
Abstract
Previous studies have shown that administration of high doses of morphine in the acute phase of spinal cord injury (SCI) significantly undermines locomotor recovery and increases symptoms of chronic pain in a rat spinal contusion model. Similarly, SCI patients treated with high doses of opioid for the first 24 h postinjury have increased symptoms of chronic pain 1 year later. Whether these adverse effects are driven by morphine only or all opioids compromise recovery after SCI, however, is unknown. Based on our previous findings we hypothesized that activation of the kappa opioid receptor (KOR) is key in the morphine-induced attenuation of locomotor recovery after SCI. Thus, we posited that opioids that engage KOR-mediated signaling pathways (morphine, oxycodone) would undermine recovery, and clinically relevant opioids with less KOR activity (fentanyl and buprenorphine) would not. To test this, we compared the effects of the clinically relevant opioids on locomotor recovery and pain in a male rat spinal contusion model. Rats were given a moderate spinal contusion injury followed by 7 days of intravenous morphine, oxycodone, fentanyl, buprenorphine, or saline, and recovery was assessed for 28 days. All opioids produced analgesia on tests of thermal, mechanical, and incremented shock reactivity. However, tolerance developed rapidly with buprenorphine administration, particularly with daily administrations of 5 morphine milligram equivalent (MME) buprenorphine. Opioid-induced hyperalgesia (OIH) also developed across days following administration of higher doses (10 MME, 20 MME) of morphine and oxycodone. Fentanyl and buprenorphine did not produce OIH. Contrary to our hypothesis, however, we found that high doses of all opioids reduced recovery of locomotor function. Unlike the other opioids, the effects of buprenorphine on locomotor recovery appeared transient, but it also produced chronic pain. Morphine, oxycodone, and buprenorphine decreased reactivity thresholds on tests of mechanical and incremented shock stimulation. In sum, all opioids undermined long-term recovery in the rat model. Further interrogation of the molecular mechanisms driving the adverse effects is essential. This study provides critical insight into pain management strategies in the acute phase of SCI and potential long-term consequences of early opioid administration.
Introduction
Opioids are a standard treatment for pain in the acute phase of spinal cord injury (SCI), with approximately 80% of patients receiving morphine within the first 24 h. 1 Unfortunately, we found that early administration of high opioid doses increases the risk for the development of long-term pain. 1 Commensurate with these clinical findings, we found that morphine administered in the acute phase of SCI increases the incidence of chronic pain symptoms and attenuates locomotor recovery in a rat model.2–5 Our previous studies suggest that these adverse effects of morphine may be mediated, at least in part, by activation of the kappa opioid receptor (KOR).2,6,7 Therefore, we hypothesized that opioids that do not engage the KOR system might produce robust analgesia in the acute phase of SCI without compromising recovery.
Clinically relevant opioids differ in their affinity for KORs, as well as their potential for indirect activation of this receptor. Although both morphine and oxycodone bind directly to KORs, 8 fentanyl does not.9,10 In addition to direct binding, however, opioids can increase the synthesis of the endogenous KOR agonist, dynorphin. Binding to any of the classical opioid receptors can engage extracellular signal-regulated kinase 1/2 (ERK1/2), the upstream kinase that is responsible for cAMP-response element binding protein (CREB) regulation of dynorphin gene expression.11,12 Supporting this, we showed that 3 days of intravenous morphine administration increases ERK1/2 and dynorphin expression in spinal microglia and macrophages. 13 Rivat et al. also found that subcutaneous fentanyl increased spinal dynorphin levels, 14 and Király et al. found that intraparietal oxycodone increased prodynorphin and dynorphin A in the hippocampus and striatum. 15 By upregulating dynorphin, even opioids, such as fentanyl, with lower affinity for KORs would indirectly activate these receptors.
Buprenorphine, however, is a unique opioid that is both a mu opioid receptor (MOR) agonist and a KOR antagonist. 16 Buprenorphine is commonly used to treat opioid dependence,17,18 but there is growing interest in its utility as an analgesic.19–22 Indeed, relative to other opioids, buprenorphine has low addiction potential, a ceiling effect on respiratory depression, and decreased withdrawal symptoms.23,24 Moreover, buprenorphine-induced KOR antagonism has been suggested to reduce the negative outcomes of repeated opioid administration, including opioid-induced hyperalgesia (OIH) and antinociceptive tolerance.25,26
Based on our previous findings we hypothesized that, because it is a KOR antagonist, buprenorphine would not undermine long-term recovery of locomotor function after SCI, even if it elevated dynorphin via the ERK1/2 pathway. To test this, we compared the effects of four clinically relevant opioids morphine, fentanyl, oxycodone, and buprenorphine on recovery after SCI. We hypothesized that opioids that directly bind to KORs (morphine, oxycodone) would increase symptoms of chronic pain and undermine locomotor recovery, whereas opioids that do not directly engage KORs (fentanyl) or have antagonistic actions at KORs (buprenorphine) would not. Surprisingly, our data did not support our hypothesis. Although buprenorphine had less effect on locomotor recovery, than morphine and oxycodone, it significantly increased long-term pain symptoms after SCI, and rats rapidly developed tolerance to this drug.
Methods
Subjects
In total, data from 100 male Sprague-Dawley rats were used in these studies. There was an n = 5–8 for SCI subjects treated with each of the opioids at each of three doses (5 morphine milligram equivalent [MME], 10 MME, 20 MME). There were also 8 saline-treated SCI subjects, and 4 sham subjects treated with 10 MME of each opioid or saline. Based on a priori exclusion criteria, an additional 11 rats were excluded from the study. Seven rats had Basso, Beattie, Bresnahan [BBB 27 ] scores greater than 6 on day 1 postinjury, and the jugular catheters lost patency in 4 rats. Mortality also occurred in SCI rats: 1 rat treated with saline, 1 with morphine (5 MME), 3 with fentanyl (one 10 and two 20 MME subjects), and 6 with oxycodone (one 5, one 10, and four 20 MME subjects) died prior to the end of the experiment. Subjects died on average 9.5 ± 4.55 days after SCI.
Surgery
Intravenous jugular catheter implantation
Five days prior to the spinal contusion injury, rats were implanted with a jugular catheter, as previously described.7,13,28 Briefly, the rats were anesthetized (isoflurane, 2–3% gas), and a catheter (0.025-mm internal diameter) was inserted into the jugular vein and threaded through a subcutaneous tunnel, to exit between the scapulae. The catheter was connected to a subcutaneously implanted back mount pedestal (Plastics Inc., Roanoke, VA). All incisions were then closed with Michel clips, and the catheter was flushed with 150
Spinal contusion injury
Five days after the jugular catheter implantation, the rats were given a moderate contusion injury as previously described. 29 Briefly, subjects were anesthetized using isoflurane (2–3% gas), and the dorsal spinous process at T12 was removed, exposing the spinal cord. A moderate spinal contusion injury was produced with an impact force of 150 kdynes and a 1 sec dwell time (Infinite Horizon [IH] spinal cord impactor). For the sham surgery, rats were anesthetized, and the incision and laminectomy were performed; however, the rats were not impacted with the IH device.
Following the contusion injury, subjects’ bladders were expressed manually every morning (between 7:00 and 9:00 a.m.) and evening (between 4:30 and 6:30 p.m.). This schedule was maintained until subjects regained bladder function (operationally defined as voiding on their own for three consecutive days).
