
Abstract
Traumatic brain injury (TBI) has been discussed as a risk factor for Alzheimer’s disease (AD) due to its association with AD risk and earlier cognitive symptom onset. However, the mechanisms behind this relationship are unclear. Some studies have suggested TBI may increase pathological protein deposition in an AD-like pattern; others have failed to find such associations. This review covers literature that uses positron emission tomography (PET) of β-amyloid (Aβ) and/or tau to examine individuals with a history of TBI who are at increased risk for AD due to age. A comprehensive literature search was conducted on January 9, 2023, and 26 resulting citations met inclusion criteria. Common methodological concerns included small samples, limited clinical detail about participants’ TBI, recall bias due to reliance on self-reported TBI, and an inability to establish causation. For both Aβ and tau, results were widespread but inconsistent. The regions that showed the most compelling evidence for increased Aβ deposition were the cingulate gyrus and cuneus/precuneus. Evidence for elevated tau was strongest in the medial temporal lobe, entorhinal cortex, precuneus, and frontal, temporal, parietal, and occipital lobes. However, conflicting findings across most regions in both Aβ- and tau-PET studies indicate the critical need for future work in expanded samples and with greater clinical detail to offer a clearer picture of the relationship between TBI and protein deposition in older individuals at risk for AD.
Introduction
Alzheimer’s disease (AD) is the most common form of dementia, with approximately 6.7 million Americans currently living with the disease, and this number is expected to grow over the coming decades as the population ages. 1 Worldwide, it is estimated that 152 million people will be living with AD by 2050. 2 There are presently no cures, though recent disease-modifying therapies have become available. 3 AD is conventionally delineated into two categories based on the reported age of symptom onset, late-onset AD (onset aged 65 years or older) and early-onset AD (onset before the age of 65). 4 Late-onset AD is more common, representing approximately 95% of all AD cases. 5
Generally, AD is clinically associated with progressive memory decline and cognitive impairment and is pathologically defined by the presence of neurodegeneration and cortical accumulation of β-amyloid (Aβ) as plaques and hyperphosphorylated tau as neurofibrillary tangles (NFTs). 6,7 A framework for describing AD as a function of the most widely used biomarkers is the A/T/N (Aβ/tau/neurodegeneration) classification system, which encompasses biomarkers of Aβ, tau, and neurodegeneration and/or neuronal injury. 8 Recently proposed updates to this framework explore the utility of additional biomarker categories, including inflammation/astrocytic activation (“I”), vascular brain injury (“V”), and α-synuclein (“S”), to better incorporate comorbid pathologies in suspected AD patients. Though this definition of AD is of great utility in a research context, a standardized protocol for assessment and diagnosis of AD in a clinical setting has been developed by the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). 9,10 The CERAD battery incorporates clinical and neuropsychological testing, neuroimaging, and pathological assessment when possible. 9,10
Aβ and tau accumulate in characteristic patterns in typical AD. Aβ pathology classically deposits in five phases known as Thal stages; this process can begin decades before the onset of cognitive symptoms in individuals who will go on to develop AD. 11,12 Stage I is characterized by Aβ deposition in the frontal, temporal, parietal, or occipital neocortex. 11 The allocortex (insula, entorhinal cortex, and CA1 of the hippocampus) is also positive for Aβ deposition in Stage II. 11 Stage III involves Aβ depositions in the diencephalon (thalamus and hypothalamus), striatum (caudate and putamen), and basal forebrain. 11 Stage IV typically sees Aβ deposition in brainstem nuclei including the substantia nigra, red nucleus, central gray, colliculi, inferior olivary nucleus, and intermediate reticular zone. 11 Finally, Stage V is characterized by Aβ deposits in the cerebellum and additional brainstem nuclei (locus coeruleus, pontine, parabrachial, reticulotegmental, dorsal tegmental, and raphe nuclei). 11
Tau deposits in six stages known as Braak stages. 13 In Stage I, tau is predominantly located in the transentorhinal region. 13 In Stage II, lesions extend into the hippocampus and/or entorhinal cortex. 13 In Stage III, tau deposits are found in the fusiform, parahippocampal, and/or lingual gyri. 13 Patients typically begin to show cognitive symptoms in approximately Braak Stage IV; at this stage, tau is present in all regions of Stages I–III but is also found in the inferior temporal gyrus and insula. 13 Stage V is defined with widespread tau deposition in regions that include those in all previous stages, as well as the peristriate area in the occipital lobe and other frontal, superolateral, and occipital directions. 13 In Stage VI, the final defined stage, tau pathology is found in all stages seen in Stages I–V, both secondary and primary neocortical areas, and the striate area of the occipital lobe. 13
There are a variety of well-known risk factors for AD, the most well-characterized of which include advanced age, family history, and genetic variations (such as the ε4 allele of the APOE gene). 14 Traumatic brain injury (TBI), also called concussion or head injury, is another frequently cited risk factor for AD. TBI is extremely common; one survey indicated over a quarter (28.9%) of American adults have experienced at least one in their lifetime. 15 Most injuries are thought to be mild, and the diagnostic criteria to establish a diagnosis of mild TBI requires that at least one of the following be present after an injury occurs: loss of consciousness for any period of time, post-traumatic amnesia, confusion, disorientation, or another sign of an altered mental state, or neurological abnormalities such as seizure. 16 –19 Common mechanisms for incurring TBIs include the head being struck by or against something or due to a trip, slip, or fall. 15 Sports and recreation, as well as motor vehicle accidents, are also frequent contexts for injury. 15 In addition, there may be gender- 20 –22 and age-based demographic differences in TBI incidence, 15,23,24 as well as disparities by racial/ethnic group identification, 15,25 and educational attainment. 15
Outside the general population, TBIs also occur regularly amongst athletes who play contact sports, as well as military service members. 26,27 For example, 5–35% of American servicemembers deployed to Iraq and Afghanistan sustained a TBI during deployment. 26 In addition, TBI has been studied as a risk factor for neurodegenerative disease in the context of the well-documented relationship between repetitive TBI and chronic traumatic encephalopathy (CTE), a neurodegenerative disease identified frequently in American football players and other populations with high rates of repetitive TBI. 28 –31 Interest in TBI as a risk factor for AD has also been growing due to reports of a link between TBI and earlier onset of AD, 32 –35 though there is a lack of consensus in the literature as some studies failed to find such an association. 36,37 Consequently, less attention has been directed toward understanding the biological underpinnings of the relationship between TBI and AD risk. However, developing technologies, including the widespread use of positron emission tomography (PET) as a tool for in vivo detection and quantification of pathological protein buildup, have better enabled the field to analyze the association of risk factors such as TBI with the spatial and temporal dynamics of protein accumulation in neurodegenerative diseases.
