
Abstract
Traumatic brain injury (TBI) is a leading cause of morbidity and disability, with mild TBI (concussions) representing over 80% of cases. Although often considered benign, mild TBI is associated with persistent neuropsychiatric conditions, including post-traumatic stress disorder, anxiety, and depression. A hallmark of these conditions is impaired fear extinction (FE), the process by which learned fear responses are inhibited in safe contexts. This dysfunction contributes to maladaptive fear expression and is linked to altered neurocircuitry, particularly in the infralimbic cortex (IL), a key region in FE. Despite extensive evidence of impaired FE in patients with mild TBI and animal models, the specific mechanisms underlying this deficit remain poorly understood. This study aimed to address this gap by combining cued-FE behavior, local field potential recordings, and whole-cell patch-clamp techniques to investigate how mild TBI affects IL network activity and excitability in a mouse model of TBI. Our results demonstrate that mild lateral fluid percussion injury significantly impairs FE memory, as evidenced by an elevated cued-fear response during extinction testing 10 days post-injury. Field potential recordings revealed decreased activation of the IL network in both layers II/III and V, which was consistent with the observed behavioral deficits. Further analysis of synaptic physiology revealed an imbalance in excitatory and inhibitory neurotransmission (E/I imbalance) in the IL, characterized by reduced excitatory input and enhanced inhibitory input to neurons in both layers. Moreover, intrinsic excitability was altered in IL neurons after mild TBI. This study provides novel insights into how mild TBI disrupts the neurocircuitry underlying FE, specifically by suppressing IL excitability. These results highlight the importance of understanding the mechanistic disruptions in IL activity for developing therapeutic strategies to address fear-based disorders in patients with mild TBI.
Introduction
Traumatic brain injury (TBI) is a leading cause of morbidity and disability in the United States, 1,2 with approximately 5.3 million people suffering long-term impairments from their injuries. 3 TBI is increasingly being described as a “chronic disease process” due to the initial event often evolving into long-term pathological alterations that require ongoing rehabilitation. 4,5 Mild TBIs, which make up over 80% of head injuries in the United States, 6 are considered a “silent epidemic” because the disabilities that manifest post-injury are predominantly neuropsychiatric and not immediately obvious. 1,7 –11 The classification of these head injuries as “mild” (i.e., concussions) once led to the perception that they are relatively benign. 12,13 However, nearly 5 million patients with mild TBI are diagnosed with neuropsychiatric conditions such as post-traumatic stress disorder, panic disorder, and generalized anxiety disorder; 14,15 and these patients report persistent, debilitating symptomatology that results in poor health and social and economic outcomes. 1,6,7,14,15 Thus, mild TBIs are a significant public health problem, and their pervasive sequelae highlight the need to evaluate how their dynamic neurological properties increase vulnerability to the development of neuropsychiatric disorders.
Notably, poor fear regulation characterizes many of the neuropsychiatric disorders comorbid with mild TBI. 14 –20 Fear optimizes survival 21 –24 and fear-evoking objects and experiences can generate lasting fear memories, enabling individuals to anticipate and respond to environmental threats. However, traumatic experiences can trigger fear memories and subsequent distress in nonthreatening situations, and excess fear expression can become maladaptive and indicate the development of an anxiety and/or stress disorder. 19,20,23,25 –28 Extinguishing fear—i.e., fear extinction (FE)—in appropriate and safe situations therefore preserves mental health. Indeed, impaired FE has repeatedly been found in various fear-based neuropsychiatric disorders associated with trauma, reputably post-traumatic stress disorder, but also phobias, panic disorder, and obsessive-compulsive disorder. 14,17,23,25,27,29 –36 All of these disorders have been reported to occur in mild TBI survivors, 10,14,15,37 and FE is impaired in both human 15,38 –42 and experimentally modeled 43 –47 mild TBI. Thus, impaired FE is a robust clinical endophenotype for these fear-based neuropsychiatric disorders, 25,27,36,48 –50 and dissecting FE and its governing neurocircuitry after mild TBI can provide invaluable insight into the development of therapy for fear-based neuropsychiatric disorders in mild injury survivors.
FE requires learning and remembering that a fear-evoking object or situation is nonthreatening (i.e., safe) after it is repeatedly presented without an aversive consequence. 21,23,51 To form a FE memory, an aversive conditioned stimulus (CS) that triggers a fearful memory is repeatedly presented in the absence of a threatening unconditioned stimulus (US), acquiring a safer new meaning. This learned experience is then consolidated to form a retrievable FE memory capable of attenuating the original conditioned fear responses (CRs) prompted by the CS. 22 –24,51 –54
In rodents, the infralimbic cortex (IL) subregion of the medial prefrontal cortex (mPFC) is essential to the consolidation and retrieval of FE memories. IL activation facilitates FE memory retrieval using electrophysiology, 55 –67 chemogenetic, 65,68,69 pharmacological, 70 –72 and immunohistochemistry 73 –75 techniques. Accordingly, lesioning the IL to diminish activity 76 –78 —as well as silencing the IL using electrophysiologic, 58,61,64,72,79 chemogenetic, 55,68,69,80 and pharmacological 71,72,79,81 –84 manipulations—blocks next-day retrieval of FE memories. Furthermore, infusing protein synthesis inhibitors that block plasticity-induced memory consolidation into the IL also prevents FE memory retrieval. 56,57,79,83 –91 The literature discussed above suggests that the potentiation of IL neurons is necessary for the retention of FE memories. Notably, FE memory retrieval in humans is governed by activation of the ventromedial prefrontal cortex (vmPFC), the human analog of the rodent IL. 32,36,54,92 –97
The IL collaborates with the basolateral amygdala (BLA) to orchestrate FE memory formation by supporting the learning process. Optogenetically enhancing the activity of IL-BLA neurons during extinction training does not affect the ability to extinguish fear over the course of the session but improves extinction memory retrieval the next day. 61,62 Conversely, suppressing IL-BLA neuronal activity during extinction training does not hinder learning but impairs recall the following day. 55,61,62 These findings are consistent with studies using optogenetic 98 pharmacological inhibition 82 or lesioning 77,78 of the IL alone, which similarly impair extinction recall without altering learning. However, in rats, optogenetic enhancement of IL activity during extinction training has been shown to reduce the level of fear expression within-session, suggesting that IL activation enhances extinction learning in this species. 58
Treatment development for mild TBI survivors burdened by fear-based neuropsychiatric disorders is hindered by our incomplete understanding of the circuit dysfunction underlying FE impairment post-injury. Numerous studies have found that FE is impaired after mild TBI in both humans and rodents. 15,38 –46,99,100 However, the differences in behavioral methods (e.g., cued vs. contextual extinction) known to engage distinct neurocircuits 101 –107 complicate comparisons and our ability to discern the specific circuitry dysfunction involved. Furthermore, far less is known about how the IL responds to mild TBI 45,108 or whether its activity correlates with injury-induced extinction impairments. Thus, a key challenge in the analysis of FE impairment after mild TBI is our imprecise understanding of how the IL network and its information-consolidating mechanisms are affected by mild TBI.
To address these outstanding gaps in knowledge in the TBI field, this work combined behavioral and electrophysiology techniques to interrogate FE and IL activation after mild TBI. Importantly, our laboratory has demonstrated that experimentally inducing mild TBI using the well-established lateral fluid percussion injury (LFPI) model 109 –111 decreases mPFC network excitation that is essential to efficacious neurotransmission, 112 highlighting the mPFC’s vulnerability to mild TBI. Here, we present evidence that mild LFPI also impairs FE memory. Consistent with our lab’s previous findings, 112 we found that mild LFPI diminishes overall IL network excitability in concert with a reduction in excitatory neurotransmission and enhanced inhibitory synaptic input onto IL neurons. Overall, this work demonstrates that mild TBI disrupts the balance between excitatory and inhibitory (E/I) neurotransmission, in turn suppressing IL activity that is necessary for adroit FE.
Methods and Materials
Subject details
A total of 120 (58 Sham, 62 LFPI), 6–12-week-old male wild-type C57/BL6 mice (Jackson Laboratory, Bar Harbor, ME, USA) were used for experiments. All experiments and procedures were performed in accordance with the guidelines published in the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Children’s Hospital of Philadelphia (CHOP) Institutional Animal Care and Use Committee (Protocol 0694). Food and water were provided ad libitum. With the exception of one animal, behavioral and physiological experiments were performed on separate cohorts of animals due to the significantly larger number of animals required for electrophysiology experiments (behavior: 16 Sham, 16 LFPI; physiology: 42 Sham, 47 LFPI).
Lateral fluid percussion injury
Mild TBI was induced using LFPI, a well-characterized model known to reproduce pathological responses and symptomology observed in human concussive TBI. 109 –111 LFPIs were performed as part of a two-day procedure, wherein a craniectomy was performed 1 day prior to injury induction to allow 24 h for the subject to recover from the procedure. 112 –115
On day 1, mice were prepared for the surgery after being anesthetized with a mixture of ketamine (2.6 mg/kg) and xylazine (16 mg/kg) via intraperitoneal injection. After confirming the animal was deeply anesthetized with the tail-pinch test, they were placed into a stereotaxic frame (Stoetling, Wood Dale, IL, USA) and the skin above the skull was surgically opened. A trephine instrument was then used to perform a 3-mm diameter craniectomy over the right parietal bone while keeping the dura intact. A modified 3 mm Luer-lock needle hub was superglued with Loctite (Düsseldorf, Germany) over the cranial window and further secured with dental cement. Then the needle hub was filled with a sterile saline solution and closed with a Luer-lock cap. Finally, the animal was placed onto a heating pad until mobile and was returned to its home cage for 24 h.