Drug administration
Drug administration began 24 h following SCI or sham surgery. Morphine (generously supplied by the National Instutute on Drug Abuse (NIDA) drug supply program), oxycodone (oxycodone hydrochloride; generously supplied by the NIDA drug supply program), fentanyl (fentanyl citrate; Millipore Sigma), and buprenorphine (buprenorphine hydrochloride; PAR Pharmaceutical) were diluted or reconstituted in ultrapure Milli-Q water. Then subjects were given 5, 10, or 20 MME (see Table 1 for conversions) of an opioid, or an equivalent volume of saline for the first 7 days following injury. Drugs were administered intravenously (IV) in 0.5 mL (Table 1) doses, separated by 1 h, up to the prescribed daily dose. For the 5 and 10 MME groups, subjects received the same dose of drug each day. For 20 MME groups, subjects received 10 MME on days 1–2, then 20 MME on days 3–7 to reduce the risk of overdose. Heparin-saline (150
Opioid Conversion Factors and Dosing
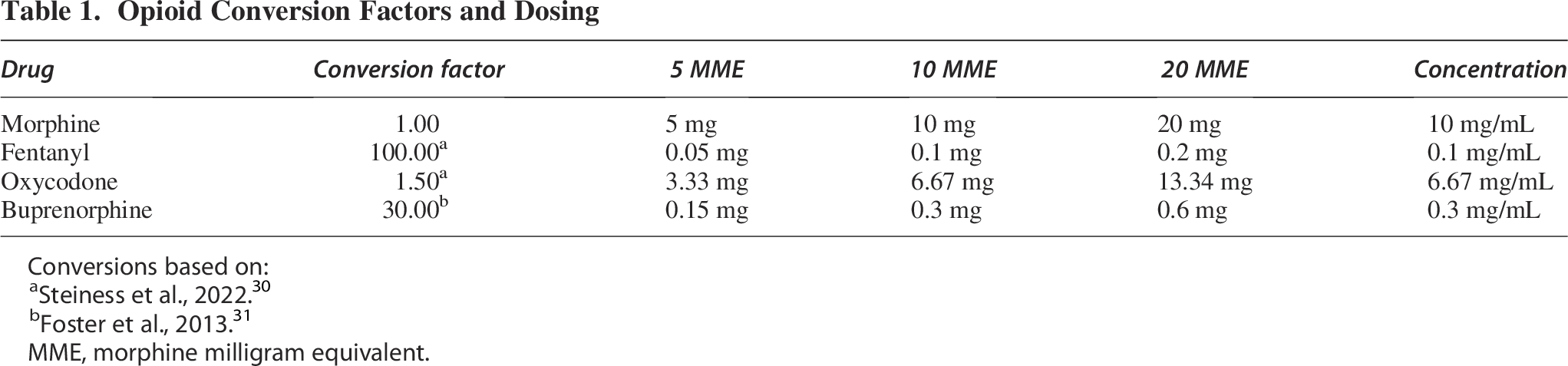
Conversions based on:
Steiness et al., 2022. 30
Foster et al., 2013. 31
MME, morphine milligram equivalent.
Assessment of sensory reactivity
Thermal reactivity was assessed with the tail-flick test prior to drug treatment on days 1, 3, and 7, as well as 30 min post-treatment. Sensory reactivity in the absence of an opioid (prior to treatment and at least 23 h after a previous dose of drug) enables assessment of OIH, and testing while the drug is active (30 min post administration) allows for evaluation of analgesic tolerance, as described in our previous publications.2,6,7,28,29 The tail-flick test was also conducted on days 14 and 28 post injury, to assess long-term pain. Briefly, the rats were placed in restraining tubes and a hot light was focused onto the rat’s tail. The latency to flick the tail away from the light was recorded. If a subject failed to respond, the test was automatically terminated after 8 sec of heat exposure.
Mechanical reactivity was measured using von Frey filaments (Semmes-Weinstein Aesthesiometer, Stoelting Co.) on days 1, 3, 7, 14, and 28 post injury, as previously described.3–5,7,28,32 Von Frey filaments of increasing diameter were applied to the L4 dermatome on the plantar surface of the hind paw and the force applied was increased until the rats exhibited both a motor (hind paw withdrawal) and vocal response. 33 If a motor and/or vocal response was not observed, testing was terminated with the filament corresponding to a force of 300 g.
Nociceptive reactivity was also assessed with an incremented tail shock stimulus. Shock thresholds were assessed using a manual shocker (H13-15, Coulbourn Instruments) that allowed for continuous variation of shock (AC, constant current) intensity. Briefly, subjects were placed in the restraining tubes and their tails were secured inside an electrode constructed from lightweight fuse clips. Test shocks were applied 7–9 cm from the base of the tail. Test shock intensity was gradually incremented at a rate of 0.1 mA every 1 sec. After both movement and vocalization responses were detected, the shock was terminated. If a subject failed to respond, the test trial was terminated when the shock intensity reached 1.2 mA. The incremented shock test was conducted on days 1, 3, 7, 14, and 28 post injury.
Assessment of locomotor recovery
Locomotor recovery was assessed using the BBB scale.27,34 BBB scores were collected daily for the first 7 days post injury, then every other day from days 9 to 15, and every third day from days 18 to 27. For this assessment, care was taken to ensure that all scorers were blind to a subject’s experimental treatment, and that investigators had high intra- and interobserver reliability (all r’s > 0.89).
Motor function was also assessed using the DigiGait Image Analysis System (Mouse Specifics). This automated system is used to quantify spatial and temporal indices of gait on a motorized treadmill. Subjects were tested prior to surgeries and 2 days prior to tissue collection (27 days following SCI injury or 6 days following sham laminectomy). Rats were acclimated to the testing room for 30 min and were allowed to explore the DigiGait apparatus for 2 min on the day prior to baseline testing. Rats were reacclimated the day before testing on day 27 post injury. In each testing period, the treadmill was held at a constant speed of 20 cm/sec with no incline/decline. Subjects were recorded for 1 min. As SCI rats often have difficulty producing coordinated, weight-supported stepping, even 4 weeks post injury, a 10 sec window of representative gait was selected for analysis. Gait symmetry was derived as an index of locomotor coordination. Gait symmetry is the ratio of forelimb-to-hindlimb stepping. Perfect gait symmetry is 1.00, with deviations from 1.00 denoting a loss of coordination. Thus, there is an inverse relationship between the deviation of the gait symmetry score from 1.00 and locomotor function (i.e., higher deviation scores are indicative of worse locomotor function).
Histology
On day 29 post injury, subjects were deeply anesthetized (100 mg/kg pentobarbital) and perfused intracardially with phosphate-buffered saline followed by 4% paraformaldehyde. A 1.5-cm segment of spinal tissue centered on the lesion site was collected for cryostat sectioning. Every 10th transverse slice (20 μm) was collected, mounted, and stained with Cresyl violet for Nissl substance and Luxol fast blue for myelin. 27 Sections ±0, 600, 1200, 1800, and 2400 μm rostral and caudal from the lesion epicenter were analyzed using Stereo Investigator software (MFB Bioscience). Three indices of lesion magnitude were derived for each section: damaged tissue, residual gray, and white matter.
A correction factor, derived from age-matched, undamaged cord sections, was used to control for variability in the section area across subjects, as previously described. 35 An index of relative lesion (% relative lesion) was derived by summing the amount of “missing” tissue (i.e., atrophy, necrosis, or apoptosis) and the measured damaged tissue area.
Statistical analysis
Three-way ANOVAs were used to compare the analgesic efficacy, and the development of tolerance, with 10 MME opioid doses in SCI and sham rats across days. Three-way ANCOVAs were used to examine the development of OIH across injuries, drugs, and days, with day 1 scores serving as covariates to control for individual variation in post surgical acute pain. For both tolerance and OIH, the effects of drug dose for each opioid were then separately explored, in SCI rats only, with two-way ANOVAs and days post injury as the repeated measure. Two-way ANOVAs were also used to assess the dose-dependent effects of each opioid on chronic pain and weight loss.
Repeated-measures ANCOVAs were used to examine the effects of opioids on locomotor recovery. Day 1 BBB scores collected prior to drug treatment served as covariates, with days post injury as the repeated measure, and dose as the independent variable in these analyses. Gait symmetry data, acquired with the Digigait, were analyzed with a one-way ANOVA, comparing doses for each opioid separately.
Similarly, two-way ANOVAs were used to analyze the effects of opioid dose and spinal location, relative to the lesion epicenter, on tissue sparing and tissue loss. For all ANOVAs, Tukey’s multiple comparison tests were used for further analyses of significant main effects or interactions (p < 0.05).
Results
All opioids produced robust analgesia on the tests of thermal, mechanical, and electrical stimulation
To determine the effects of SCI on analgesic efficacy and the development of tolerance, post administration reactivity thresholds from 10 MME sham and SCI groups were compared for each drug. Not surprisingly, SCI increased reactivity thresholds relative to shams. There was a main effect of surgery for both motor and vocal [F(1, 43) = 11.98, 15.99, respectively, p < 0.001; Fig. 1] responses to von Frey stimulation, as well as drug–surgery interactions [both Fs (4, 43) > 6.67, p < 0.001]. Rats with SCI had increased motor withdrawal and vocal responses relative to shams, reflecting decreased sensory function with injury. There was also a main effect of surgery on motor and vocal responses to incremented tail shock [F(1, 43) = 5.08, 5.44, respectively, p < 0.05; Fig. 1], and drug–surgery interactions for both motor and vocal responses to tail shock [both Fs (4, 43) > 2.66, p < 0.05]. Sham saline rats were more sensitive to stimulation than all other groups.
