limited animal studies have integrated models of experimental TBI and infection. Yet animal modeling of this “two-hit” insult, to closely mimic the human condition, offer an invaluable opportunity to further dissect the underlying neurological and immunological mechanisms that result from this complex clinical scenario. Such insights are urgently needed to provide a path forward towards novel therapeutic intervention strategies aiming to improve outcomes for patients.
Drawing upon our own experiences in the field, as well as the published literature, this section will highlight some of the key variables for consideration when designing two-hit insult experiments (as summarized in Fig. 2). With infections of the respiratory tract and lungs, including pneumonia that is the most prevalent in patients after moderate and severe TBI, we focus on the incorporation of lung infection models with experimental TBI.
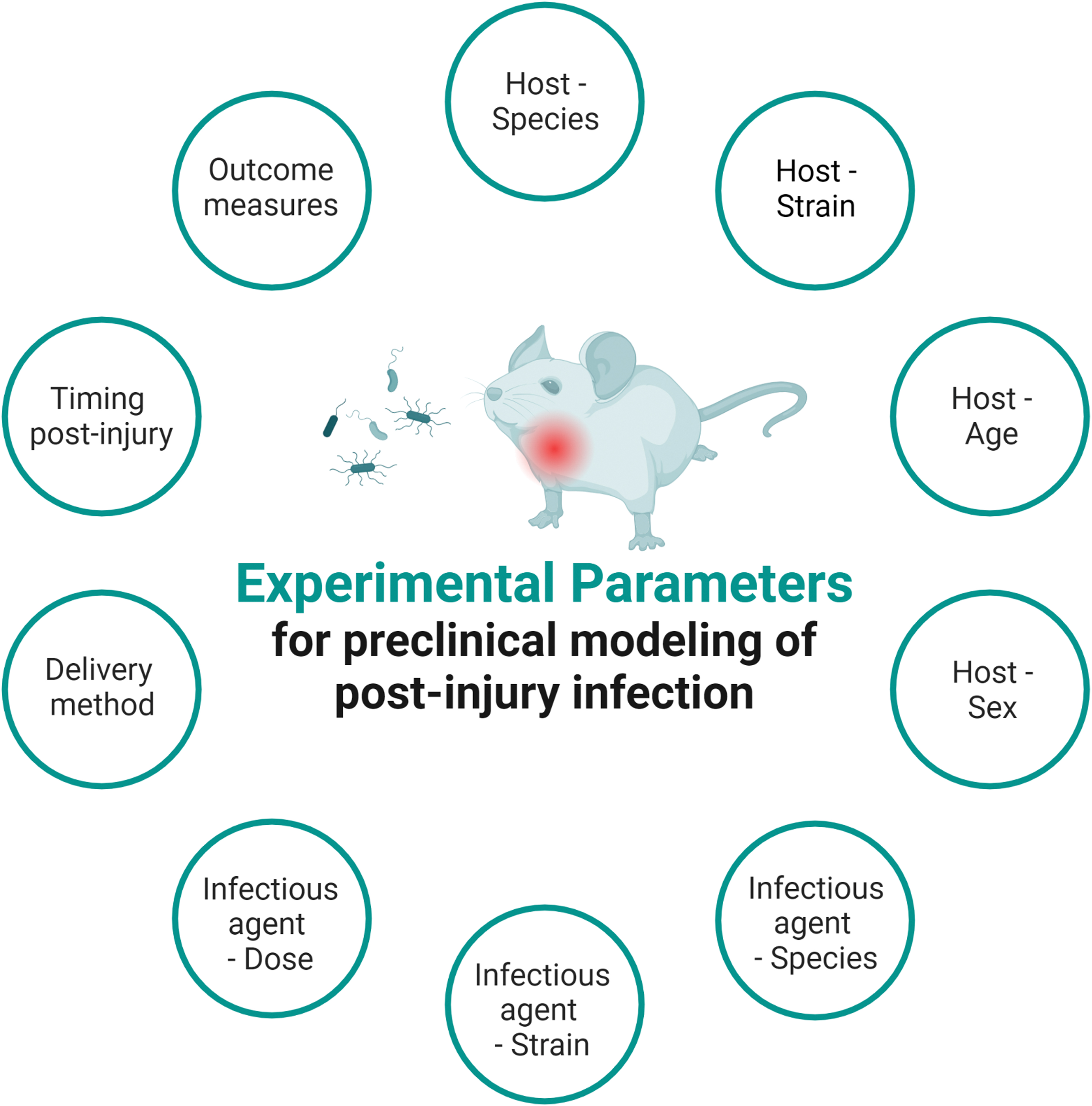
Experimental parameters to be considered when designing an animal study of post-TBI infections. “Infectious agent” may be a synthetic pathogen-associated molecular pattern such as Gram-negative lipopolysaccharide (LPS), or an attenuated or live bacterial or viral pathogen of relevance to human disease.