On day 2, mice were randomly assigned to either the Sham or LFPI group. Mice were anesthetized intranasally with isoflurane (2% oxygen in 500 mL/min) until their breathing reached a rate of one respiration every 2 sec, after which isoflurane administration was stopped. The Luer-lock caps were removed from the hub, and the saline was replaced with new sterile saline. The needle hub was connected to the LFPI device (Department of Biomedical Engineering, Virginia Commonwealth University, Richmond, VA, USA; Custom Design and Fabrication, Sandston, VA, USA) via high-pressure tubing. After normal breathing resumed, but before becoming sensitive to toe pinch, LFPI mice then received a 10–15 ms fluid pulse onto the intact dura of the brain increasing intracranial pressure peaking between 1.4 and 1.8 atm to generate a mild injury. Next, mice were immediately placed in the supine position, and self-righting times, the length of time after injury until a mouse spontaneously rights itself, were measured to assess the severity of the injury. 116,117 The mouse was then placed under isoflurane anesthesia again, the needle hub was removed, and the wound was sutured. All mice were then placed on a heating pad until fully recovered and returned to their home cage. Sham mice also underwent all of the above procedures, including being attached to the LFPI device, except for receiving the fluid pulse. Mice with an excessive righting time (over 6 min), indicating a more severe injury, were excluded from the study. 118 –120 Any mice with a breached dura, herniation, or other gross tissue damage were excluded (Supplementary Fig. S1A and B). Any mice in which bleeding was observed immediately after injury (approximately 1 mouse in 20) were also excluded. If any indication of past or present bleeding was visible when brain slices were prepared 7–10 days following injury, those mice were excluded as well. The incidence of such bleeding 7–10 days after injury is low enough that we do not keep a careful record of it, but we estimate it conservatively at 1 mouse per 50. Motor, coordination, and balance were observably uncompromised both prior to and throughout experiments. All experiments and data analyses were conducted while blinded to group conditions.
Cued-fear extinction paradigm
Cup-handling
To alleviate the stress and aversion associated with human contact, mice underwent a 5-day cup-handling protocol prior to behavior experiments. 121,122 Briefly, mice arrived housed in groups of 3–5 to CHOP’s Department of Veterinary Resources animal welfare facility and acclimated to their holding room for 1–2 weeks. Then, group-housed mice were individually cup-handled in their home cages for 2 min on days 1 and 2. On days 3–5, mice were single-housed in transfer containers with clean bedding and nesting materials and transported to the experimental-procedure room where they habituated to their containers for 45–60 min before being cup-handled for 2 min. Finally, mice were returned to their containers and transported to the holding room before regrouping in their home cages. Mice that did not voluntarily interact with the handler by day 5 were excluded from experiments. This protocol trained mice to disassociate handling from harm, reducing experimenter-induced stress during fear response testing. 121,122
Behavioral apparatuses
Mice were cued-fear conditioned in Context A (Fig. 1B), a fear conditioning chamber (Med Associates, St. Albans, VT, USA) featuring two aluminum walls opposite two clear Plexiglass walls (21.6 × 17.8 × 12.7 cm), a clear roof, and stainless-steel barred floors that delivered the electric foot shock. An LED light provided dim white illumination. The CS consisted of an auditory tone (80 dB, white noise, 20 s) generated by a removable audio stimulus generator, and the sound intensity was confirmed by a sound pressure level measurer during experiments from the same manufacturer. The US intensity was confirmed with an aversive stimulation current test package prior to introducing any mouse to the chamber (Med Associates). Mice underwent cued-FE training and testing in Context B (Fig. 1B), a custom-made cylindrical chamber with Plexiglas walls modified to contain blue and orange stripes, a perforated, opaque black plastic floor, and scented with vanilla extract.
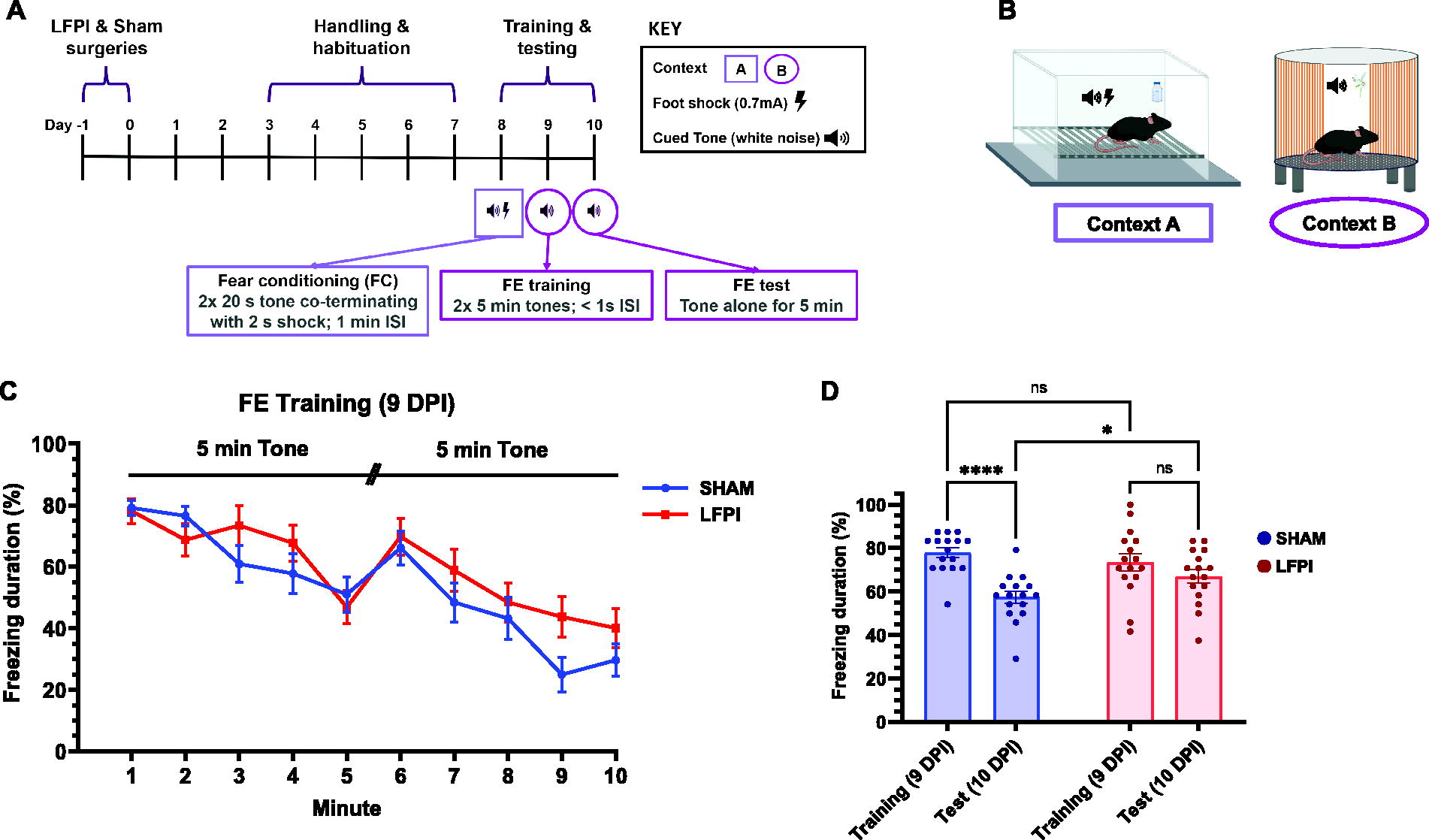
Mild LFPI spares initial extinction learning but impairs extinction memory recall.
Cued-fear conditioning
A cue-based extinction paradigm was used due to its critical dependence on BLA circuitry, a region critical for fear regulation and expression. 54,101,123 To establish the fear memory, mice underwent cued-fear conditioning and extinction experiments 8–10 days post-injury (DPI; Fig. 1A). On the eighth DPI, mice freely explored Context A for 3 min, after which mice were subjected to a 20 s auditory tone cue (CS; 80 dB white noise) that co-terminated with a 2 s electric foot shock delivered from the barred floor (US; 0.7 mA) 18 s into the CS tone. The CS–US pair occurred every 60 s (60 s interstimulus interval, ISI) for three total trials. Mice then freely roamed Context A for 60 s before being removed from the chamber.
Cued-fear extinction training and testing
To establish a cued-FE memory, mice were introduced to novel and distinct Context B, where the CS was presented in the absence of the US for 2 days. On the ninth DPI, mice freely explored Context B for 3 min. Mice were then exposed to the CS alone for two 5 min-trials (<1 s ISI) for a total of 10 min. Finally, mice freely roamed the chamber for 60 s and then returned to their transfer container. On the 10th DPI, mice returned to Context B and again freely explored the chamber for 3 min, after which they were subjected to the CS alone for 5 min before being removed from the chamber.