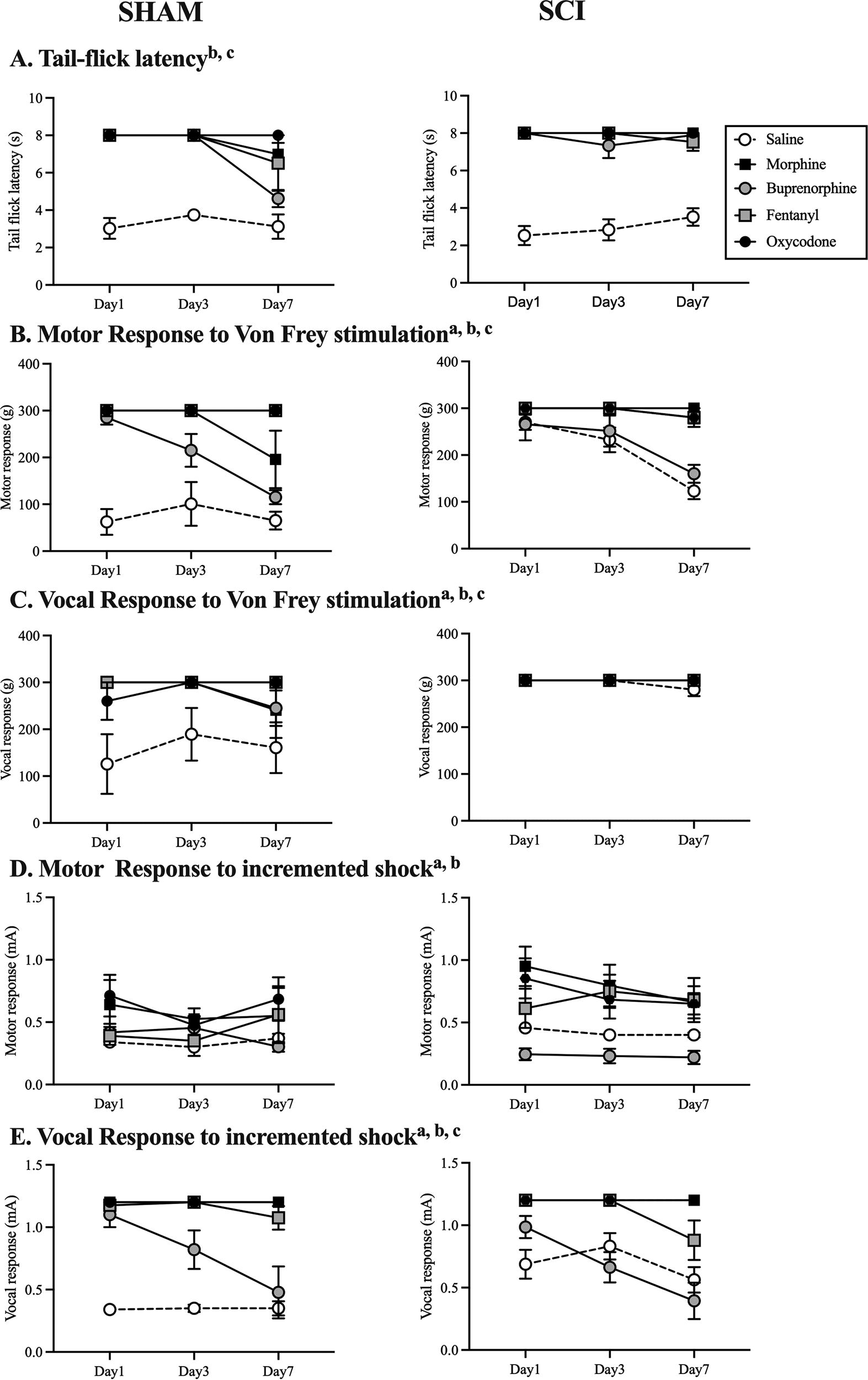
Acute SCI decreased sensory function, increasing reactivity thresholds 30 min post opioid and saline administration with thermal (tail-flick,
Irrespective of injury, however, all opioids produced robust analgesia compared with saline controls (Fig. 1). There was a main effect of drug for each sensory test: tail-flick [F(4, 43) = 6.04, p < 0.0001], motor [F(4, 43) = 46.33, p < 0.0001], and vocal [F(4, 43) = 9.04, p < 0.001] response to von Frey, and motor [F(4, 43) = 11.73, p < 0.0001] and vocal [F(4, 43) = 45.33, p < 0.0001] response to incremented shock. All opioids produced analgesia (all p’s < 0.05), but buprenorphine had significantly less efficacy than the other drugs and did not differ from saline on the incremented shock test. There was also a main effect of day on each of the tests [all Fs (2, 86) > 4.01, p < 0.05], except for the motor response to incremented shock, driven primarily by buprenorphine-treated SCI rats decreasing reactivity thresholds with time post injury. There were significant day–drug interactions for the motor response to von Frey stimulation [F(8, 86) = 4.77, p < 0.001] and vocal response to incremented tail shock [F(8, 86) = 5.33, p < 0.001]. Tolerance was evident in the buprenorphine-treated rats by day 7 post injury on all tests. There was also evidence of breakthrough pain in shams by day 7 for morphine on the tail-flick test and von Frey test, and for fentanyl with incremented tail shock.
Next, we examined the effects of dose on analgesic efficacy across days in the SCI rats only. Two-way ANOVAs revealed main effects of dose for each of the opioids on all tests except for vocal responses to von Frey stimulation. Main effects of day, indicative of tolerance, were also found for all opioids. With morphine treatment, there was a significant effect of day on all tests [all Fs (2, 44) > 4.19, p < 0.05; Supplementary Fig. S1], except for the tail-flick test. These effects were primarily driven by the saline-treated rats. Saline-treated rats displayed increased sensitivity, with lower motor reactivity thresholds, from days 1 to 7 (p < 0.0001) on the von Frey test. For vocal responses, however, the 5 MME morphine dose also lost efficacy across days (p < 0.001). Both the 5 and 20 MME morphine doses had reduced analgesic efficacy on day 7 (p < 0.05) for motor responses to incremented shock. The saline-treated and the 5 MME morphine rats also had decreased vocal reactivity thresholds to incremented shock on days 3 and 7 (all p’s < 0.05). Overall, tolerance appeared to develop with the 5 MME, but not the 10 or 20 MME, dose of morphine.
There was little evidence of tolerance with oxycodone and fentanyl administration. There was a main effect of day for both oxycodone and fentanyl for motor responses with von Frey stimulation [F(2, 44) < 10.48, p < 0.0001 for both drugs; Supplementary Figs. S2 and Figs. S3, respectively]. As described for morphine, this effect of day was driven by the saline controls (p < 0.0001). With oxycodone, there was also a significant effect of day on the incremented shock test [F(2, 44) = 5.99, p < 0.005]. Tolerance developed with 5 MME oxycodone, with motor reactivity thresholds decreasing from day 1 to 7 (p < 0.005). Similarly, tolerance developed for the vocal response to incremented shock stimulation [F(2, 44) = 6.82, p < 0.001] with the 10 MME dose of fentanyl. Overall, however, these opioids remained highly effective at blocking evoked pain responses across the 7 days of administration.
In contrast, tolerance developed to buprenorphine on all tests [main effect of day, F(2, 44) > 5.58, p < 0.001], except for the tail-flick test (Supplementary Fig. S4). Motor thresholds with von Frey stimulation decreased for all doses across days (p < 0.001). Vocal reactivity thresholds also decreased across days for the 5 MME dose (p < 0.0001). On the incremented shock test, tolerance developed with the 5 MME dose for motor responses (p < 0.0005), and for all doses for vocal reactivity thresholds (p < 0.05). Therefore, despite producing acute analgesia, buprenorphine was highly susceptible to tolerance.