PET is a radiological technique that can be used to visualize the concentrations and distributions of radiolabeled molecules across the body in vivo. 38,39 It is widely used in AD research for the purpose of detection and quantification of pathological changes associated with Aβ and tau, as well as other molecules in the brain. PET uses a small amount of a radioactive tracer that binds to a specific protein target. 40 The tracer emits positrons as a function of radioactive decay, and the positrons are detected by the scanner and computationally mapped back to their place of origin, enabling visualization of both the relative quantity and spatial positioning of the target protein. 40 There are a multitude of different tracers that have been developed to label Aβ and tau. The Aβ tracers used by the studies covered in this review include Pittsburgh Compound B ([11C]-PiB), Florbetapir ([18F]AV-45), Florbetaben ([18F]FBB), 18F-FPYBF-2, and Flutafuranol ([18F]-NAV4694) (Table 1, Supplementary Data S2). The tau tracers include Flortaucipir ([18F]AV-1451) and Florquinitau ([18F]-MK-6240) (Table 1, Supplementary Data S2). Of the tracers used in the studies included in this review, the U.S. Food and Drug Administration (FDA) has approved [18F]Florbetapir (Amyvid), [18F]Florbetaben (NeuraCeq), and [18F]Flortaucipir (Tauvid).
Characteristics of Included Studies
ADNI, Alzheimer’s disease neuroimaging initiative; PTSD, post-traumatic stress disorder; TBI, traumatic brain injury; UCSF, University of California, San Francisco; MVA, motor vehicle accident; MCI, mild cognitive impairment; mTBI, mild traumatic brain injury; DOD, Department of Defense; CN, cognitively normal.
There is an urgent need to better understand the long-term consequences of TBI, including biological pathways related to AD, as TBI affects many people across many demographics. However, few studies have used Aβ- and tau-PET to detect changes in protein deposition after TBI in individuals at increased risk for AD due to age. These studies are critical in characterizing the effects of TBI on biomarkers and AD risk in vivo. The purpose of this review is to synthesize literature that used Aβ and tau-PET to explore associations between TBI and pathological biomarkers in aging populations at risk for AD.
Methods
Eligibility criteria
We included studies that used tau and/or Aβ-PET in humans to study the impact of TBI in populations either with or at risk for AD due to advancing age. Gray literature (i.e., conference abstracts, and meeting reports) were eligible for inclusion. If a peer-reviewed and published article was substantially similar to a gray literature report, the gray literature was excluded, and the peer-reviewed article was included. As this review’s purpose is to consider TBI as a risk factor solely for AD, we excluded articles that mainly focused on other neurodegenerative dementias such as CTE. Additional exclusion criteria were: (1) no use of either Aβ or tau PET, (2) not primarily focused on TBI, (3) studies using preclinical models, (4) studies not available in English, and (5) review or meta-analysis articles. In addition, while articles used varying terminology to describe participants’ injuries (e.g., TBI, head injury, concussion), we hereafter exclusively use the term TBI. Finally, we applied an age cutoff to include studies with participants who were on average >40 years of age. In doing so, we avoided studies focused primarily on younger adults or pediatric populations who would not be considered at increased risk for AD due to age.
Information sources, search procedure, and study selection
A medical librarian composed and conducted comprehensive search strategies in MEDLINE (Ovid), Embase (Ovid), and the Cochrane Database of Systematic Reviews on January 9, 2023. Full search terms are available in the Supplementary Data S1. The systematic review management tool Covidence removed duplicate records. A total of 1476 unique citations were initially identified. The research team screened in two stages, first combined title and abstract screening and then full-text review. At each stage of the screening process, two reviewers independently screened each article and conflicts were resolved by consensus. After title and abstract screening, 81 articles were deemed appropriate for full-text review. After a full-text review, the research team identified 26 articles that matched the inclusion criteria; data from these articles was then extracted using Covidence. Detailed data on the number of articles excluded during each step of the screening process can be found in Figure 1, and detailed data on the selected articles can be found in Table 1. No additional statistical analyses were performed on the selected citations, chiefly because of a large variation in sample sizes across studies and the inability to identify individuals in commonly used datasets (e.g., the Alzheimer’s Disease Neuroimaging Initiative [ADNI]) that may have been analyzed in more than one citation.