Electrophysiology
Brain slice preparation
Animal dissection was performed as previously described. 10,112,113,124 Brains were vibratome-sectioned (VT 1000S; Leica Microsystems, Deerfield, IL, USA) at 0.06 mm/s speed to 350 μm thickness in an ice-cold, sucrose-containing aCSF cutting solution containing (in mM): sucrose 202, KCl 3, NaH2PO4 1.25, NaHCO3 26, d-glucose 10, MgCl2 1, and CaCl2 2. Coronal slices containing IL were placed in an aCSF storage solution containing (in mM): NaCl 130, KCl 3, NaH2PO4 1.25, NaHCO3 26, d-glucose 10, MgCl2 1, and CaCl2 2. Stored slices were then incubated in a water heater set to 32°C for 30 min, after which the storage bath was removed and left to reach room temperature for at least 1 h before being used for electrophysiology recordings. Slices were transferred to the recording bath one at a time. Slices were used 1.5–4 h after preparation at 32°C and aerated with 95% O2/5% CO2 throughout preparation and recordings.
Extracellular recording
Coronal slices were transferred to an extracellular recording chamber for field potential recordings. The chamber was equipped with an upright microscope (MZ7.5; Leica Microsystems). Slices were visualized through a 4× objective, and photomicrographic images were taken of the first two recordings and referenced to ensure electrode placement was consistent between experiments. The stimulating electrode was a non-concentric bipolar (#ME12206; World Precision Instruments, Sarasota, FL, USA). Field recording electrodes were fabricated from borosilicate glass pipettes with filaments (#1B150F-4; World Precision Instruments, Sarasota, FL, USA) and pulled to a tip resistance of 2–4 MΩ using a horizontal puller (P-97; Sutter Instrument Company, Novato, CA, USA). The stimulating electrode was positioned onto IL layer II/III, and the recording electrode was placed into IL layer V, angled diagonally and 100 μm apart.
During field potential recordings, brain slices were continuously superfused with room temperature, aerated, standard aCSF at a rate of approximately 1.8 mL/min. Pipettes for field excitatory post-synaptic potential (fEPSP) recordings were filled with standard aCSF. fEPSPs were evoked using a stimulus isolator (ISO-Flex; A.M.P.I., Jerusalem, Israel), acquired by a MultiClamp 700B amplifier and Digidata 1440A digitizer (Molecular Devices, Sunnyvale, CA, USA), and recorded from IL layer V with the pClamp10.7 data acquisition software (Molecular Devices). Field potentials were generated to evaluate their input-output relationship (30–300 µA stimulus intensity, 30 µA increments, 8 s ISI, filtered at 10 kHz). In the presence of 50 μM of NMDA-receptor antagonist (2R)-amino-5-phosphonovaleric acid and 6 μM of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-receptor antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), the fiber volley persisted but the fEPSP was completely abolished, confirming the fEPSP trace was indeed a post-synaptic event (Fig. 2B). Subsequent addition of the voltage-gated Na2+ channel blocker tetrodotoxin (TTX; 4 μM) was used to extinguish the fiber volley and verify its identity in the trace (Fig. 2B).
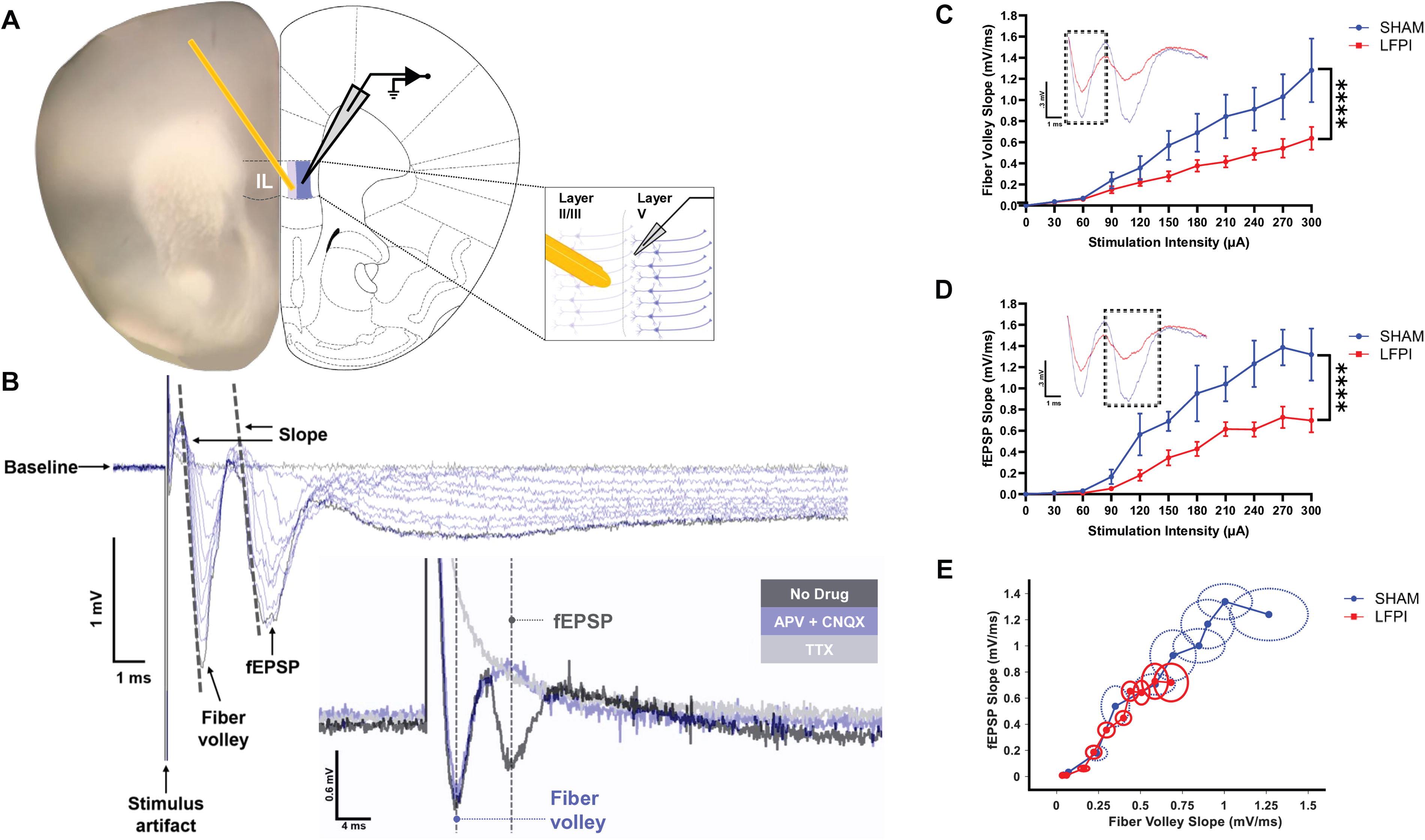
IL network is less excitable after mild TBI.
Whole-cell patch-clamp recording
Following incubation, slices were transferred to a recording chamber and anchored in place with stainless-steel harps (SHD-26H/2; Warner Instruments, Holliston, MA, USA) for whole-cell patch-clamp recordings. Bath aCSF was superfused (1.8 mL/min) and heated (31°C) with an inline heating system (TC324B, Warner). Recordings were made from IL layers II/III and V pyramidal-shaped neurons 60–100 μm deep. Cell layers and pyramidal neurons were visualized with an upright microscope (BX51, Olympus) and differential contrast video microscopy. Cell layers were easily identifiable at 60× magnification: layer I lacks cells, layer IV is absent in the medial cortex, layer II/III contains a dense band of wedge-shaped cell bodies with large apical dendrites, and layer V features similar cells with fewer and larger somata. The identity of pyramidal neurons was further confirmed by their firing properties in current-clamp mode. Series resistance was monitored and compensated at 60% and only recordings with a series resistance <35 MΩ were included in the dataset. Recording pipettes were fabricated as described and pulled to a tip resistance of 2–8 MΩ. All recordings were made using MultiClamp 700B amplifier and a Digidata 1440A digitizer (Molecular Devices). Electrophysiology data were recorded with the pClamp 10.7 data acquisition software (Molecular Devices) sampled at 20 kHz and filtered at 3 kHz.
Recordings of excitatory post-synaptic current (EPSC) and intrinsic neuronal properties were conducted with a K-gluconate internal solution composed of (in mM): K-gluconate 130, ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic Acid (EGTA 1), HEPES 10, MgCl2 1, CaCl2 0.01, KCL 10, Mg-ATP 5, Na2-phosphocreatine 10, Na-GTP 0.5, and titrated with KOH to a final PH between 7.25 and 7.35. Inhibitory post-synaptic current (IPSC) recordings were undertaken using a CsCl internal formulated to achieve isotonic chloride recordings, containing (in MM): CsCl 130, EGTA 1.1, HEPES 10, MgCl2 2, QX-314 5, Mg-ATP 5, Na2-phosphocreatine 10, Na-GTP 0.5, and titrated with CsOH to a final pH between 7.25 and 7.35. QX-314 was added to the CsCl internal to block voltage-gated Na+ currents and avoid post-synaptic action potentials (APs) from obscuring events. EGTA was included in both internal preparations to both facilitate seal formation and to reduce cytosolic Ca2+ elevations. Internal solutions were prepared to an osmolality between 280 and 290 mmol/kg. All measurements were corrected for liquid junction potentials of 17.3 mV (K-gluconate) and 9.3 mV (CsCl), calculated using the Clampex Liquid Junction Potential AgCl Calculator (Molecular Devices).