Repeated morphine and oxycodone administration drives OIH after SCI
To examine the effect of SCI on OIH, evoked pain thresholds were assessed prior to opioid (0 or 10 MME) administration in the sham and SCI rats and analyzed with three-way ANCOVAs. To account for effects of acute SCI on sensory function per se, day 1 preadministration values were used as covariates in all analyses. There was a main effect of SCI on motor [F(1, 42) = 12.85, p < 0.001] and vocal [F(1, 42) = 12.58, p < 0.001] responses to von Frey filament stimulation, as well as motor and vocal responses to incremented tail shock [F1, 42) = 4.58, 4.78, respectively, p < 0.05; Fig. 2]. SCI significantly decreased reactivity thresholds from days 7 to 1, irrespective of drug treatment. The decrease was not evident in shams.
There were also significant main effects of drug on the tail-flick and von Frey tests [across all tests, F(4, 42) < 3.19. p < 0.05]. There was a tendency for an effect of drug on the incremented shock test, but it was not significant for either motor or vocal responses (p < 0.06). Effects of drug on the tail-flick test and in response to von Frey stimulation were driven by the SCI rats. There was no evidence of OIH in shams. In the SCI group, both morphine and oxycodone decreased motor [F(1, 11) = 4.98, 5.83, respectively, p < 0.05] and vocal [F(1, 11) = 7.52, 29.57, respectively, p < 0.05; Fig. 2B and C] response thresholds with von Frey stimulation. Rather than OIH, buprenorphine significantly increased tail-flick latencies relative to saline controls [F(1, 12) = 12.83, p < 0.05] suggesting decreased sensory function with buprenorphine treatment. Overall, these results suggest that SCI increases the potential for OIH, relative to shams.

OIH developed in SCI rats with morphine and oxycodone treatment, but not in shams. SCI significantly increased reactivity thresholds on the von Frey
Examining the effects of repeated morphine administration in SCI rats only, there were increases in tail-flick latencies across days [F(2, 66) = 4.14, p < 0.05] in the 10 and 20 MME groups. Rather than reflecting OIH, these data suggest reduced spinal reflexes with repeated administration of the higher doses of morphine. For motor reactivity with von Frey stimulation, there was also a significant effect of day [F(2, 66) = 22.83, p < 0.0001] (Supplementary Fig. S1). Rats treated with saline, 10, and 20 MME morphine displayed significant decreases in motor thresholds on day 7, relative to day 1 and 3 (p < 0.05). These data suggest that nociceptive sensitivity was developing irrespective of morphine administration, but it is noted that the magnitude of effects was greater in the 10 and 20 MME groups, relative to saline controls. There were also significant main effects of day on vocal reactivity to von Frey stimulation [F(2, 66) = 13.82, p < 0.0001] and incremented tail shock [F(2, 66) = 7.34, p < 0.005]. OIH developed in the 10 and 20 MME groups, with the rats displaying significantly lower thresholds on day 7 relative to day 1 in both groups (p < 0.05 in all comparisons). On the incremented tail shock test, rats treated with 5, 10, or 20 MME morphine displayed evidence of OIH, with reduced reactivity thresholds on day 7 relative to days 1 and 3 in all groups (p < 0.05). There was no effect of day for motor reactivity on the incremented tail shock test. Overall, the data indicate that higher doses of morphine lead to the development of OIH, with reduced vocal reactivity thresholds for mechanical and electrical stimulation, after SCI.
Similarly with oxycodone treatment there was evidence of OIH (Supplementary Fig. S2), specifically in rats treated with the 10 MME dose. There was a main effect of dose on motor and vocal reactivity with von Frey stimulation [F(2, 66) = 16.39, 20.11, respectively, p < 0.0001]. As found for morphine, the saline as well as the 5 and 10 MME oxycodone groups displayed decreased motor reactivity thresholds with von Frey stimulation across days, reflecting nociceptive sensitization after SCI that might be independent of drug on this task. There was a selective and robust decrease in vocal reactivity thresholds on day 7 relative to days 1 and 3, however, in the 10 MME group on this task. There was no effect of oxycodone on the tail-flick test or incremented tail shock test.
In contrast to morphine and oxycodone, there was little evidence of OIH with repeated fentanyl administration (Supplementary Fig. S3). There was an effect of day for motor reactivity on the von Frey test [F(2, 66) = 14.43, p < 0.0001]. The saline, 5, and 10 MME dose groups displayed lower reactivity thresholds across on day 7, relative to days 1 and 3 (p < 0.05 in all cases). As noted for morphine, these data suggest that mechanical nociceptive sensitivity was developing after SCI irrespective of fentanyl administration. There was no evidence for OIH for vocal or motor reactivity on any other test.
Buprenorphine administration also produced little evidence of OIH. There was a main effect of dose on tail-flick responses [F(3, 21) = 7.47, p < 0.001]. As can be seen in Supplementary Figure S4, the 10 MME group displayed longer tail-flick latencies across the days [F(1, 12) = 12.83, p < 0.01], relative to vehicle-treated controls, suggesting loss of sensory function in this group rather than OIH. Similarly, there was an effect of dose on motor thresholds on the incremented shock test [F(3, 21) = 9.79, p < 0.001]. All of the rats treated with buprenorphine displayed lower reactivity thresholds, relative to vehicle-treated controls across the 3 days of testing, including prior to treatment [5 MME (F(1, 11) = 10.91, p < 0.01; 10 MME (F(1, 12) = 11.93, p < 0.01; 20 MME, F(1, 10) = 35.96, p < 0.001]. For vocal reactivity on the incremented shock task, rats treated with 10 MME buprenorphine also displayed a transient increase in reactivity thresholds on day 3, relative to days 1 and 7 (p < 0.05 in both cases). Dose did not significantly affect motor or vocal reactivity thresholds on the von Frey test.
Despite producing analgesia, all opioids undermined locomotor recovery
BBB scores were balanced across groups prior to morphine treatment [F(3, 22) = 1.17, p > 0.05], with average BBB scores ranging from 0.83 ± 0.38 for 10 MME treated subjects to 2.08 ± 0.57 for 20 MME treated subjects. Despite starting with similar BBB scores, all doses of morphine undermined recovery (Fig. 3A). The 5 MME [F(1, 11) = 17.91, p = 0.001], 10 MME [F(1, 11) = 22.36, p < 0.001], and 20 MME [F(1, 11) = 48.25, p < 0.001] groups significantly differed from the saline group. There was no effect of morphine dose on gait symmetry. Across all opioids and dose groups, however, gait symmetry scores were correlated with BBB scores (r2 = 0.103, p = 0. 023).