The experimental host animal
There are obvious practical benefits to the use of rodents for these types of studies, alongside the relative ease of measuring bacterial load, immunological responses, and morbidity and mortality in rats and mice. Indeed, while large animal models of both TBI and pneumonia exist (e.g., pigs, sheep, and rabbits), 133,150,151 most published studies to date have used rodents in the common experimental TBI models, FPI, and CCI. The choice of injury severity in the model is important, given that clinically, infections are most often associated with severe TBI. Ideally, models should also incorporate common secondary insults such as hypoxemia and hypertension, which are known risk factors both for hospital-acquired infections as well as poorer outcomes more broadly post-injury. 19,152,153 Such secondary physiological insults can be more readily incorporated and monitored in large animal models, to mirror the complex ICU experience of severe TBI patients. 154
It is also worth noting several limitations that stem from host species-specific responses to infections. The rodent lung and respiratory tract differ considerably from humans in terms of their anatomy, distribution of cells, and connective tissue, 155,156 which can influence the distribution of bacteria infecting the lungs. 157 Species-specific physiological and immune responses should also be considered. The causes of mortality in the context of pneumonia also differ between humans and laboratory animals—while humans may succumb to cardiac dysfunction, stroke, or pulmonary embolism associated with pneumonia, these events are typically rare in mice. 158 As alluded to earlier, even within the same species, strain-specific differences in susceptibility to bacterial infections have been reported. 158 For example, C57BL/6 mice show marked resistance to lung infection with 104 CFU K. pneumoniae (strain ATCC 43816 serotype 2), while 129/Sv mice are highly susceptible to the same strain/dose. 141 There are also important differences in how bacteria interact with the host species (e.g., how virulent a particular strain is in the rodent vs. human).
Another consideration for the study design and choice of host animal is whether to include sex as a biological variable. Experimental TBI studies have historically utilized mostly male animals. However, this bias has shifted in recent years with increased awareness of sex-specific differences in injury responses, including neuroinflammation. 159,160 By contrast, in the microbiology literature, the majority of studies inducing pneumonia to assess antimicrobial efficacy have been conducted in female mice. 150 Sex is likely to be an important variable in dual-hit insult scenarios as host-pathogen interactions may be sex-dependent, mediated at least in part via the effects of steroidal sex hormones. 161 Limited evidence has alluded to differential vulnerability to bacterial infections in male and female patients after TBI, 46 while a recent experimental study of pulmonary P. aeruginosa infection in CCI mice, as described above, reported that males had higher mortality, impaired bacterial clearance, and an altered immune response compared with females. 136 Further, the potential effect of age on outcomes after TBI and infection also represents a knowledge gap in the field.
The infectious agent
For optimal clinical relevance, experimental models should focus on microorganisms that drive the infections most frequently reported in TBI patients. For example, P. aeruginosa, K. pneumoniae, S. aureus, and A. baumannii are among the most common, treatment-resistant opportunistic pathogens in nosocomial infections such as ventilator-associated pneumonia. 46,150 Therefore, the specific strain of interest must be decided upon, including whether to use a clinical isolate or laboratory-derived strain. The choice of bacterial strain can be a key determinant of the manifestation of a pneumonia phenotype in the model. For example, different bacterial strains may have different degrees of pathogenicity depending upon the host species, through variable expression of different virulence factors depending on the microenvironment. 133 Clinically-isolated strains can be adapted for increased virulence in rodents; yet, such manipulations may result in the infection eliciting an altered immune response of reduced relevance to the clinical scenario. 162,163 Inoculations are typically performed with bacteria in logarithmic phase due to their increased ability to survive and expand during this growth phase, although such a detail is rarely reported. 150 A dose–response curve is likely required to determine the optimal concentration of live bacteria to be inoculated to elicit the desired infection-associated symptoms and pathology in each new experimental paradigm.