Neurons were voltage- or current-clamped to a −70 mV holding potential. EPSCs were recorded in the presence of the gamma-aminobutyric acid (GABA) receptor antagonist (−)- bicuculline methiodide to isolate spontaneous excitatory events (Abcam, 30 μM, Cambridge, UK). To isolate spontaneous inhibitory events, IPSCs were recorded in the presence of both N-methyl-d-aspartate (NMDA) channel antagonist DL-2-amino-5-phosphonopentanoic acid sodium salt (DL-AP5, sodium salt; Abcam, 50 μM, Cambridge, UK) and AMPA channel antagonist (1,2,3,4-tetrahydro-7-nitro-2,3-dioxoquinoxaline-6-carbonitrile disodium; DS-CNQX; Abcam, 20 μM, Cambridge). To isolate miniature, AP-independent EPSCs and IPSCs, Na+ channel antagonist TTX citrate (Abcam, 0.4 μM, Cambridge) was added. To test drug efficacy, DS-CNQX and DL-AP5 were added to block EPSCs, and bicuculline methiodide was used to abolish IPSCs (data not shown).
Measurements and statistical analyses
The percent of time spent freezing, a defensive response characterized by complete immobility except breathing, was measured as a proxy for fear to quantify fear reduction over time. 125,126 Freezing behavior was scan-sampled every fifth second throughout their duration in the chamber, and the total instance of freezing was divided by total observations to generate a freezing percentage per animal. 127,128 FE training data were analyzed using a two-way repeated measures analysis of variance (ANOVA) to evaluate the effects of condition (Sham vs. LFPI), time (within extinction training, per minute; Fig. 1C; between training and test days, initial 2 min; Fig. 1D) and their interaction. Only the first 2 min were assessed when evaluating fear memory and FE memory since within-session learning can occur that confounds the interpretation of extinction memory over the course of the paradigm. Significant interactions were followed by Fisher’s LSD post hoc tests for multiple comparisons.
Fiber volley and fEPSP slopes were determined as the slope of the initial linear portion of the response using Clampfit 10.7. Voltage slopes from slices were pooled to calculate the average response for each animal. Fiber volley and fEPSP data were analyzed using a two-way repeated measures ANOVA test to assess the effects of condition, stimulus intensity and their interaction on voltage slope. Significant interactions were followed by Tukey’s multiple comparisons test.
Whole-cell patch-clamp data were analyzed using Clampfit software. Spontaneous and miniature post-synaptic current events were identified using the Template Search algorithm in Clampfit 10.7, and event amplitudes and interevent intervals (IEIs) extracted via Clampfit 10.7. The Kolmogorov–Smirnov test was used to compare the distributions of Sham and LFPI data sets by calculating the cumulative distribution function and measuring the maximum absolute difference between the two distributions. To ensure equal weighting, 75 random current events were selected from each cell and pooled to create a cumulative probability histogram. Since 75 events per cell provides a high-powered test, the Kolmogorov–Smirnov test was only considered significant when p < 0.0001.
AP firing frequency was determined by counting the number of APs within a 500 ms pulse for each current step and multiplying the count by two to calculate the frequency in Hertz. Frequency data were analyzed using a two-way repeated measures ANOVA to evaluate the effects of group condition, current step intensity, and their interaction. Resting membrane potential was measured within the first 5 sec after break-in; this occurred prior to the dialysis of the internal solution and the holding current injection (I = 0). All other intrinsic excitability properties were assessed from current-clamp recordings with 500 ms current steps ranging from −50 to 175 pA in 25 pA increments. Input resistance was calculated from the steady-state, passive voltage responses to −50, −25, and 0 pA steps using Ohm’s law (R = ΔV/I). Afterhyperpolarization (AHP) latency was defined as the time course between the AP threshold during repolarization and the AHP trough. The AP threshold was determined as the membrane voltage where the rate of change first exceeded 30 mV/ms. 129 The first APs generated were selected for analyses. AP amplitude was measured from baseline-to-peak voltage of the AP, while AP half-width was defined as the duration at the voltage level halfway between the peak and threshold. Apart from frequency data, all intrinsic excitability properties were analyzed using an unpaired two-tailed t-test to compare group means and determine significant differences between the Sham and LFPI conditions. All statistical analyses were performed in Prism 10.4.1 (GraphPad, San Diego, CA, USA).
Results
Mild LFPI spares extinction learning but impairs extinction memory retrieval
Before evaluating FE memories, mice underwent cued-fear conditioning training in Context A to establish a fear memory (Fig. 1A and B). Twenty-four hours later (9 DPI), mice were re-exposed to the cue in separate and distinct Context B to undergo FE training (Fig. 1A and B). Both Sham and LFPI groups exhibited within-session decrease in freezing through the course of extinction training, suggesting LFPI does not impair cued-FE learning or eliminate the capacity for within-session FE (two-way repeated measures ANOVA: injury effect, F(1, 30) = 1.368, p = 0.2514; minute effect, F(3.911, 117.3) = 20.97, p < 0.0001; interaction, F(9, 270) = 1.469, p = 0.1592; Fig. 1C). However, when testing FE memory during the initial 2 min of cue exposure the next day (10 DPI), LFPI mice maintained a heightened freezing response relative to Sham mice (Sham vs LFPI, test day) and demonstrated a deficit in the ability to extinguish fear after training (training vs. test), indicating impaired FE memory retrieval (two-way repeated measures ANOVA: injury effect, F(1, 30) = 0.6094, p = 0.4411; day effect, F(1, 30) = 23.98, p < 0.0001; interaction, F(1, 30) = 6.465, p = 0.0164; Fisher’s LSD multiple comparisons: Sham, training vs. test, p < 0.0001; LFPI, training vs. test, p = 0.1064; test day, Sham vs. LFPI, p = 0.0299; Fig. 1D).
Network excitability in IL is diminished after mild LFPI
FE memories are associated with increased output from the IL to the BLA, and a decrease in output from the IL would be expected to disrupt FE. To determine whether the FE impairment post-injury is associated with reduced IL activity, we performed ex vivo field potential recordings in IL (Fig. 2A). Specifically, IL layer II/III was stimulated (30–300 μA current steps) to evoke fiber volleys that reflect presynaptic APs, and the resultant evoked fEPSPs in IL layer V. A two-way repeated measures ANOVA revealed a significant reduction in the fiber volley slope in response to stimulus intensity (injury effect, F[1, 11] = 4.136, p = 0.0668; stimulus intensity effect, F[10, 110] = 39.72, p < 0.0001; interaction, F[10, 110] = 4.591, p < 0.0001; Fig. 2C), and multiple comparisons identified a significant shift in fiber volley response at higher stimulation intensities (210 μA, p = **0.0087; 240 μA, **p = 0.0094; 270 μA, **p < 0.003; 300 μA, ***p = 0.0001), suggesting IL layer II/III is less excitable post-injury. Additionally, as hypothesized, we observed a significant reduction in fEPSPs from IL layer V after LFPI, with multiple comparisons revealing a significant decrease in the fEPSP response at higher stimulation intensities (two-way repeated measures ANOVA: injury effect, F[1, 11] = 9.555, p = 0.0103; stimulus intensity effect, F[10, 110] = 43.74, p < 0.0001; interaction, F[10, 110] = 4.240, p < 0.0001, Fig. 2D; multiple comparisons: 120 μA, p = 0.0197; 150 μA, p = 0.0372; 180 μA, p = 0.0017; 210 μA, p = 0.0105; 240 μA, p = 0.002; 270 μA, p < 0.0001; 300 μA, p = 0.0002). An unpaired t-test did not detect any significant group differences for fEPSP slopes compared with fiber volley slopes (F[5, 6] = 1.184, p = 0.5312; Fig. 2E), suggesting reduced excitatory input from IL layer II/III may be driving the decreased excitability in IL layer V. Overall, these data demonstrate that mild LFPI decreases IL network excitability.