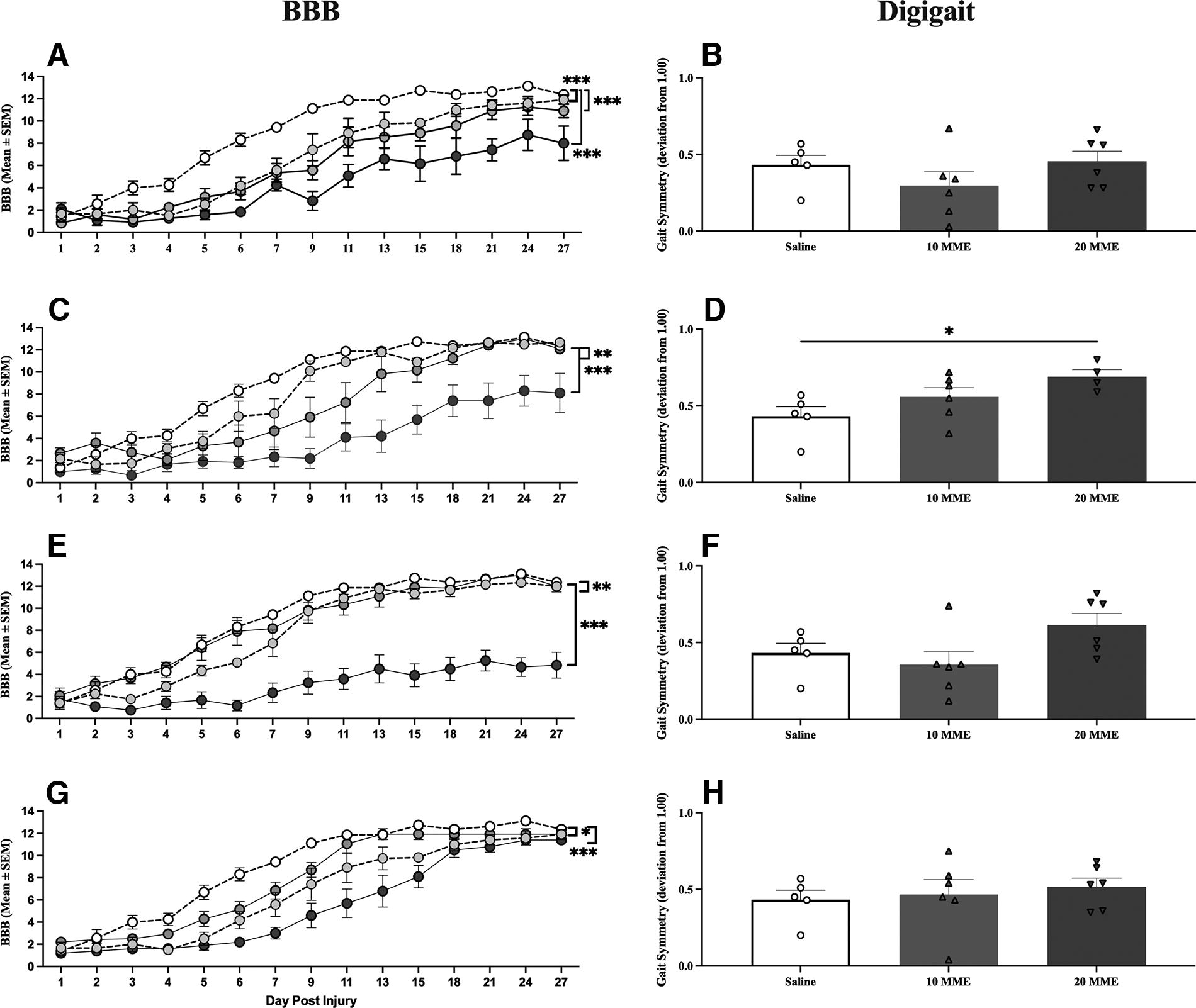
All opioids significantly undermined locomotor recovery. There was a dose-dependent effect of
There was also a negative effect of oxycodone on locomotor recovery. There were no significant group differences in BBB scores prior to drug treatment [F(3, 22) = 2.58, p > 0.05], with average BBB scores ranging from 1.37 ± 0.31 for 20 MME treated subjects to 2.66 ± 0.49 for 10 MME treated subjects. Post-treatment, however, there was a main effect of dose [F(3, 20) = 16.07, p < 0.001; Fig. 3C]. The 5 MME [F(1, 11) = 11.88, p < 0.005], 10 MME [F(1, 11) = 15.06, p = 0.005], and 20 MME [F(1, 10) = 46.44, p < 0.001] groups had significantly worse locomotor recovery than saline controls. Notably, the effects in the 5 and 10 MME groups appeared to be driven by the first 2 weeks of recovery. A one-way ANOVA of gait symmetry scores also revealed a main effect of dose on gait symmetry [F(2, 14) = 4.15, p < 0.05; Fig. 3D]. Rats treated with 20 MME of oxycodone performed significantly worse than saline controls (p < 0.05).
For fentanyl, BBB scores ranged from 1.1 ± 0.62 for 5 MME treated subjects to 2.1 ± 0.67 for 10 MME treated subjects on day 1 postinjury, with no significant differences across groups [F(3, 22) = 0.30, p > 0.05]. Again, however, there was a main effect of dose on locomotor recovery [F(3, 21) = 47.63, p < 0.001; Fig. 3E]. The 5 MME [F(1, 11) = 16.24, p < 0.005] and 20 MME [F(1, 11) = 169.28, p < 0.001] groups, but not the 10 MME [F(1, 11) = 0.80, p > 0.05] group, significantly differed from saline-treated controls. As found for oxycodone, the effects seen in the 5 MME group appear to be driven by the first 2 weeks following injury. There was also a trend for a dose-dependent decrease in gait symmetry [F(2, 14) = 3.14, p = 0.074; Fig. 1F].
Buprenorphine also appeared to transiently undermine recovery. There were no differences between groups prior to drug administration [F(3, 22) = 2.32, p > 0.05], with average BBB scores ranging from 1.22 ± 0.25 for 20 MME treated subjects to 2.16 ± 0.33 for 5 MME treated subjects. After treatment, there was a main effect of dose [F(3, 21) = 15.75, p < 0.001; Fig. 3G]. All doses undermined recovery relative to saline-treated subjects; 5 MME [F(1, 11) = 13.87, p < 0.01], 10 MME [F(1, 12) = 9.99, p < 0.01], and 20 MME [F (1, 10) = 59.20, p < 0.001]. In contrast to what was seen with higher doses of morphine, oxycodone, and fentanyl, however, all buprenorphine-induced deficits appeared to be due to delayed recovery rather than long-term locomotor deficits. There was no effect of dose of buprenorphine on gait symmetry [F(2, 14) < 1.0, p > 0.05; Fig. 3H).
Irrespective of KOR activation, opioids increased pain in the chronic phase of SCI
Replicating our previous studies,3–5,7,13,28,29,32 morphine administration increased the expression of chronic pain. There was a main effect of morphine dose on vocal responses to von Frey stimulation [F(3, 22) = 3.72, p < 0.05; Fig. 4A]. This effect occurred with all doses of morphine, but only the 5 MME group significantly differed from saline (p < 0.05). There were no significant main effects or interactions on any other test (all Fs < 3.00, p’s > 0.05).
Oxycodone had a more robust effect, than morphine, on chronic pain. There was a main effect of dose on motor [F(3, 21) = 6.00, p < 0.005; Fig. 4B] reactivity with von Frey stimulation and motor [F(3, 21) = 3.83, p < 0.05; Fig. 5B] reactivity to tail shock. Rats treated with 10 MME of oxycodone displayed lower reactivity thresholds than those treated with saline or 5 MME oxycodone (p < 0.05) on the von Frey test, and lower reactivity thresholds than saline controls on the incremented shock test (p < 0.05). These results suggest that oxycodone produced significant symptoms of chronic pain evoked with mechanical and electrical stimulation.
As found for acute pain, fentanyl did not affect the development of chronic pain. There were no main effects of dose or any interactions between dose and day post injury on any pain test (all Fs < 2.76, p’s > 0.05).
However, contrary to our hypothesis, and despite only transient effects on locomotor recovery, buprenorphine produced chronic pain. There was a main effect of dose on motor responses to von Frey stimulation [F(3, 22) = 5.28, p < 0.01; Fig. 4D]. Both the 5 MME and 20 MME buprenorphine group had significantly lower reactivity thresholds than saline controls (both p’s < 0.05). There was also a main effect of dose on the motor [F(3, 22) = 11.07, p < 0.001] and vocal [F(1, 22) = 3.94, p < 0.05] responses to incremented shock (Fig. 5D). Irrespective of dose, buprenorphine-treated rats had significantly lower motor reactivity thresholds than those treated with saline (all p’s < 0.05). The 10 MME treatment group also had lower vocal thresholds than the 5 MME group. There was no effect of dose for von Frey vocal reactivity thresholds, but there was an effect of time [F(1, 22) = 10.99, p < 0.01]. All buprenorphine-treated rats displayed lower vocal thresholds on day 14 relative to day 28 of testing. Irrespective of treatment all groups, including saline, became less sensitive to tail-flick across days [F(1, 22) = 5.014, p = 0.036, data not shown], but treatment with the 10 and 20 MME doses increased reactivity levels relative to the vehicle-treated controls on this task (p < 0.07).