Administration of bacteria
As noted earlier, a bacterial infection can be induced via multiple routes into the animal host. For lung infection models, the bacterial inoculum may be delivered intranasally or intratracheally as liquid or powder aerosol, with the choice of delivery method being an important factor for the dose of bacteria that reaches the lower respiratory tract and lungs, and the likelihood of off-target effects. 150,164 A single bolus inoculation is most common, 150 involving forced aspiration of a high concentration of bacteria in suspension, although this carries the caveat that this mode of administration typically induces acute infection in rodent models that differs from what is seen clinically. 158,165 Intratracheal inoculation, involving direct delivery of the bacterial suspension into the trachea, results in a reproducible dosing strategy. However, both intratracheal and intranasal inoculation routes typically require a skilled operator and anesthesia to perform, which represents an additional stressor to the experimental animals.
Some studies of ventilator-associated pneumonia incorporate mechanical ventilation of the animal in addition to delivery of a bacterial inoculation, as the ventilation process itself is considered to be an important driver of lung inflammation. While not yet examined in combination with experimental TBI, the addition of mechanical ventilation compared with animals with bacterial inoculation alone results in exacerbated lung infections and higher mortality in mice. 57,166 In future, other comorbidities and/or host factors that can influence the response to infection and/or TBI should also be incorporated into such models. For example, prior exposure to illness, the role of genetics and epigenetics in outcomes after injury and infection, and the influence of medications such as antibiotics warrant investigation. 158
Another decision is whether to induce neutropenia in the experimental animals prior to inoculation. Often used in studies to evaluate the efficacy of antibiotics, neutropenia induced by drugs such as cyclophosphamide or vinblastine allows for more rapid bacterial growth and evaluation of the killing effect of the antibiotic itself, and minimizes spontaneous resolution of the infection. 150 In addition, some opportunistic pathogens (e.g., P. aeruginosa, K. pneumoniae, and A. baumannii) may not be able to establish reproducible lung infections in immunocompetent animals.
To date, most animal studies of pneumonia (and all studies of TBI plus a bacterial infection) have used just a single microorganism. 167 However, future studies might consider the incorporation of polymicrobial infection to better reflect the human scenario. Micro-aspiration to sample patients' oropharyngeal secretions have found that numerous species of bacteria (both colonized microorganisms and causative pathogens) are typically present, at least for community-acquired pneumonia, which creates a uniquely complex immune response. 158,168 For example, in an adult population of community-acquired pneumonia cases in Norway over a 3-year period, multiple pathogens were detected in 41% of cases, with S. pneumoniae being most commonly identified in co-infected cases. 169
In summary, there are many experimental variables to consider when establishing a model of hospital-acquired infection in rodents. Bacterial inocula should be prepared, and infections performed in a rigorous, standardized manner; methodology should be reported in depth for optimal transparency and reproducibility. Pilot studies are usually required to determine the most appropriate inoculum for each strain of bacteria and host strain/species. In addition, parameters of the experimental TBI model must be determined, including which model and injury severity. Finally, the timing of infection relative to TBI (i.e., pre-injury, concurrent, or at x time point post-injury) is another critical determinant of the model phenotype, as demonstrated by Doran and colleagues, when differences in timing of post-injury infection dramatically altered the lung immune state (i.e., immune suppression versus hyper-reactive) and mortality outcomes following TBI. 139
Outcome measures
Once the two-hit insult model is established, a strong interdisciplinary team to evaluate outcomes is likely required, ranging from clinicians to neuroscientists, microbiologists, and immunologists, to ensure relevant expertise in considering the impact of an infection post-TBI on the whole host animal. For TBI and pneumonia combined models, outcome measures typically include neuroinflammation, brain histology, and neurobehavioral outcomes relative to the TBI insult, accompanied by lung histology and lung inflammation read-outs, and mortality. 170,171 Some practical and logistical considerations can influence the choice of outcome measures. For example, due to the use of live infectious material in these models, most of the live animal work must be performed in an appropriate biocontainment environment (e.g., Class 2 Biosafety Cabinet or facility), which may limit the extent of neurobehavioral testing that can be performed.