Mild LFPI decreases the amplitude and frequency of excitatory synaptic input while augmenting the amplitude and frequency of inhibitory events onto layer II/III pyramidal neurons
Brain network E/I balance reflects the summed effects of complex synaptic connections between neurons, individual neuron excitability, and the network’s overall architecture. Diminished IL network activation post-injury may emerge from the disruption of delicately balanced E/I neurotransmission fundamental to proper circuit function. To detect putative synaptic alterations contributing to IL network excitability changes after mild LFPI, we performed a series of whole-cell patch-clamp recordings to measure spontaneous and miniature post-synaptic currents in IL pyramidal neurons. Given the reduction in IL layer II/III fiber volley slope, we first examined synaptic inputs onto pyramidal cells in IL layer II/III. Here, we observed a significant decrease in spontaneous EPSC (sEPSC) amplitude (Kolmogorov–Smirnov test: significance set at p < 0.0001, D[3148] = 0.2814; Fig. 3B). There was also a significant increase in the IEI between events indicative of a decrease in event frequency (Kolmogorov–Smirnov test: significance set at p < 0.0001, D[3148] = 0.1301; Fig. 3C). We did not observe any statistically significant differences in miniature EPSC (mEPSC) amplitude or frequency between groups (amplitude, Kolmogorov–Smirnov test: p = 0.0860, D[2323] = 0.05206; Fig. 3E; IEI, Kolmogorov–Smirnov test, p = 0.03922, D[2323] = 0.3335; Fig. 3F). Altogether, these data illustrate a significant reduction in AP-dependent excitatory input onto IL layer II/III cells following mild LFPI.
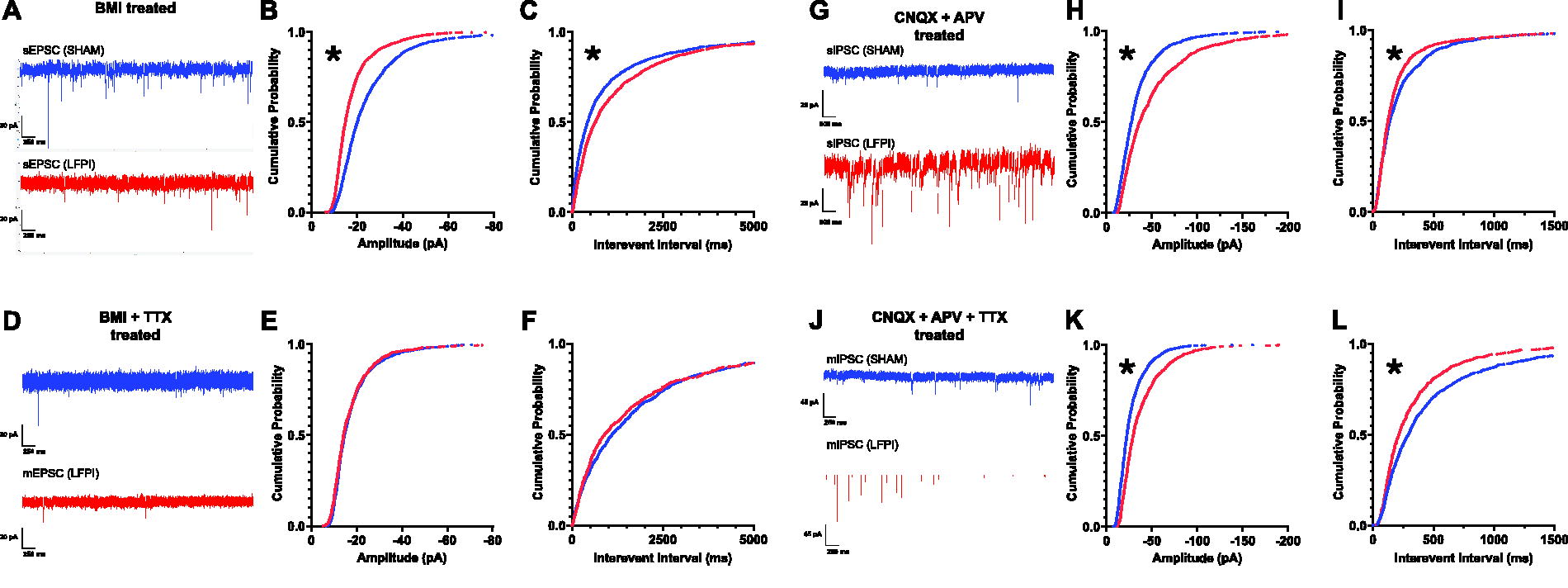
Mild TBI decreases the amplitude and frequency of excitatory synaptic input and increases the amplitude and frequency of inhibitory synaptic input onto IL layer II/III pyramidal neurons.
Conversely, sIPSC data analyses revealed a significant enhancement in amplitude in the LFPI group compared with Sham (Kolmogorov–Smirnov test: significance set at p < 0.0001, D[2998] = 0.2013; Fig. 3H). Additionally, a significant decrease in the sIPSC IEI was found, indicating an increase in frequency of afferent inhibitory input (Kolmogorov–Smirnov test: significance set at p < 0.0001, D[2998] = 0.08928; Fig. 3I). Similarly, there was a significant increase in mIPSC amplitude (Kolmogorov–Smirnov test: significance set at p < 0.0001, D[2398] = 0.2145; Fig. 3K), and a significant decrease in the mIPSC IEI (Kolmogorov–Smirnov test: significance set at p < 0.0001, D[2398] = 0.1298; Fig. 3L). These findings suggest inhibitory input onto IL layer II/III cells is augmented following mild LFPI. Combined with the reduction in AP-dependent excitation, these findings support the hypothesis that mild LFPI induces a shift toward inhibition in the E/I ratio that suppresses IL network activity.
Mild LFPI increased input resistance and AHP latency in layer II/III neurons
Next, the intrinsic properties of IL layer II/III neurons were evaluated to further interrogate their contribution to reduced IL network activation. In IL layer II/III neurons derived from LFPI brain slices, the input resistance was significantly increased (unpaired two-tailed t-test, t[33] = 2.751, p = 0.0096; Fig. 4D). However, we also detected a significant increase in AHP latency post-injury (unpaired two-tailed t-test: t[32] = 2.058, p = 0.0478; statistical outliers identified by the ROUT test Q=1% were removed prior to analysis; Fig. 4E). No significant differences in AP firing or other AP properties were detected between mild LFPI and Sham (AP frequency, two-way repeated measures ANOVA: injury effect, F[1, 33] = 0.01146, p = 0.9154; current step effect, F[2.131, 70.32] = 130.1, p < 0.0001; interaction, F[7, 231] = 0.7742, p = 0.6095; Fig. 4B; membrane potential, unpaired two-tailed t-test: t[29] = 0.7281, p = 0.4724; Fig. 4C; AP threshold, unpaired two-tailed t-test: t[33] = 1.825, p = 0.0771; Fig. 4F; AP amplitude, unpaired two-tailed t-test: t[33] = 1.089, p = 0.2842; Fig. 4G; AP half-width, unpaired two-tailed t-test: t[33] = 1.374, p = 0.1788; Fig. 4H). Together, these results identify alterations in the intrinsic excitability of IL layer II/III neurons that do not affect their activity patterns.

Mild TBI increased input resistance and AHP latency in layer II/III pyramidal neurons, resulting in no net change to intrinsic excitability.
IL layer V pyramidal neurons receive fewer sEPSCs, smaller mEPSCs, and a net increase in both spontaneous and AP-independent IPSCs following mild LFPI
We began our investigation into the source of the decreased excitability in layer V by examining the excitatory input onto layer V pyramidal neurons. In line with the reduction in fEPSPs recorded from IL layer V, we hypothesized that spontaneous and miniature excitatory currents onto layer V neurons were diminished. There was a significant difference in the distributions of sEPSC amplitudes between Sham and LFPI cells, with the slight rightward shift denoting a small but significant increase in sEPSC amplitude post-injury (Kolmogorov–Smirnov test: significance set at p < 0.0001; p < 0.0001, D[3223] = 0.09213; medians: Sham, −15.54 pA, LFPI, −16.69 pA 1.15 pA difference; means: Sham, −18.13 pA, LFPI, −19.71 pA, 1.58 pA difference; Fig. 5B). Mild LFPI also increased the time between events, indicating a decrease in the frequency of afferent excitatory activity onto IL layer V neurons (Kolmogorov–Smirnov test: significance set at p < 0.0001; p < 0.0001, D[3223] = 0.1825; Fig. 5C). The AP-independent EPSC amplitude data were reciprocal when compared with the spontaneous data, such that mEPSCs decreased in amplitude post-injury (Kolmogorov–Smirnov test: significance set at p < 0.0001; p < 0.0001, D[1723] = 0.1718; Fig. 5E). No changes in the IEI of these events were observed (Kolmogorov–Smirnov test: significance set at p < 0.0001; p = 0.0013, D[1723] = 0.09313; Fig. 5F). Overall, these findings reflect a net reduction in synaptic excitatory input onto IL layer V neurons after injury.