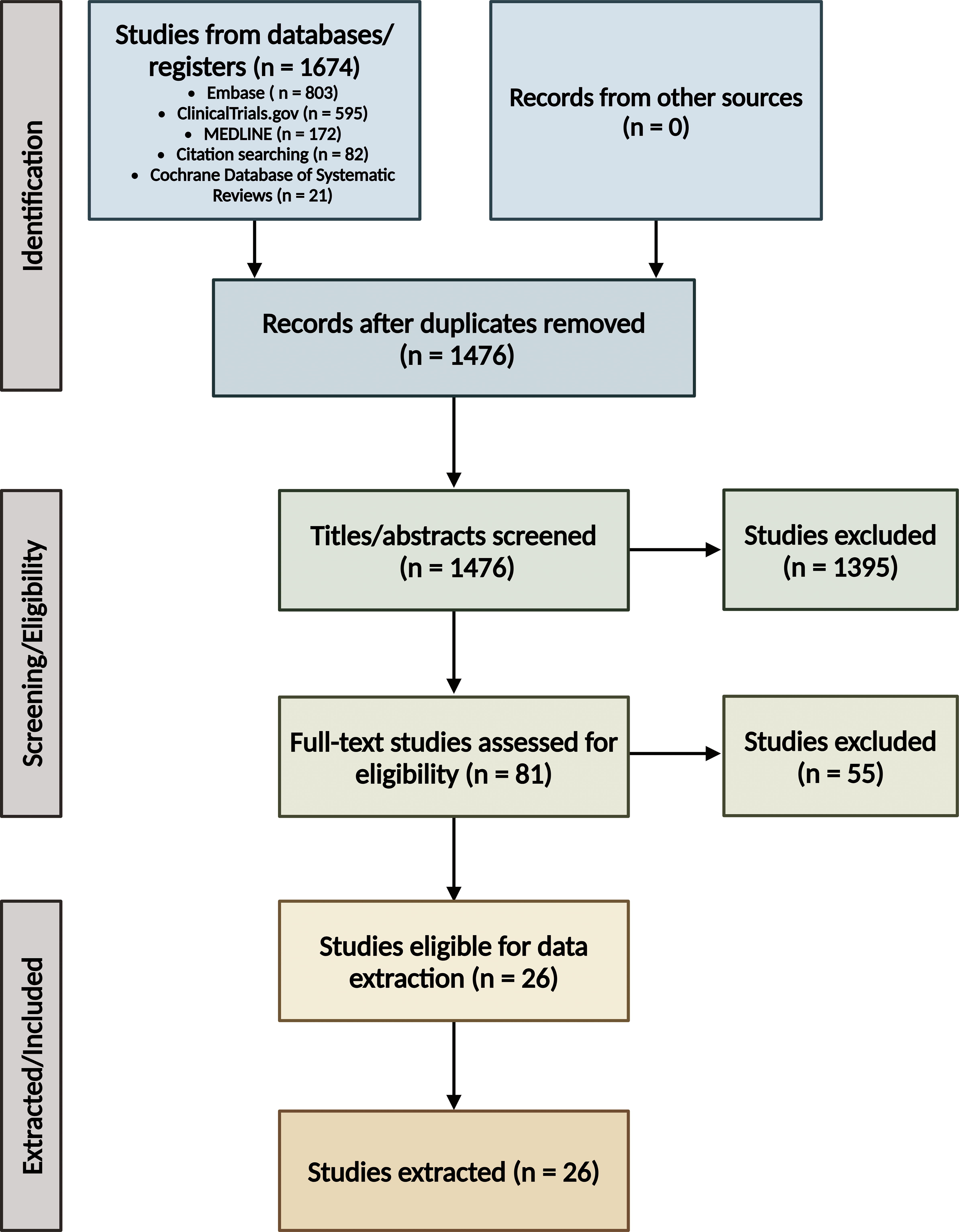
Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) 65 flow diagram. Created with BioRender.com.
Results
Aβ
Twenty of the included studies used at least one Aβ tracer. Florbetapir was used in 12 articles, [11C]-PiB was used by four, and [18F]-FPYBF-2 and [18F]-Flutafuranol were each used in one article. In addition, one study used both Florbetapir and [11C]-PiB, and one used both Florbetapir and Florbetaben. The regions that had the strongest supportive evidence for increased Aβ tracer uptake after TBI were the cingulate gyrus and cuneus/precuneus. Commonly identified limitations of the citations were small sample size, limited clinical detail about TBI, recall bias, the potential for biased recruitment strategies, and an inability to establish causation due to cross-sectional study designs.