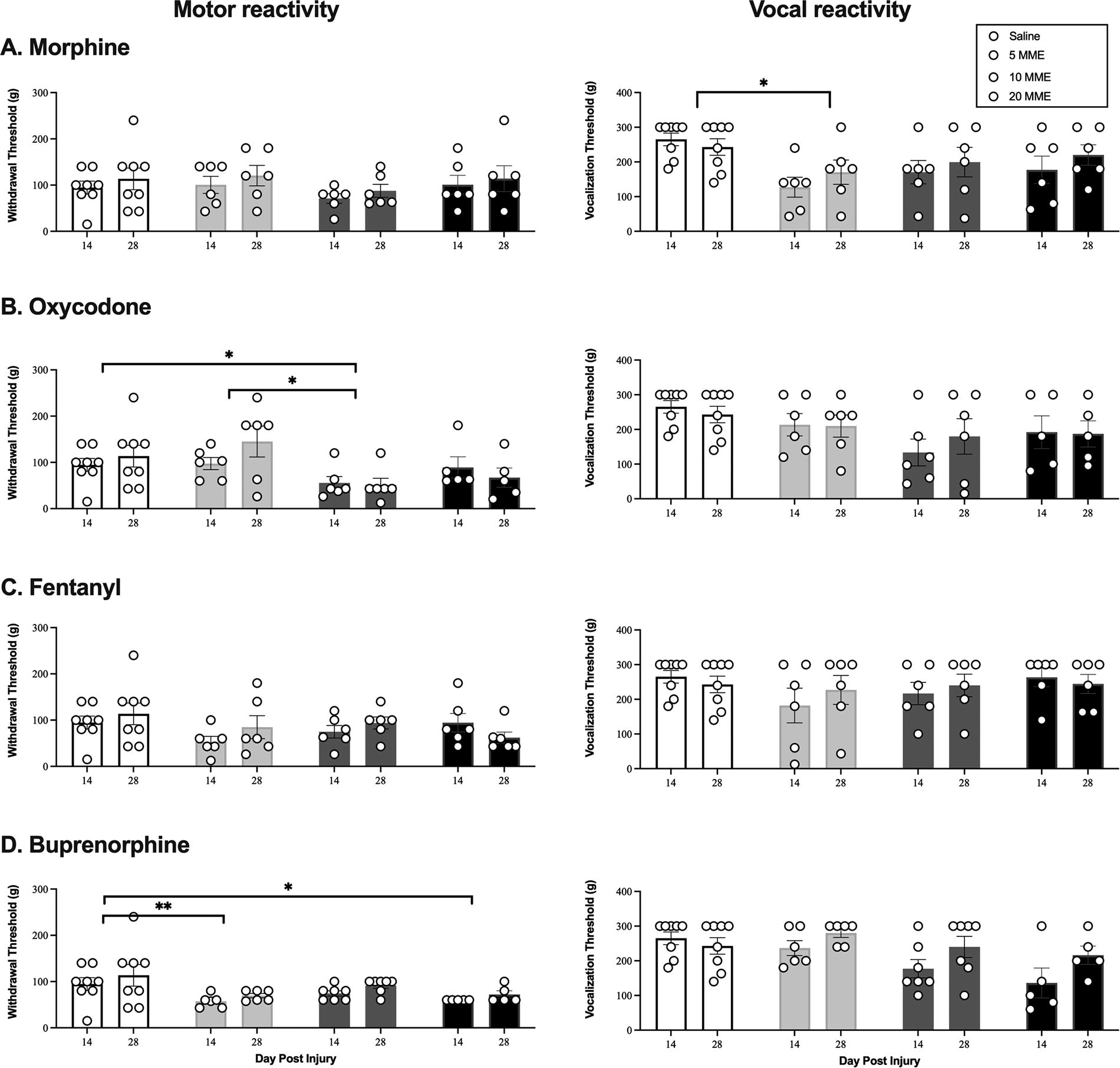
Morphine, oxycodone, and buprenorphine decreased mechanical reactivity thresholds in male rats, relative to saline-treated SCI controls, in the chronic phase of injury. There was a significant effect of dose of morphine
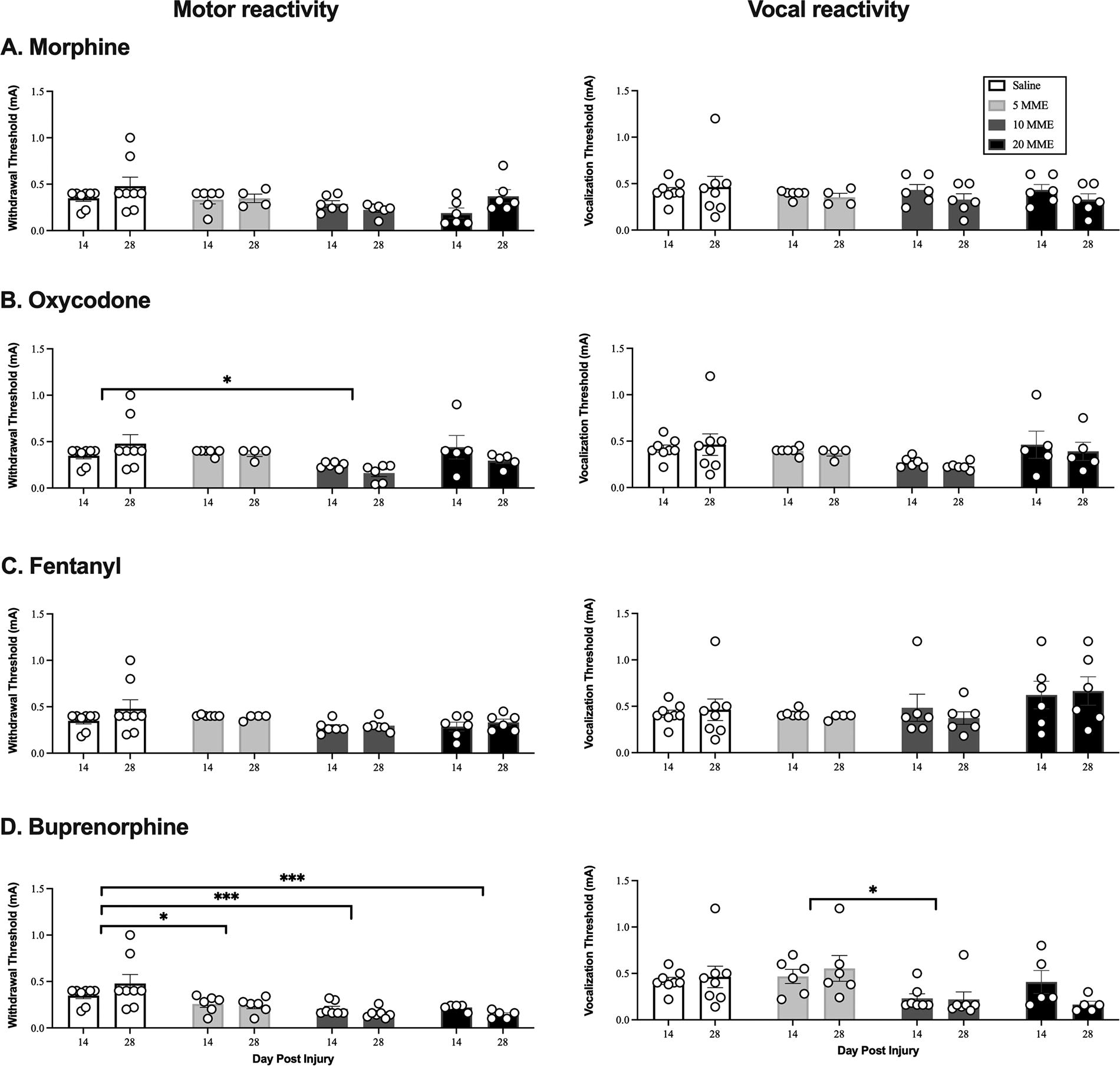
Oxycodone and buprenorphine increased reactivity to incremented shock in the early chronic phase of injury. Morphine
Early opioid treatment reduced weight gain following SCI
Effects were greatest with higher opioid doses (10 and 20 MME), but all opioids and doses increased weight loss (Fig. 6) and increased the amount of time that rats took to return to baseline weights after SCI, relative to saline controls: morphine [F(3, 22) = 36.17, p < 0.0001], oxycodone [F(3, 21) = 10.11, p < 0.001], fentanyl [F(3, 22) = 8.99, p < 0.001], and buprenorphine [F(3, 22) = 28.80, p < 0.001]. In the 20 MME morphine and oxycodone groups, subjects never returned to presurgery body weights.