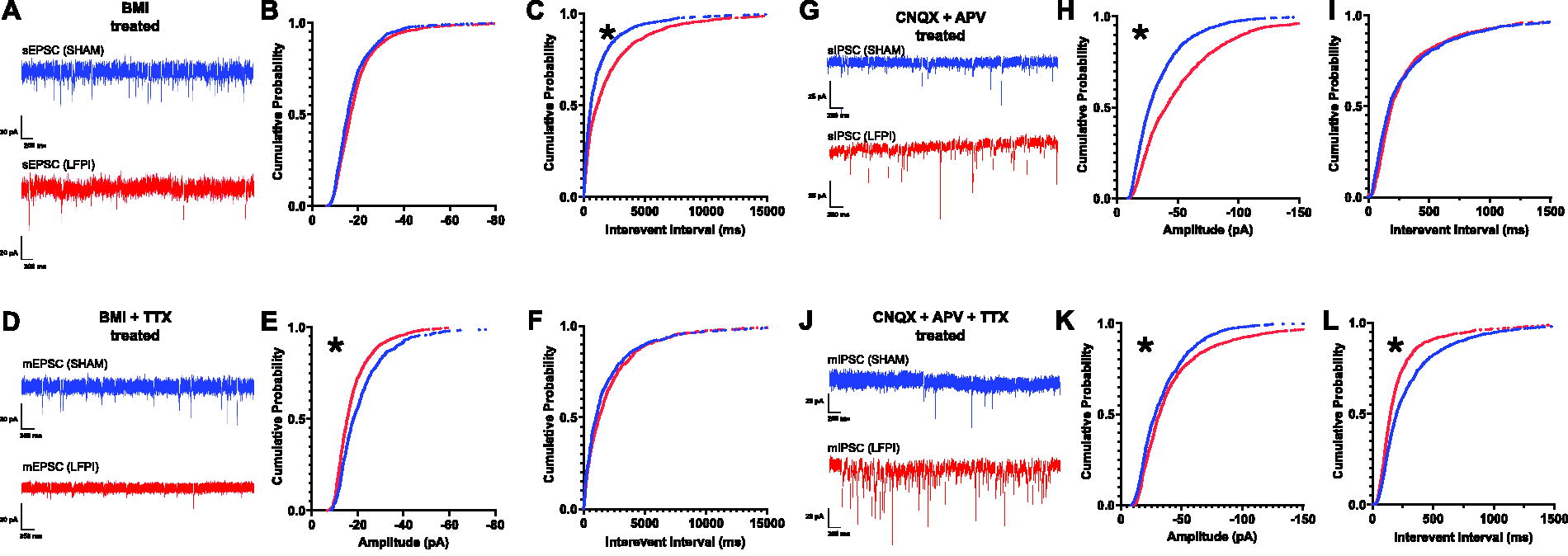
Mild TBI decreases the frequency of sEPSCs and amplitude of mEPSCs, while augmenting sIPSC frequency and AP-independent inhibitory synaptic input onto IL layer V pyramidal neurons.
We next examined whether mild LFPI also alters inhibitory synaptic inputs onto layer V pyramidal neurons. sIPSC data analyses revealed a significant increase in amplitude after injury (Kolmogorov–Smirnov test: significance set at p < 0.0001, D[2773] = 0.2273; Fig. 5H). However, there was no significant change in the sIPSC IEI (Kolmogorov–Smirnov test: significance set at p < 0.0001, p = 0.0013, D[2773] = 0.08035; Fig. 5I). When assessing AP-independent events, there was a significant increase in both mIPSC amplitude (Kolmogorov–Smirnov test: significance set at p < 0.0001, D[2473] = 0.1006; Fig. 5K), and IEI (Kolmogorov–Smirnov test: significance set at p < 0.0001, D[2473] = 0.1630; Fig. 5L). Together with the reduction in AP-dependent excitation, these results support the hypothesis that mild LFPI induces net inhibitory functional changes in IL layer V, decreasing IL layer V excitatory input while increasing inhibitory input, resulting in an E/I imbalance that favors the suppression of IL neuronal excitability.
Mild LFPI increased the resting membrane potential and AHP latency in layer V pyramidal neurons
Finally, we investigated the effect of mild LFPI on IL layer V neurons in current-clamp to determine whether injury-induced alterations in their intrinsic excitability played a role in the reduction in IL network activation. Group data did not find any statistically significant differences in the AP firing frequency after mild LFPI (two-way repeated measures ANOVA: injury effect, F[1, 30] = 1.247, p = 0.2731; current step, F[1.694, 50.81] = 22.01, p < 0.0001; interaction, F[7, 210] = 0.7039, p = 0.6687; Fig. 6B), despite these neurons having a significantly more depolarized resting membrane potential post-injury that would theoretically improve the cell’s ability to propagate APs (unpaired two-tailed t-test: t[14] = 3.026, p = 0.0091; Fig. 6C). Alternatively, a more depolarized resting membrane potential can cause cells to adapt to sustained depolarization by inducing molecular changes that aid to impede excitotoxicity. Similar to IL layer II/III, IL layer V presented a significant increase in AHP latency after mild LFPI (unpaired two-tailed t-test: t[28] = 2.784, p = 0.0095). Statistical outliers were determined by the ROUT test Q=1% and removed before analysis (Fig. 6E). Finally, no other components of intrinsic excitability differed between groups, although AP half-width duration was approaching statistical significance (input resistance, unpaired two-tailed t-test: t[30] = 0.6492, p = 0.5211; Fig. 6D; AP threshold, unpaired two-tailed t-test: t[30] = 0.5591, p = 0.5802; Fig. 6F; AP amplitude, unpaired two-tailed t-test: t[30] = 0.8716, p = 0.3904; Fig. 6G; AP half-width duration, unpaired two-tailed t-test, t[30] = 1.993, p = 0.0554; Fig. 6H).
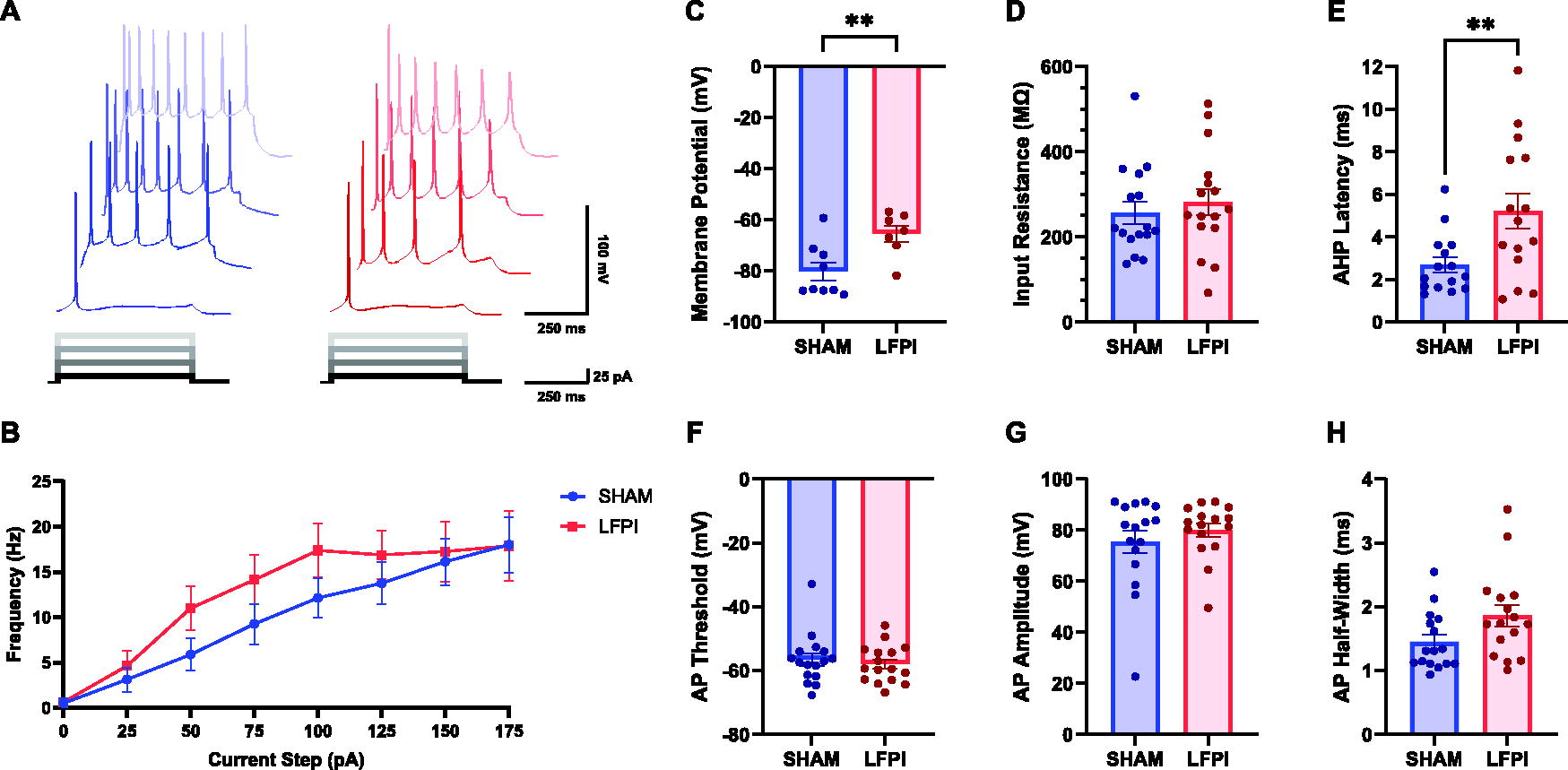
Mild TBI increased the resting membrane potential and AHP latency in layer V pyramidal neurons.