Evidence for limbic Aβ deposition
Four articles analyzed Aβ tracer uptake in limbic regions of interest (ROIs), including the medial temporal lobe (MTL)/hippocampus, temporal pole, and entorhinal cortex (Table 2, Supplementary Data S3). 46,48,51,53 In the hippocampus, one article found evidence for altered Florbetapir uptake at multiple post-injury timepoints, 48 while an article using [11C]-PiB found no change. 53 In addition, one study suggested there may be increased Florbetapir in the temporal pole of TBI cases, 51 but another found no change in the temporal pole or entorhinal cortex. 46
Amyloid Deposition in Limbic Regions
— = Not studied; NS = not significant; x = significant.
Evidence for subcortical or white matter Aβ deposition
Seven articles investigated Aβ tracer uptake in subcortical ROIs, including the thalamus, globus pallidus, caudate and putamen (striatum), and cerebellum; the corpus callosum and global white matter (WM) analyses were also included in this category (Table 3, Supplementary Data S4). 42,48,50,51,53,55,56 Four articles using different Aβ tracers (Florbetapir, 48 [11C]-PiB, 42,53 and [18F]-FPYBF-2, 55 ) reported no significant differences in thalamic Aβ burden in TBI. In addition, there was no significant difference in [18F]-FPYBF-2 binding in the globus pallidus. 55 In the striatum (caudate and putamen), two studies found some evidence of altered Florbetapir uptake, with the caveat that alterations were time-dependent, 66 and potentially related to more severe diagnoses and/or higher APOE ε4 allele frequency in the TBI group. 56 Similarly, another study showed significantly higher [11C]-PiB distribution volume ratios in the striatum of TBI patients relative to controls. 42 However, two articles using [11C]-PiB, 53 and [18F]-FPYBF-2, 55 both found no evidence for altered tracer binding in the striatum of TBI cases relative to non-TBI. Next, of the four articles that examined cerebellar ROIs, three found evidence for altered Florbetapir and [11C]-PiB binding, 50,51,53 while another found no change in Florbetapir binding in TBI relative to controls. 56 Two studies investigated Aβ burden in the global WM (using [18F]-FPYBF-2 55 and [11C]-PiB 42 ) but saw no difference between focal TBI patients and controls. Finally, one study saw elevated Florbetapir in the corpus callosum of TBI patients, 50 but another reported no change using [18F]-FPYBF-2. 55
Amyloid Deposition in Subcortical Regions
Evidence for cortical Aβ deposition
Twenty articles assessed at least one measure of cortical Aβ tracer uptake, including global Aβ status/burden, cortical lobes, the cingulate gyrus, and cuneus/precuneus (Table 4, Supplementary Data S5). 41 –56,58 –60,67 14 articles assessed a global cortical ROI, and five found evidence for altered uptake of [11C]-PiB or Florbetapir. 42,49,52,54,56 However, nine articles saw no change in global uptake of [11C]-PiB, Florbetapir, Florbetaben, or [18F]-NAV4694 in TBI patients. 41,43 –45,47,58 –60,67
Amyloid Deposition in Cortical Regions
Many articles considered individual lobar ROIs. In the frontal lobe, four found evidence for altered Florbetapir uptake, 50 –52,56 which might have been related to ongoing cognitive impairment, 56 and/or multiple injuries. 52 Three additional papers using Florbetapir, 29 [11C]-PiB, 31 and [18F]-FPYBF-2, 55 found no change. In the parietal lobe, two studies observed altered Florbetapir uptake in TBI, albeit in opposing directions where one showed lowered uptake, 50 and another elevated uptake. 56 However, five additional studies saw no differences. 46,48,51,52,55
Temporal lobe ROIs were examined by the same seven articles that studied parietal ROIs. Similar to the parietal lobe, four reports noted altered Aβ tracer uptake in TBI cases, 50,51,55,56 with comorbid post-traumatic stress disorder (PTSD) 50 and more severe injuries (diffuse axonal injury [DAI]) 55 as potential contributing factors. However, three articles saw no change in uptake. 46,48,52 In the occipital lobe, results were similarly split. Two studies using Florbetapir, 29,30 and one using [18F]-FPYBF-2, 34 found altered Aβ deposition. 48,51,55 Two studies using Florbetapir, 35,40 and one using [11C]-PiB, 53 found no change in TBI cases. 52,53,56
Seven articles investigated the cingulate gyrus. In the anterior cingulate, two studies from the same group showed elevated Florbetapir uptake, 50,51 while three articles from other groups saw no change. 48,52,53 In the posterior cingulate, four studies observed altered Aβ tracer uptake, 50 –53 but three did not. 46,48,56 Finally, ROIs of the cuneus and/or precuneus were analyzed in seven studies, of which five showed altered tracer uptake, 48,50,51,53,56 albeit in an inconsistent pattern. Though four of the five showed elevated uptake, 50,51,53,56 a case report suggested there was evidence of lowered Aβ. 48 Furthermore, two additional studies found no change in cuneus/precuneus Aβ deposition in TBI. 46,52
Tau
Of the 10 articles that used a tau tracer, eight utilized Flortaucipir, and two used [18F]-MK-6240. The regions with the most concordant evidence for elevated tracer uptake in TBI patients were the MTL and entorhinal cortex, precuneus, and cortical lobes. The major limitations described were a small number of participants with TBI, a paucity of clinical detail, and the potential for recall bias.