Broadly, an experimental TBI typically causes a neuroinflammatory response involving the activation of resident glial cells, alongside neuronal and axonal damage. 172 In general, pneumonia models in rodents cause a phenotype of vascular congestion, alveolitis and alveolar collapse, together with significant neutrophil infiltration into the lung tissue, coinciding with a peak in sickness symptoms up to 24 h post-infection and resolution within days. 133 Lung pathology assessments should be performed in a blinded fashion by a trained individual, preferably a pathologist, with expertise in infectious disease and lung pathology in rodents. In addition, assessment of bronchoalveolar lavage fluid and sera for immune cells and cytokines can provide important evidence linking the injury consequences with the infection. Assessment of the bacterial burden in the blood and lungs of infected mice, to gauge disease progression or severity, is usually achieved by postmortem homogenization of lung tissue and plating of serial dilutions onto nutrient-rich agar.
Finally, while immunological and pathological markers of TBI-induced lung injury and the consequences of respiratory infection provide mechanistic insight, there is also a need to test pulmonary function in mice in vivo in the two-hit insult models. This is possible using several approaches to assess pulmonary function in mice, 173,174 but there is a trade-off between accuracy, invasiveness, and convenience between approaches. Nevertheless, investigating physiological and functional responses in the lung following post-traumatic respiratory infection (e.g., lung resistance and dynamic compliance) will be needed in the future to mimic the clinical scenario more accurately.
As should be the case for all experimental TBI studies, those that also incorporate an infection model are recommended to provide detailed reporting of all data and methodology that could influence the results (e.g., by following the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines). 175 Key variables associated with establishment of the infection model, such as a clear description of consistent culture conditions and methods, are important yet often omitted details essential for reproducibility and transparency.
Conclusion
TBI is one of the world's leading causes of injury-related death and disability, with profound and widespread consequences for patients, their families, and society. 176 While it is indisputable that TBI patients are at increased risk of infections, particularly after moderate and severe TBI requiring hospitalization, treatment approaches such as the use of prophylactic antibiotics lack robust evidence of efficacy. 177,178 Despite several decades of laboratory-based research into the biological mechanisms of TBI, morbidity remains high among survivors. We postulate that a lack of consideration of co-morbid factors, such as acute post-injury infections, may contribute to the difficulties of translating animal studies into clinical practice. Increasing evidence from both patients and experimental models indicates that such a “dual-hit” insult poses a unique, multi-faceted, multi-organ challenge to the patient, due to complex interactions between the brain and peripheral organs such as the lung and gut, via multiple mechanisms. While recent studies have provided greater understanding of how lung-related bacterial infections can influence neurological injury, research into other infections such as UTIs and sepsis is lacking.
Therefore, we advocate for more pre-clinical studies to investigate the added burden of an infection on TBI, to generate essential new understanding of this complex insult and how an infection can alter TBI outcomes. Specifically, pre-clinical studies can be particularly useful to address knowledge gaps evident in the clinical literature—for example, what are the risk factors for poor outcomes? How do specific secondary insults (e.g., hypoxemia) influence vulnerability to infection? How exactly does an infection alter the post-injury immune response? Could targeted interventions to modulate the immune response improve outcomes? Such insights may allow for the design and development of novel therapeutic strategies to target either the infection itself, or to augment host immunity to ameliorate the combined effects of TBI and infection on the body.
Transparency, Rigor, and Reproducibility Summary
This narrative review was designed to critically appraise the available published research pertaining to animal models of TBI and infection, to determine the scope of existing literature on the topic and provide a thoughtful evaluation of key experimental design elements for the consideration of those who might seek to establish such models. Relevant literature was identified by searching electronic databases PubMed and Ovid Embase between March 2022 and July 2023 (NG, DJL and BDS), using keywords including variations of the following: traumatic brain injury (e.g., “neurotrauma,” “TBI,” “brain injury”) and infection (e.g., “hospital-acquired infection,” “bacteria,” “pneumonia,” etc.). Only studies using rodent models (mice and rats) were included, from English language publications (see Supplementary File S1 for search strategy). The review was not registered prior to commencement.
Footnotes
Acknowledgments
The authors would like to thank Professor Stefanie Vogel (University of Maryland School of Medicine, Baltimore, USA) for critical feedback on the manuscript. Figures were created using
Authors' Contributions
Project conceptualization: NG, DJL, BDS. Manuscript draft: NG, JL, DJL, BDS. Manuscript edits: NG, JL, DJL, BDS.
Funding Information
The authors acknowledge funding support to BDS via an Epilepsy Research Program Idea Development Award (#W81XWH-19-ERP-IDA) from the US Department of Defense, an Establishment Grant from Monash University's Central Clinical School, and a Near-Miss Grant from Veski, and to DJL via a Science Foundation Ireland grant (SFI17/FRL/4860).
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary File S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.