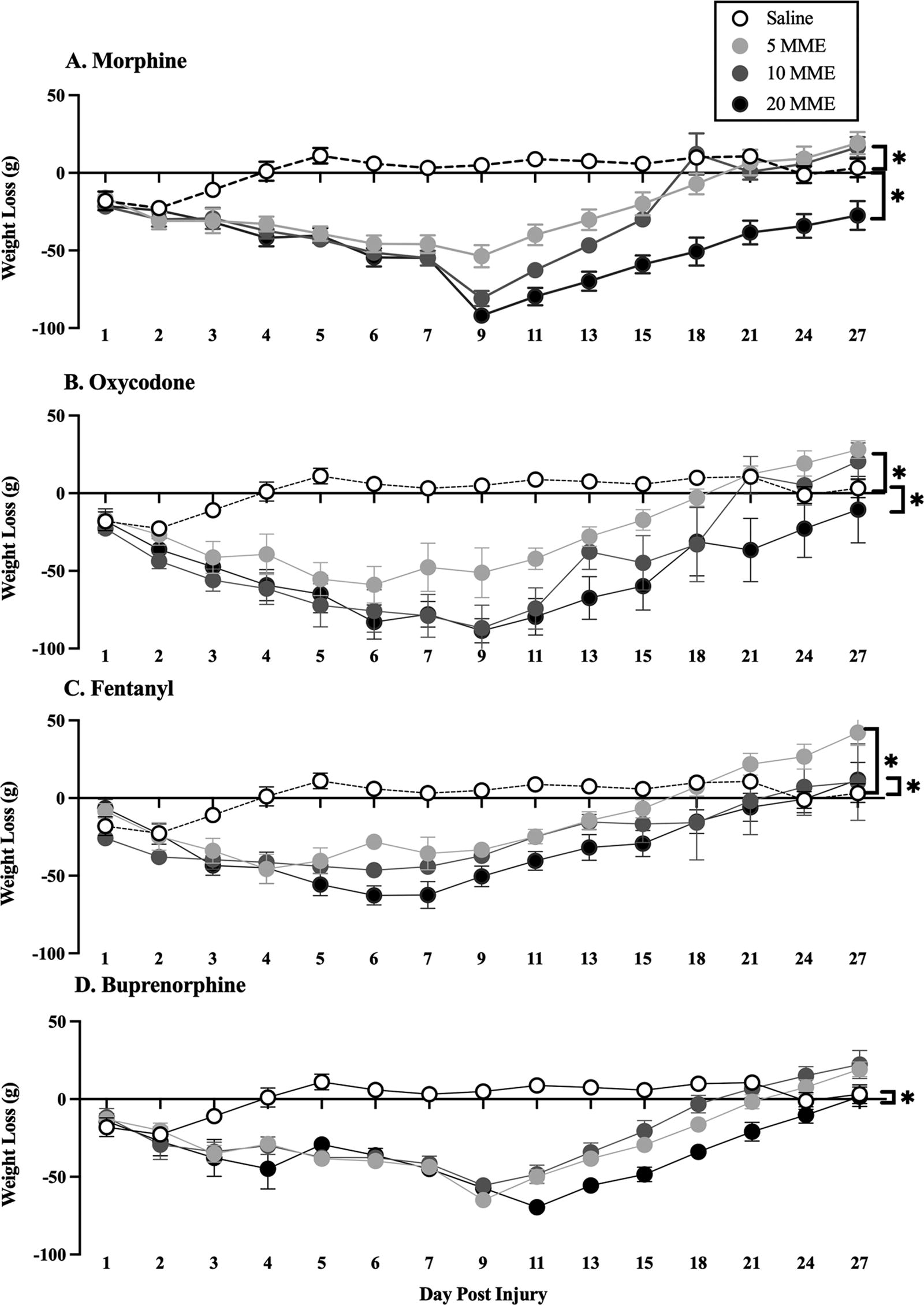
Opioid administration reduced weight gain following SCI. Seven days of
Acute opioid treatment increased lesion size
There was no effect of morphine, oxycodone, or fentanyl on % relative lesion rostral, caudal, or at the center of the lesion (Fig. 7). There was, however, a dose-dependent effect of buprenorphine on lesion size [F(3, 19) = 3.34, p < 0.05]. Post hoc analysis showed that 20 MME of buprenorphine increased lesion size (p = 0.05; Fig. 7) relative to the 10 MME group. There was no effect of 5 and 10 MME buprenorphine relative to saline controls, on lesion size rostral, central, or caudal to the lesion site (all F < 3.0, all p’s > 0.05).
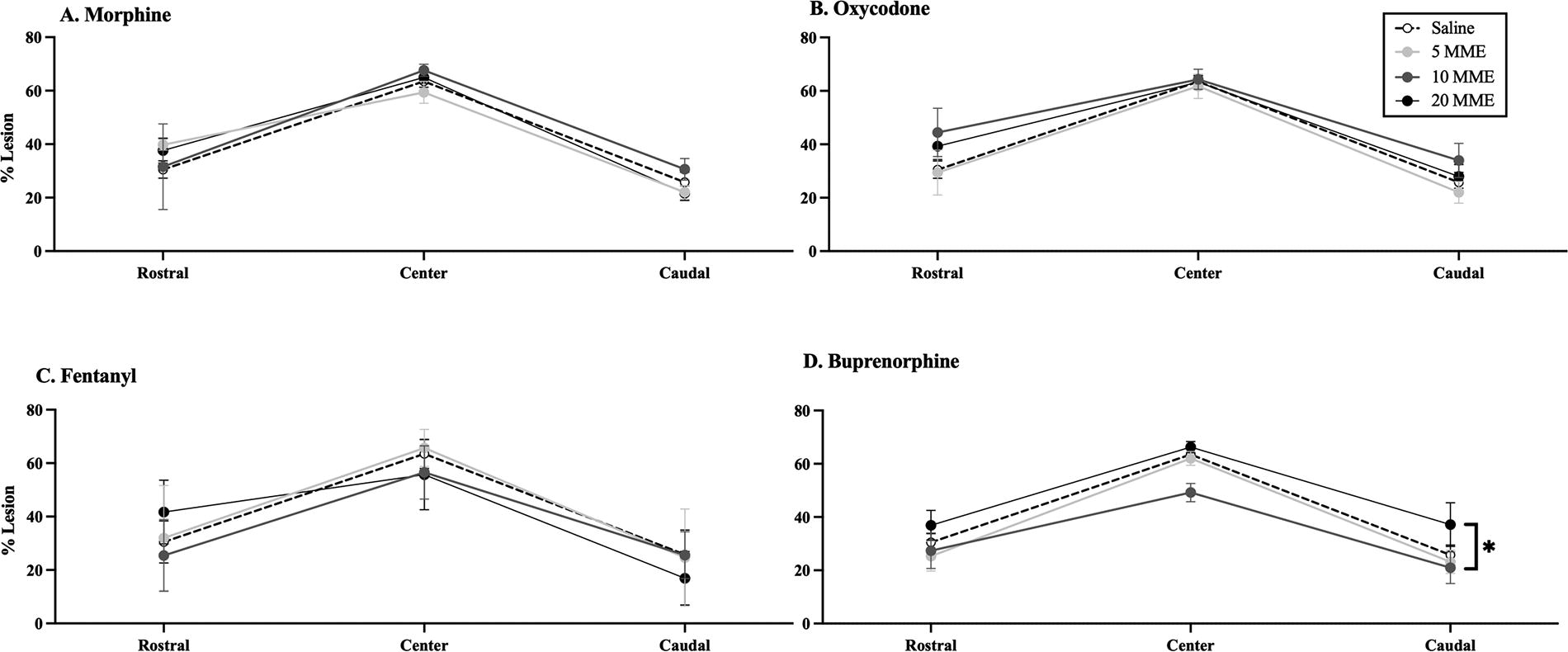
Buprenorphine administration increased lesion size. Although there were similar tendencies for an increased lesion size rostral to drug treatment,
Discussion
Irrespective of KOR binding profiles, there was a dose-dependent effect of all opioids on locomotor recovery. Replicating our previous results, high doses of morphine significantly undermined recovery of locomotor function and increased the development of pain.4–7,13,28,29,32 Supporting our hypothesis, oxycodone, which directly binds to KORs, also undermined locomotor recovery. Surprisingly, however, a high dose of fentanyl also reduced recovery and there were transient effects of buprenorphine on locomotor function. Morphine, oxycodone, and buprenorphine also increased symptoms of chronic pain, and all drugs slowed weight gain post injury with long-term effects of the high doses of morphine and oxycodone. These data indicate that all opioids have the potential to undermine recovery.