Evidence for limbic tau deposition
Six articles examined tau deposition in at least one limbic ROI, including the MTL/hippocampus, entorhinal cortex, and temporal pole (Table 5, Supplementary Data S6). 59 –63,68 In an MTL/hippocampus composite, there was no evidence for altered Flortaucipir uptake. 61 However, results from three studies of noncomposite MTL/hippocampus ROIs suggested elevated Flortaucipir binding in TBI. 60,63,68 In the entorhinal cortex, one study saw no cross-sectional or longitudinal changes to Flortaucipir uptake in TBI-exposed participants, 59 while another article observed elevated Flortaucipir in the transentorhinal cortex of TBI participants. 68 Finally, two studies showed elevated Flortaucipir Standardized Uptake Value Ratio (SUVR) in the temporal pole of TBI participants. 62,68
Tau Deposition in Limbic Regions
Evidence for cortical and/or subcortical tau deposition
Ten articles analyzed tau tracer binding in at least one cortical and/or subcortical ROI, including composite (mesial-temporal, temporoparietal, metatemporal, and neocortex) ROIs, cortical lobes, cingulate gyrus, precuneus, insula, basal ganglia, and substantia nigra (Table 6, Supplementary Data S7). 57 –64,67,68 In composite ROIs, two reports observed elevated [18F]-MK-6240 binding in AD cases compared to controls, but no change in TBI patients. 57,58 Intriguingly, controls demonstrated higher SUVRs than TBI cases. 58 Another article also showed no difference in Flortaucipir binding between TBI and non-TBI participants in mesial-temporal, temporoparietal, and neocortex composites. 67
Tau Deposition in Cortical and Subcortical Regions
Of the three studies that considered frontal lobe/gyrus ROIs, all found elevated Flortaucipir uptake in TBI, 60,62,63 which authors hypothesized could be related to cognitive impairment, 60 and/or comorbid PTSD. 62 Similarly, four studies demonstrated altered Flortaucipir uptake in temporal ROIs, 60,62,63,68 although another reported neither cross-sectional nor longitudinal changes in a temporal composite. 59 Next, five studies saw consistent evidence for elevated Flortaucipir SUVRs in parietal ROIs of TBI cases. 60,62 –64,68 Similarly, four reports of occipital ROIs found elevated Flortaucipir in TBI participants, 61,63,64,68 though one study reported no differences. 62
Five articles showed elevated Flortaucipir in the cuneus/precuneus of TBI participants relative to controls. 60,62 –64,68 Multiple injuries, 64 and comorbid PTSD, 62 were discussed as potential contributing factors. Only one article investigated the insula but found higher Flortaucipir SUVR in TBI participants relative to controls. 62 Similarly, two studies reported elevated Flortaucipir SUVRs in the basal ganglia of TBI participants. 62,63 Finally, the same group observed elevated Flortaucipir in both the substantia nigra, 63 and cingulate gyrus, 64 of TBI cases.
Discussion
Findings from the current body of literature are incongruent regarding an association between TBI and Aβ and/or tau deposition on PET in older adults at risk for AD. The areas with the most concordant evidence for elevated Aβ deposition in TBI patients were the cingulate gyrus and the cuneus/precuneus. However, findings were mixed in most limbic, cortical, and subcortical ROIs. Many studies reported at least some evidence for altered Aβ deposition in TBI, but the patterns were inconsistent and other studies saw no change.
Studies of tau deposition were generally in better agreement than the Aβ studies. This may be a consequence of fewer tau tracers being available, as Flortaucipir was used in all but two reports, but five Aβ tracers were employed. In limbic ROIs, there was fairly consistent evidence for elevated tau in the MTL/hippocampus and temporal pole of TBI participants. In cortical composite ROIs, results were mixed, with some studies finding elevated tracer binding in TBI and others finding no change. The consensus was improved when individual lobes/gyri were considered, which suggests greater utility of cortical parcellations as opposed to composite ROIs. In the cortical lobes, most reports saw increased SUVRs in TBI. Jointly, these studies suggest there may be elevated cortical tau in older individuals with TBI history.
There was also concordant evidence for increased tau in the cingulate, insula, basal ganglia, and substantia nigra of TBI cases, though these ROIs were subject to limited analysis and require confirmatory replications. Similarly, in the precuneus, five studies showed elevated tau in TBI. Together, these articles suggest the precuneus may be vulnerable to pathological Aβ and tau deposition in older adults with TBI history.
Though currently available PET tracers may not be highly sensitive to early Thal stage amyloid deposition, the findings from studies in this review are somewhat consistent with an AD-like pattern of early Aβ deposition in showing the strongest evidence for elevated Aβ in the cuneus/precuneus and cingulate gyrus. 11 Similarly, increased tau SUVRs in the MTL and entorhinal cortex, precuneus, and cortical lobes of TBI cases are also relatively consistent with an AD-like pattern of tau deposition based on the Braak stages. However, the ROIs with higher Aβ deposition tended to reflect early Thal stages, but the ROIs with elevated tau represented nearly all the Braak stages. For example, there was elevated tau in early-depositing regions such as the entorhinal cortex, which typically shows tau in Braak Stage I, but there was also elevated tau in later-depositing regions such as the cortical lobes, which typically demonstrate widespread tau only in Stages V and VI. 13 Though the evidence is not conclusive, these patterns suggest TBI in older adults may be associated with AD-like Aβ deposition, but a more unique pattern of tau deposition. This raises the question of whether elevated tau is consistent with AD or another neurodegenerative disease, such as CTE, 69 –71 which will be discussed later.