Discussion
This work combined LFPI, a cued-FE behavior paradigm, local field potential recordings, and whole-cell patch-clamp techniques and generated data supporting the hypothesis that mild TBI diminished FE memory and decreases IL network excitability. First, we demonstrated that mild TBI results in elevated cued-fear response during FE testing 10 DPI, indicating a reduction in FE in mice that underwent mild TBI. Next, we identified injury-induced decreases in IL network activation in layer II/III and layer V, both of which would be expected to diminish FE. 55,130 –133 Consistent with the IL network data, further experiments evaluating excitatory and inhibitory neurotransmission onto IL neurons post-injury revealed E/I imbalances with a specific deficit in excitatory input together with augmented inhibitory input onto neurons in both layers II/III and V. Finally, we characterized injury-induced changes in the intrinsic excitability in cells from both IL layer II/III and IL layer V. This study is the first to demonstrate a deficit in FE memory and a reduction in IL excitability after mild TBI. We further elucidate mechanistic perturbations in synaptic physiology that potentially underly reduced FE and increased vulnerability to neuropsychiatric disorders, post-injury.
The mild LFPI-induced FE memory impairment demonstrated here is congruent with previous reports from animal models of TBI. 43 –47,99 Parallel findings of deficient FE memory in human TBI survivors 15,38 –41 together with evidence of impaired FE as a clinical endophenotype for an amalgam of trauma and anxiety disorders (e.g., post-traumatic stress disorder [PTSD], phobias, panic disorder, and obsessive-compulsive disorder 25,27,36,48 –50 ) have encouraged exploring the utility of animal models to characterize the shared etiology among these conditions. 15,47,134 –136 This is especially true for the comorbidity of TBI and PTSD: FE impairment is a hallmark of PTSD, 25,27,33,36,137,138 is nearly two to three times more likely to be diagnosed in patients with TBI, 139,140 and has received marked attention over the last decades due to high rates of concurrent diagnoses among U.S.–Iraq war veterans. 15,136,139 –143 It should nonetheless be noted that animal models of fear-based neuropsychiatric disorders have clear practical limitations, including an inability to collect verbal self-reports that disclose intrinsic emotional disturbances (i.e., dissociative symptoms, intrusive thoughts or nightmares) or detect preexisting risk factors and diatheses that help shape the clinical presentations of these disorders (i.e., gene-environment interactions, resilience 144 ); capturing these discrepancies is further complicated by the inherent heterogeneity of TBI. 2,12,14,135,141,145 –149 Nonetheless, animal models of FE have furthered our understanding of human neuropsychiatric symptoms by reliably producing non-verbal behaviors and/or physiological responses that complement verbal reports, and FE is the fundamental conceptual framework for exposure therapy, which is currently recognized as the most effective treatment for PTSD and anxiety disorders. 33,150 –152 Moreover, rodent-TBI studies employing FE protocols and other fear assessment tasks have revealed neurobehavioral outcomes linked to biological mechanisms analogous to clinical research results, and these findings have helped refine affective treatment strategies for injury survivors. 12,14,15,30,47,134 –136,141,145,146,148 Thus, pre-clinical behavioral testing post-TBI remains relevant and essential. The injury-induced FE impairment described here supplements our current understanding of mild TBI and comorbid neuropsychiatric disorders, and, in turn, can be utilized to further demystify the mechanisms underlying injury-induced fear-based neuropsychiatric pathologies.
An intriguing behavioral outcome of this study is the preservation of cued-fear memory and extinction learning preceding FE impairment in injured animals (Fig. 1C and D), showing that while mice retain the ability for within-session FE, they fail to recall their extinction memory. This phenomenon has also been observed in individuals with PTSD and anxiety disorders, 32,35,38,39,145,150,153 –157 including patients receiving exposure therapy treatment. Individuals with PTSD and fear-based anxiety disorders sometimes demonstrate the ability to “learn” FE during exposure therapy (i.e., reduce their fear responses), but will often struggle to recall the extinction memory when re-exposed to the original fear-inducing stimulus, leading to a resurgence of fear. 26,53,158,159 Similar findings have been found in TBI studies evaluating fear memory and extinction. While some clinical 160 and pre-clinical 44 –46,161 –163 studies have demonstrated sustained cued-fear memory and/or extinction learning, others in humans 38 and rodent-TBI models 44,99,124,161,164 –168 have not. The disparity in fear memory and extinction deficits observed in TBI subjects can be explained by several factors, including the brain’s capacity for plasticity and spontaneous recovery, 169 the type of brain injury endured (e.g., focal, diffuse; blast, midline or lateral impact 102,134,149 ), timing of fear memory and extinction assessment after the injury (e.g., same-day or multiday protocol, within or outside of the therapeutic window 30,40,107,170 –172 ) injury-induced dysregulation of overlapping fear circuits, 14,15,141,173 and differences in fear conditioning and extinction methods (e.g., cued or contextual, trace or delayed conditioning, brief or continuous CS exposure 24,107,174 ).
Activation of the BLA is associated with the storage and retrieval of fear memories and plays a central role in driving fear expression. 53,125,175 –178 Early-phase FE learning is also driven by BLA activity, which shifts toward inhibition over time as disynaptic projections from IL promote feedforward inhibition and ultimately suppress fear expression. 59,66,179 –187 Our lab previously found a diminished cued-fear response that was attributed to decreased BLA network excitability following mild-to-moderate LFPI. 124 Provided the same deficits in BLA network activation exist in the current study, why did this cohort of mild LFPI mice successfully recall the cued-fear memory and reduce freezing at a rate comparable to Sham during extinction training? The discrepancies in cued-fear may be partially explained by our different behavioral paradigms. To test the cued-fear memory, Palmer et al.’s 124 behavior experiment was designed to deliver three, brief bouts of CS exposure (20 s) with varied interstimulus intervals (ISI) that interfere with memory recall to test the lability of the cued-fear memory. In contrast, the FE training paradigm utilized here prolonged the CS exposure (10 min of continuous exposure) with minimal interference (1× ISI, <1 s) to stabilize the fear memory and promote fear inhibition to assess FE memory the next day. 105 –107,188 –191 It is plausible that extensive cue exposure was able to rescue BLA activation, in turn meeting the threshold for an adequate cued-fear response. Thus, the extended CS exposure may have occluded an existing impairment in fear memory post-injury, while supporting fear inhibition over time.
Previous data from our lab demonstrated opposite alterations after injury in mPFC circuitry neighboring the IL113 that complement our understanding of the mPFC’s role in fear regulation. Within the rodent mPFC, the decision to express or suppress fear requires the engagement of prelimbic (PL) and IL subdivisions, respectively, which bidirectionally regulate fear expression via differential recruitment of amygdala neuronal subpopulations. 65,182,192 –195 Whereas the PL promotes fear expression during fear conditioning and early extinction training through reciprocal connections with the BLA and does not participate in extinction memory processes, the IL suppresses fear expression by inhibiting BLA activity and plays a critical role in extinction memory consolidation and retrieval. 55 –57,59 –65,67 –85,87 ,89 –91,98,196 This has resulted in the PL and IL being conceptualized as fear “go/stop” circuitry that competitively mediate downstream threat detection networks to produce or suppress fear responses. 53,137,196 –199 In the PL, LFPI induced a decrease in the layer V fEPSP, without a significant change in the fiber volley from layer II/III, suggesting the decrease in the fEPSP was not due to a reduction in the afferent input. 112 In contrast, here we demonstrate diminished IL network activation as shown by a directly proportional reduction in both the layer II/III fiber volley and layer V fEPSP that does suggests an impairment in AP initiation and/or propagation. Additionally, in mPFC layer II/III neurons, Smith et al. determined an increase in the frequency of spontaneous and miniature EPSCs onto PL neurons and a decrease in the amplitude of spontaneous and miniature IPSCs, suggesting a shift toward enhanced excitatory input and reduced inhibitory input onto PL layer II/III neurons. Conversely, IL layer II/III neurons demonstrate a decrease in both the amplitude and frequency of sEPSCs, and a significant increase in the amplitude and frequency of both spontaneous and miniature IPSCs, denoting diminished excitation and enhanced inhibition onto IL layer II/III neurons. Whereas Smith et al. did not observe any changes in neurotransmission onto PL neurons in layer V, we observed a pattern of decreased excitation and augmented inhibition in IL layer V similar to that in IL layer II/III: less frequent sEPSCS, smaller mEPSCs, larger sIPSCs and mIPSCs, and more mIPSCs. Altogether, the data suggest that there is a significant E/I imbalance within the mPFC after mild LFPI, consisting of a shift toward enhanced excitation in the PL and augmented inhibition in the IL. Both shifts in E/I balance result in memory impairment and theoretically promote fear expression, though future experiments are needed to investigate PL circuitry in the context of fear regulation. Nonetheless, we are the first lab to evaluate the neurocircuitry of both mPFC subregions after mild LFPI, and future work aims to interrogate IL-BLA and PL-BLA circuitry post-injury.