We expected morphine and oxycodone to undermine recovery, but we did not expect fentanyl and buprenorphine to have adverse effects. Although all of these opioids activate the MOR our previous studies suggest that MOR activation is not directly mediating adverse effects on pain and locomotor recovery after SCI. DAMGO, a selective MOR agonist, did not produce adverse consequences when administered intrathecally after SCI, 2 and CTOP, a MOR antagonist, did not block the adverse effects of morphine on locomotor recovery. 36 It is possible that indirect effects on KOR activity may underlie the attenuation of locomotor recovery seen with these drugs. As described previously, morphine, fentanyl, and oxycodone have been shown to increase dynorphin expression.13–15 Intrathecal dynorphin has been demonstrated to induce motor dysfunction even in intact rats,37–40 and the immunoreactivity of dynorphin within a SCI lesion has been shown to be correlated with the degree of injury and subsequent motor recovery. 38 Thus, despite different opioid receptor binding affinities, many opioids, including fentanyl, could increase spinal dynorphin expression and undermine locomotor recovery. Notably, buprenorphine could also elevate dynorphin via the ERK1/2 pathway, and with a half-life of 2.2 h,41,42 may not be sufficient to block secondary KOR activation by dynorphin. Nonetheless, while buprenorphine slowed recovery in this study, by day 28 buprenorphine-treated rats had recovered to levels commensurate with vehicle-treated controls. These data support the idea that KOR activation may be key in the long-term attenuation of locomotor recovery induced by opioids after SCI.
Although buprenorphine did not undermine long-term recovery of locomotor function, it did produce symptoms of chronic pain. Similar to morphine and oxycodone, buprenorphine decreased mechanical and electrical stimulation reactivity thresholds. As seen in our previous studies,13,28 the most robust effects of opioids administered IV were on the von Frey test of mechanical stimulation with no effects on thermal reactivity. The location of spinal circuitry engaged by stimulation of the hind paw versus the tail, with the lumbar circuitry mediating the hind paw withdrawal reflex located closer to the injury site than the sacral circuits mediating the tail flick, may underlie the test-dependent effects of opioids observed in the current study. At the receptor level, our data also suggest that activation of the KOR may not underlie the development of opioid-induced pain. Indeed, in our previous studies we have shown that the development of OIH with repeated morphine administration is not blocked by the KOR antagonist, nor-binaltorphimine. 7 In the current study, morphine and oxycodone produced acute OIH and, as reported by others, 43 we saw rapid development of tolerance with repeated buprenorphine administration, perhaps reflecting the recruitment of additional pronociceptive pathways. Indeed, rather than KOR-mediated, the negative effects of opioids on pain may be elicited by dynorphin-mediated NMDA activation. 7 As noted previously, binding to any of the classical opioid receptors can engage CREB regulation of dynorphin gene expression.11,12 Dynorphin at high levels can bind and activate NMDA receptors,44,45 and excessive NMDA activation can lead to both central sensitization and cell death.46–51 With buprenorphine, the KOR is blocked which would leave higher levels of glial-derived dynorphin available for NMDA receptor binding. Counterintuitively, therefore, buprenorphine may increase susceptibility to the development of pain after SCI, as seen in the current study. NMDA-mediated cell death would also negatively impact tissue sparing as we saw with the higher dose of buprenorphine. Alternatively, the reduced efficacy of buprenorphine in attenuating acute pain may have contributed to the development of chronic pain. Studies have shown that uncontrolled nociceptive input can sensitize pain pathways, increase blood–spinal cord barrier permeability, and reduce recovery after SCI.52–56 Further investigation into the mechanisms driving the development of chronic pain with each of these drugs, including acute pain, KOR-antagonism, and dynorphin levels, is necessary.
This study takes an important step in investigating the effect of various clinically relevant opioids on SCI recovery, but there are limitations. To reduce animal numbers, a single saline-treated group served as a control for all opioids. Although we do not expect that additional groups would elicit different results, this should be acknowledged. Furthermore, we did not analyze the effects of opioid treatment on long-term pain outcomes in shams. Future studies should investigate whether morphine and oxycodone increase the incidence of long-term pain outside the context of SCI. The doses used in this study are also higher than those used clinically. However, doses used in rodent studies cannot be equated to those applied in the clinic. Although the plasma half-life of morphine, for example, is very similar for rats and humans,57,58 the Kp,uu, active influx/efflux processes, and metabolites differ across species and conditions.59–61 As each of these variables will affect the efficacy and dose that reaches the central nervous system, we have based our doses on those self-administered by T11 SCI rats in the acute phase of injury.62,63 There are also differences in the half-life of each of these drugs used, and differential cumulation might impact outcomes. For example, morphine has a half-life of 115 min whereas fentanyl has a half-life of approximately 50 min in rats.64,65 The protracted half-lives of some of the drugs may produce a cumulative effect that must be considered when making direct comparisons in analgesic efficacy. Finally, both the age and sex of people experiencing SCI are changing over time. The incidence of SCI among females is increasing 66 and the percentage of individuals 60 years or older increased from 4.6% in the 1970s to 13.2% in the 2000s. 67 Future studies must investigate the effects of age and sex on opioid-induced attenuation of SCI recovery.
Conclusions
The present study suggests that any opioid administered in the acute phase of SCI can negatively impact recovery. However, some opioids appear to have worse long-term outcomes than others. We suggest, for example, that fentanyl may be able to be used at low doses without producing sustained effects on locomotor recovery or increasing chronic pain. Nevertheless, caution is warranted because at high doses fentanyl undermines locomotor recovery and repeated use is associated with addiction. By contrast, any dose of morphine appears to have significant adverse effects, either increasing pain or attenuating locomotor function. These data suggest that all opioids should be used with caution following SCI, until the molecular mechanisms mediating unwanted effects are identified, addressed, and incorporated into pain management strategies.
Transparency, Rigor, and Reproducibility Statement
The rat is a widely used model for SCI with many similar injury and opioid responses to humans. Subsequently, a large comparative database exists for SCI rat models, as well as validated tests for monitoring recovery of function (motor and sensory). A power analysis performed as part of the study design, as well as our previous studies, indicated that a sample size of 6 is sufficient to reveal adverse effects of repeated opioid treatment on locomotor recovery (effect size (f) = 0.25, DF = 3, 20, power = 0.95, p = 0.05) with a three-way repeated measures ANOVA. With unexpected mortality some sample sizes were reduced to 5. Nonetheless, in the analysis of the data collected for dose-dependent effects of buprenorphine on locomotor recovery, we found significant effects of dose with a large effect size (ηp2 = 0.692) despite a sample size of 5 in the 20 MME group. Therefore, despite the predetermined sample size not being attained, the sample size employed in the experiment proved sufficiently robust to yield powered statistically significant findings. Rats were randomly assigned to groups, with BBB scores balanced across groups prior to opioid or saline administration. Investigators who administered the therapeutic intervention were blinded to group assignment by use of an identically appearing vehicle treatment, and identification of rats based on a sequential numbering system that was independent of treatment conditions. Administration of the opioids began 24 h after injury. Controls used included sham and vehicle-only SCI rats. The statistical tests used were based on the assumptions of normal distributions, using two- or three-way repeated measures ANOVA, with day 1 scores as covariates when appropriate. Tukey’s multiple comparison tests were used for further analyses of significant main effects or interactions (p < 0.05). There is no analytical code associated with this study. The raw data acquired in this study will be made available on ODC-SCI following publication.
Authors’ Contributions
J.R.: Methodology, validation, formal analysis, investigation, resources, writing—original draft, visualization, supervision, and project administration; R.J.: Investigation. L.W.: Investigation. J.B.: Investigation. J.W.: Investigation. A.K.: Investigation. L.D.: Investigation. M.A.H.: Conceptualization, methodology, writing—review and editing, supervision, project administration, and funding acquisition. All authors read and approved the final article.
Footnotes
Funding Information
This study was supported by the NIDA Drug Supply Program and by the Office of the Assistant Secretary of Defense for Health Affairs, through the Spinal Cord Injury Program under Award No. W81XWH-22-1-0522. Opinions, interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the Department of Defense.
Acknowledgements
Authors Disclosure Statement
The authors declare that they have no competing interests or financial conflict interest to disclose.