The largely contradictory nature of the findings highlights the need for confirmatory replications in large prospective datasets to better understand the relationship between TBI and AD-related protein deposition. Several key factors likely contributed to inconsistency between studies. First, there is significant participant-level heterogeneity, including differing TBI severity, time since injury, mechanisms of injury, and comorbid diagnoses such as PTSD or cognitive impairment. Similarly, there is substantial methodological variation between studies, including inclusion/exclusion criteria, radiotracer and ROI selection, and analysis pipelines.
Variations in injury severity, both between participants in the same study and between samples enrolled across studies, likely contributed to the inconsistency of the findings. Only one study explicitly investigated DAI, but patients with DAI may have shown different patterns of Aβ deposition compared to focal injury patients. 55 Specifically, there was increased Aβ tracer binding in the WM of DAI patients compared to focal injuries. 55 As DAI by definition is associated with widespread WM damage, this finding is logical; however, off-target WM binding is a known issue and may contribute to false positive findings. 72 –74 Nonetheless, there was also widespread variation in injury severity across studies that did not consider DAI. Some studies utilized cohorts with only self-reported TBI, such as ADNI (participants would likely be classified as mild TBI), 46,50,51,60 while others included moderate to severe TBI. 48,53 This variation limits the validity of direct comparison between studies, as injury severity may be associated with differences in protein deposition. 60,66 However, additional investigation of whether severe injuries are associated with greater protein load is needed to clarify if this is a meaningful confounding variable.
Next, the chronicity/acuity of TBI relative to the time of imaging is another likely contributor to the ambiguity of the findings. Previous studies have demonstrated changes in Aβ levels in biofluids, on imaging, and upon pathological examination acutely after TBI, particularly in severely injured individuals with short survival time. 75 –82 Additional evidence of acute post-TBI biomarker changes comes from sports medicine, where biofluid biomarkers to aid in TBI evaluation have been investigated. There is evidence of acute alterations in glial fibrillary acidic protein (GFAP), ubiquitin C-terminal hydrolase L1 (UCH-L1), and neurofilament light chain (NfL) after TBI. 83 –86 However, there is little longitudinal data beyond the injury recovery period and limited evidence of acute changes on tau- or Aβ-PET that could be detectable years after injury. 87,88 In addition, biofluid protein levels return to baseline relatively quickly, which does not support the presence of long-term associations between TBI and protein deposition on PET. 75,83,84,87 As such, in older participants with remote TBI history, biomarkers may have returned to pre-injury levels before participants undergo PET scans for AD biomarkers. Furthermore, most studies included in this review did not measure any fluid biomarkers, so persistent changes to fluid biomarkers could not be assessed to indicate whether concurrent PET changes are expected. Together, it is unclear whether alterations in AD-related protein levels after TBI are detectable on PET years or decades after injury. That evidence notwithstanding, many studies covered in this review lacked the necessary clinical detail to even consider time since injury as a factor. In studies that did characterize time since injury, there were widely varying injury-to-imaging intervals. For example, some participants were >15 years out from their TBI, 42 while others were assessed only weeks to months after injury. 48 This discrepancy limits the utility of direct comparison between studies and likely contributes to the uncertainty of the findings.
Along the same lines, varying mechanisms of injury are another plausible contributing factor to the inconsistency. Many studies could not identify mechanisms of injury, and in others, mechanisms were widely variable, encompassing military service, car accidents, sports participation, and others. This is a significant limitation, as each mechanism of injury may impact the brain in unique ways. For example, TBI resulting from blast exposure may affect the entirety of the brain and body, whereas blunt-force TBIs often result in localized damage. 89,90 If these injuries are reflected in differential tau and/or Aβ deposition patterns, this could partly explain why studies found such widespread results. To improve coherence, it could be informative to compare PET findings from participants whose TBI(s) occurred through similar mechanisms. This would be most feasible for injuries incurred due to sports, military service, or falls, as these are prominently discussed in the literature. 24,27,91 –93 However, another important mechanism for incurring TBI is intimate partner/domestic violence (IPV/DV). 94 Consideration of IPV/DV raises concerns related to demographic differences in the mechanism of injury, as the prevalence of TBIs resulting from IPV/DV may have sex-, age- and/or racial/ethnic group differences. 95 These demographic confounds could represent another source of bias if not carefully controlled. None of the analyzed studies explicitly discussed IPV/DV, but there may be similar demographic confounds in other sources of TBI, such as military service or sports participation, which were part of this review. 20 –22,25,96 Future studies should thoroughly document mechanisms of injury whenever possible, as meta-analyses between participants with similar injury mechanisms may result in improved consensus.