The experiments demonstrating a net decrease in the excitatory drive onto layer II/III and V neurons are consistent with the shifts in IL network fiber volley and field responses, respectively. However, the field experiments involved stimulating IL layer II/III to assess evoked responses, whereas the patch experiments examined tonic events that likely reflect input from numerous brain regions. Indeed, the IL lies at the intersection of limbic brain structures vulnerable to TBI, with tonic innervation from the PL, BLA, hippocampus, hypothalamus, and paraventricular nucleus of the thalamus. 57,69,74,75,179,200 –205 However, no study has evaluated the synaptic connectivity of these regions onto IL after TBI. Thus, verification is needed to assess the extent to which dysfunction in the limbic system contributes to the observed changes in spontaneous synaptic input onto IL neurons post-injury.
Both IL layer II/III and V neurons exhibited alterations in their intrinsic excitability, with notable laminar specificity in the presentation of passive membrane property alterations, suggesting these changes were likely due to compensatory adjustments rather than global extracellular changes. For example, we observed a significant increase in the input resistance of IL layer II/III neurons post-injury (Fig. 4D) that was not observed in layer V neurons (Fig. 6D). This change typically indicates reduced ion channel density that typically improves AP initiation, but consistent firing frequency suggests the altered resistance may instead be compensatory to counteract diminished excitatory input. Additionally, layer V neurons exhibited a significantly depolarized resting membrane potential post-injury. This is possibly due to altered function or quantity of ion channels that regulate the membrane voltage at rest, such as a reduction in inwardly rectifying K+ (IRK) channels, which are more concentrated in layer V neurons. 206
In contrast to the passive membrane properties, neurons from both IL layers exhibited increased AHP latency (Figs. 4E and 6E), suggesting a shared vulnerability in their AP properties after mild LFPI. This change may be attributed to ion channels that influence AHP shape in both layers. For example, AHP latency can be prolonged by the activation of small conductance Ca2+-dependent K+ (SK) channels, which open in response to a rise in intracellular Ca2+ concentrations, hyperpolarizing the cell during the AP’s falling phase to produce the AHP. IL neurons treated with SK channel agonist 5,6-dichloro-1-ethyl-1,3-dihydro-2H-benzimidazol-2-one (DCEIBO) exhibit enhanced AHP current and prolonged AHP duration, while infusing DCEIBO into the IL in vivo prior to FE training impairs extinction memory retrieval without affecting learning. 207 While SK channels have not been studied in the IL after TBI, previous research has shown that Ca2+ homeostasis mitigates memory deficits and pathology in rodent LFPI models of TBI. 208 Additionally, M-Type K+ (MK) channels, modulated by muscarinic M1 receptors, may contribute to AHP changes observed post-LFPI. Similar to SK channels, MK channels extend the AHP during repolarization and are inhibited by muscarinic M1 receptor activation. In the IL, M1 receptor agonists, such as acetylcholine 209 and cevimeline 210 enhance neuronal excitability by reducing AHP duration and increasing firing rate, playing a key role in FE memory consolidation. Numerous studies report a reduction in muscarinic receptors after TBI. 211 –213 Additionally, cholinergic projections—which densely innervate the PFC 214 —are also susceptible to damage after TBI, including LFPI. 215 –217 However, as with IRK and SK channels, the function of MK and other ion channels in the mPFC post-TBI remain unexplored.
This study possesses several limitations. First, behavioral and physiological experiments were conducted on separate cohorts of animals. Separate cohorts were utilized in order to limit the number of animals required for the study when factoring both the large sample size required for electrophysiology experiments, and the potential for attrition or other challenges possible after experimental TBI. While this approach is necessarily given the methodological constraints, it prevents direct correlation between fear responsivity and IL network activation within the same animal. Second, this study was designed to assess injury-induced alterations during the clinically relevant therapeutic window of TBI, i.e., after acute transient changes involving secondary pathology have resolved. 218,219 During this period, timely therapeutic interventions (e.g., administration of branched-chain amino acids 220,221 ) can mitigate pathology and promote recovery. However, this window does not capture the full temporal dynamics of TBI impairments, which can evolve over a longer period of time. Patients with TBI report lasting fear-based neuropsychiatric disorders and emotional health impacts that significantly affect quality of life 1-year post-injury. 222 Of relevance to the current study, FE memory impairments induced by mild LFPI persist 14 100 and even 28 45 DPI, and decreased spine density in IL layer II/III neurons is observed at 28 DPI, 45 suggesting neuroplastic deficits in IL could extend beyond the acute window. These findings emphasize the merit of evaluating the long-term trajectory of behavioral and physiological alterations, particularly in the context of FE impairment and cortical contributions to symptom maintenance. Longitudinal studies with extended time points would greatly supplement our understanding of how early brain disruptions may contribute to chronic disorders and inform therapeutic interventions. Third, the E/I imbalance measured in the IL following mild TBI can arise from several mechanisms. On the presynaptic side, mild TBI could affect afferent inputs from upstream brain regions vulnerable to TBI (e.g., hippocampus area CA1, BLA, PL), influencing vesicular release and presynaptic calcium dynamics, which may alter neurotransmitter release. 44 Alternatively, post-synaptic changes, such as reduced dendritic spine density or alterations to transmembrane proteins (e.g., receptors, transporters, pumps, channels) can further contribute to impaired excitatory and/or inhibitory synaptic signaling. Moreover, increased activity of inhibitory neurons could exacerbate the injury-induced E/I imbalance, as seen in the rise of GABAergic signaling in the mPFC 44 and augmented cholecystokinin-dependent inhibition in the hippocampal CA1 region. 223 Understanding the interplay between pre- and post-synaptic mechanisms will be essential for identifying the specific alterations and therapeutic targets that should inform future studies.
Overall, this study integrates behavior with electrophysiology and establishes that alterations in IL circuit physiology are associated with functional consequences for FE. To our knowledge, this is the first study to demonstrate an injury-induced decline in IL network activation and corresponding shifts in synaptic input to IL neurons in both layer II/III and V. Furthermore, the cortical E/I imbalance identified here suggests that mild TBI is linked to maladaptive cue fear expression. TBI is a significant disorder for which there are no neuroprotective treatments; a better understanding of circuitry dysfunction allows for a mechanistic readout of neuronal activity following TBI and targeted intervention. This work advances our understanding of the mechanisms contributing to neuropsychiatric disorders after mild TBI, providing a crucial foundation for developing treatments that could improve health outcomes, productivity, and quality of life for those afflicted.
Transparency, Rigor, and Reproducibility Summary
This study, designed for a doctoral thesis, was not formally pre-registered. Animal exclusions were based on the following criteria: hemorrhage, herniation, motor deficits, delayed righting time post-injury, aversion to the experimenter on day 5 of handling, lack of fear response during US delivery. Cell exclusions were made for access resistance ≥35 MΩ, unstable baselines, noisy signals, and capacitance <20 pF. For post-synaptic current recordings, cells with fewer than 75 events were excluded. For current-clamp recordings, cells exhibiting nonadaptive, spontaneous, or burst firing patterns were also excluded. Data from 120 male, wild-type C57/BL6 mice (58 Sham, 62 LFPI) were eligible for analyses. Behavioral and physiology experiments were conducted in separate cohorts of animals with the exception of one LFPI mouse, whose data were used for both behavior and layer V sEPSC analyses, and whose data did not significantly differ from that of other LFPI animals within each cohort. EPSC and IPSC data were collected from separate cohorts of animals to avoid contamination from drug washes. Mice were 6–12 weeks old at the start of experiments (behavior, 8 DPI; electrophysiology, 6–10 DPI). Behavioral and field potential data were analyzed per animal. For field potential recordings, data were averaged per slice to represent one animal. For patch-clamp experiments, a minimum of 10 cells per injury condition was set, based on the Cohen Lab’s extensive prior experience. Post-synaptic current amplitudes and IEI data were extracted from the same event files for each cell and not paired for analyses. Statistical analysis treated all cells as independent, with AP frequency analyzed as a repeated measure using a mixed model. Outliers (Q = 1%) were identified using the ROUT test and excluded. Current-clamp data were analyzed using means with the assumption of normal distribution, while medians were used for voltage-clamp outlier tests due to the non-parametric nature of cumulative probability data. Voltage-clamp data, based on 75 random currents per cell, were analyzed using cumulative probability histograms. Since 75 events per cell provides a high-powered test, the Kolmogorov–Smirnov test was considered significant only for p < 0.0001. Cells with fewer than 75 events were excluded. The experimenter was blinded to the injury condition until after data analysis. All raw data are available upon request.
Footnotes
Acknowledgments
The authors would like to thank Dr. Steven A. Thomas of the University of Pennsylvania for offering advice that proved to be pivotal to the design and initiation of behavioral experiments. The authors would also like to thank Dr. Brian N. Johnson of the Children’s Hospital of Philadelphia for providing invaluable support with electrophysiology experiments, generous time to assist with troubleshooting, and thoughtful input.
Authors’ Contributions
C.E.U.: Conceptualization, methodology, data formulation, investigation, visualization, writing—original draft, formal analysis, and project administration. A.M.F.: Investigation and visualization. A.S.C.: Conceptualization, methodology, resources, writing—reviewing and editing, supervision, and funding acquisition.
Author Disclosure Statement
The authors have no competing interests to disclose.
Funding Information
NIH-NICHD Grant R37HD059288 to A.S.C., supplement 206180424-S to C.E.U.
Supplementary Material
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.