Furthermore, we cannot exclude the possibility that altered protein deposition on PET represents CTE rather than AD in some older adults with TBI. This possibility is of particular concern for participants with numerous injuries, as repetitive TBI is the most significant risk factor for CTE. 30,31,97 CTE is characterized pathologically by hyperphosphorylated tau inclusions in the sulcal depths around blood vessels in the brain. 30,98,99 CTE and AD also show near-inverse patterns of tau accumulation. 69 –71 While tau in AD spreads according to the Braak stages and does not diffusely reach the neocortex until later in the disease, 13 tau in CTE tends to originate in the neocortex and spread interiorly in later disease stages. 30,69,98,99 In this review, studies frequently observed elevated tau in the MTL and entorhinal cortex, which is consistent with early tau deposition in AD. However, there was also elevated tau in the precuneus and throughout the cortical lobes, which cannot be specifically attributed to either AD or CTE. CTE can only definitively be diagnosed post-mortem, though a differential diagnosis may be established based on both the distribution and magnitude of tau tracer binding in suspected CTE patients, as well as clinical symptoms and history of brain injury exposure. 100,101 However, it is impossible to distinguish what unique tau fold isoforms are present solely based on a positive tau-PET scan. Work from cryogenic-electron microscopy (cryo-EM) has demonstrated that tau folds differently in CTE as opposed to AD, 102 but affinities of the available tau radiotracers to these disease-specific tau conformations is unclear. Some reports have attempted to clarify whether radiotracers have higher affinities for specific tau conformations, but more work is needed, as there is currently no known tau tracer that binds only to CTE-type tau and not AD-type tau. 103,104 A number of tau-PET tracers have been evaluated and compared head-to-head for use in non-AD dementias, including suspected CTE. 103,104 These tracers, including Florzolotau ([18F]-PM-PBB3), [18F]-PI-2620, and [18F]-CBD-2115, may be more optimized for use in suspected CTE than previous-generation tracers such as Flortaucipir, but none are specific to CTE pathology. 103 –109 As such, the inability to determine whether a positive tau-PET scan is consistent with CTE, AD pathology, or some combination of AD, CTE, and/or other neurodegenerative diseases is challenging, particularly when mixed pathology is suspected. These limitations further prevent a definitive statement of whether the evidence supports a link between TBI and AD-related protein deposition in older adults.
Though a CTE diagnosis cannot be made without pathological examination, some comorbid conditions that can be diagnosed during life, such as PTSD or cognitive impairment, may also be associated with protein deposition in individuals with TBI. PTSD is of particular interest, given that PTSD rates are high in military servicemembers, 110 and survivors of IPV/DV, 111 populations that also have a high prevalence of TBI. 91,92,95 Potential associations of PTSD with Aβ and tau are even more intriguing given that studies cited in this review found conflicting evidence for altered tau and/or Aβ deposition in participants with PTSD. 47,50 In addition, animal and human studies have found mixed evidence for associations between PTSD alone and AD biomarkers, 112 –114 and PTSD has also been linked to risk for cognitive impairment and dementia. 115 –119 Though conflicting, the evidence to date indicates there may be unique effects of PTSD on AD biomarkers independent of TBI. However, particularly in veteran populations, which were the only population with PTSD included in studies cited by this review, it may be challenging to identify the unique effects of TBI on AD biomarkers due to the high comorbidity of PTSD. As such, in individuals with PTSD and TBI where there is evidence for alterations to AD biomarkers, it is near-impossible to determine whether those changes are specifically attributable to TBI. PTSD as a comorbid diagnosis alongside TBI thereby contributes to the difficulty of directly comparing studies and likely also contributed to ambiguity in the findings.
The potential effects of concurrent cognitive impairment on the ambiguity of the findings also cannot be discounted, but as with many previously discussed confounding variables, associations are very difficult to identify. Nine of the cited studies did not include any impaired participants 39,40 (Table 1). In the remaining 17 studies that included impaired individuals, there were multiple levels of impairment. 49,60 (Table 1). For example, some participants were impaired due to AD, while others who did not meet the criteria for AD were classified as mild cognitive impairment, in line with clinical guidance for diagnosing AD. 120 In addition, it is possible that impairment in some participants is due to non-AD dementia (potentially CTE, as discussed earlier). 121,122 Finally, some participants were acutely impaired as a consequence of their TBI, as opposed to AD or another dementia. 43,48 Unless impaired participants are carefully matched based on diagnosis and reasons for impairment, comparison of AD protein deposition on PET has little value, particularly in the context of TBI where misdiagnosis is of concern. 121,122 As such, it is incredibly challenging to parse out the relationship between cognitive impairment and changes to Aβ- and/or tau-PET in older adults with TBI history when participants with widely varying diagnoses are combined for analysis. Too much of the variability in PET findings could be attributed to variation in cognitive status as opposed to TBI, which prevents the establishment of reliable conclusions.
Next, methodological differences across studies almost certainly contribute to the mixed findings. Participant recruitment and inclusion/exclusion criteria are critical points in which bias can be introduced into studies. For example, in some cohorts, particularly those whose primary focus is aging or AD and not TBI, participants with severe TBI history may be excluded from the sample during the medical history screening. 123 This exclusion criterion could bias the findings in favor of lower associations between TBI and protein deposition.
Another important methodological element that introduces variability is the challenge of comparing studies that used different tau and Aβ tracers, as each tracer may produce unique findings. Previous studies indicate Aβ tracers have only slight variability of sensitivity and specificity in AD, which suggests this concern may be minimal. 65,124 However, tau exists as two isomers, 3R and 4R, identified based on the number of microtubule-binding repeats present, 125 and currently available tau tracers may bind with distinct affinities to these isomers. 104 In addition, cryo-EM studies have identified unique tau folds specific to certain diseases, such as CTE, to which the sensitivity of tau tracers has not yet been conclusively determined. 102,126,127 Though differential tracer binding may explain some variability in the findings, only two tau tracers were used, and results from tau studies were more consistent than the Aβ studies, where five different tracers were used. This suggests radiotracer selection may indeed impact the findings and replicability of studies, as coherence seemingly improved when studies used the same tracer.
Similarly, ROI selection and definition may have contributed to the discrepancy in the results. As noted in the results, studies that used summary or global ROIs were challenging to compare with studies that used individual lobar or gyrus ROIs, which may have also resulted in differing conclusions (Table 6). The use of summary or global ROIs severely limits consideration of regional changes to protein deposition, as the summarization of information results in the loss of nuances between smaller parcellated brain regions. In addition, summary ROIs limit the ability to detect localized tau and/or Aβ deposits, which is especially concerning given that tau deposits in CTE may be confined to specific regions. 30,98,99 Outside the use of summary ROIs, variation could also have been introduced if labs used different segmentation methods to identify ROIs. For example, a lab using Freesurfer could generate different results than a lab using SPM (or any other image processing suite) if ROI boundaries are defined slightly differently. 128 –130 ROI selection and definition may be specific to image processing and analysis pipelines, which represents another opportunity for variability to be introduced across studies.
Unique image processing and analytical pipelines utilized by separate labs can introduce significant variation in findings, even if the exact same dataset is analyzed. This failure to replicate phenomenon has been demonstrated previously in neuroimaging modalities, particularly in fMRI. 131 As neuroimaging and most scientific fields in general face a replication crisis, the impact of image processing and analysis pipelines on diverging results cannot be ignored. This is a highly probable source of variability in the included studies and is emphasized by the observation that findings from studies published by the same group (and thereby using the same or substantially similar methods) tended to share more similar findings than did the whole body of literature. 32,47,50,68 Careful documentation of methods, in addition to the potential adoption of standardized pipelines, is necessary to improve replicability across multiple labs.
Considering the numerous potential sources for variation between studies, it is not surprising that there is little concordance in the field regarding an association of TBI with AD-related protein deposition on PET. Given the contrasting nature of the findings and the wide variation in methods used, there is not enough evidence to conclusively state whether an association exists between TBI and Aβ and/or tau deposition in older individuals, though there was more consistent evidence for elevated tau. Such associations may be present, as many of the included studies suggest, but the evidence is far from consistent, and additional data and replications are needed.
Moving forward, analyses of new and/or expanded samples will be of significant value, but replications in smaller datasets that have been previously analyzed may identify major methodological contributors to discrepancies in the results. Significant attention should be devoted to improving standardization and alignment of PET processing and analysis pipelines so it is easier to determine whether there is a consistent signal in the data or whether findings are spurious or coincidental. This is especially important when investigating associations with a small effect size and wide variation between participants, such as the association of TBI with Aβ and/or tau deposition in older individuals.
Conclusion
There was poor consensus across studies that utilized Aβ and/or tau PET to investigate AD-related protein deposition in older adults with TBI. The regions that showed the most robust evidence for increased Aβ deposition after TBI were the cingulate and cuneus/precuneus, while the strongest evidence for elevated tau was in the MTL, entorhinal cortex, precuneus, and cortex. However, inconsistent findings highlight the need for replication in large and prospective datasets to better characterize the association of TBI with AD-related protein accumulation in aging populations.
Transparency, Rigor, and Reproducibility Summary
No review design or strategy was preregistered for this review. We uploaded the searches conducted for each database to searchRxiv as a search repository and followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines for systematic reviews to ensure reproducibility and rigor. In addition, we took steps to ensure rigor during the screening process, including using Covidence to blind the screening process, having at least two reviewers screen each article at both steps of the screening process, and discussing conflicts in the screening process to come to a consensus decision for each article.
Footnotes
Acknowledgments
The authors thank Drs. Sujuan Gao, Karmen Yoder, Kelly Nudelman, Tom McAllister, Yu-Chien Wu, and Wei Wu for valuable discussions.
Authors’ Contributions
K.M.D.: Contributed to the conception, design, literature search, screening process, and writing of the article. C.J.V.: Conducted the literature search and contributed to the writing of methods section. S.C. and D.A.D.: Contributed to the screening process. S.L.R. and A.J.S.: Contributed to the conception and revision of the article. All authors read and approved the final version of the article.
Funding Information
AS and SR receive support from multiple NIH grants that provided funding for this project (T32 AG071444, P30 AG010133, P30 AG072976, R01 AG019771, R01 AG061788, R01 AG057739, U19 AG024904, R01 LM013463, R01 AG068193, U01 AG068057, and U01 AG072177).
Author Disclosure Statement
KD, CV, DD, SC, and SR do not have any relevant disclosures. AS has received support from Avid Radiopharmaceuticals, a subsidiary of Eli Lilly (in kind contribution of PET tracer precursor); Bayer Oncology (Scientific Advisory Board); Eisai (Scientific Advisory Board); Siemens Medical Solutions USA, Inc. (Dementia Advisory Board); NIH NHLBI (MESA Observational Study Monitoring Board); Springer-Nature Publishing (Editorial Office Support as Editor-in-Chief, Brain Imaging and Behavior).
Supplementary Material
Supplementary Data S1
Supplementary Data S2
Supplementary Data S3
Supplementary Data S4
Supplementary Data S5
Supplementary Data S6