
Abstract
Short interfering RNAs (siRNAs) are valuable reagents for sequence-specific inhibition of gene expression via the RNA interference (RNAi) pathway. Recently, it was suggested that 16-bp siRNAs are effective RNAi triggers and superior to “classical” 19-bp siRNAs. This contradiction with generally accepted knowledge prompted us to reinvestigate this issue. Here, in a series of experiments performed with siRNA duplexes of various lengths (from 19 to 15 bp) designed to silence either overexpressed enhanced green fluorescent protein or endogenously expressed CDK9, we demonstrate that 19-bp siRNAs are more active silencers than shorter corresponding duplexes. The discrepancy between our results and those questioned appears to be due to different modes of shortening the duplex (either at the 3′-end or at the 5′-end, with respect to polarity of the guide strand). Importantly, duplexes with intact 5′-ends but shortened at their 3′-ends retain target site specificity, whereas those shortened at the 5′-end are complementary to different target sites located upstream.
Introduction
In 2001, Tuschl and colleagues proved that RNAi can be activated by exogenously delivered siRNAs (Elbashir et al., 2001a, 2001b). This discovery paved the way for the characterization of RNAi machinery, as well as the identification of the structural requirements of siRNA effectors. Dicer-derived siRNA molecules contain a 19-bp RNA duplex with typical A-form helical structure and 2-nucleotide (nt) overhangs at both 3′-ends (Elbashir et al., 2001c). One strand is designated the antisense (guide) strand and the other the sense (passenger) strand. The guide strand is chosen for incorporation into RISC on the basis of thermodynamic “asymmetry” of the duplex (Khvorova et al., 2003; Schwarz et al., 2003; Sipa et al., 2007). Once RISC binds the siRNA, the nucleolytic activity of Ago2 is triggered. The sense strand is cleaved and released from the complex (Matranga et al., 2005; Rand et al., 2005), while the remaining antisense strand operates as a guide, leading RISC to the complementary sequence on the target mRNA. The seed region of the guide strand (nt 2–8 from the 5′-end) is responsible for the initial recognition of the mRNA molecule. Formation of a fully complementary duplex of the antisense strand and mRNA activates the nucleolytic activity of RISC. Cleavage of the mRNA occurs precisely at a position marked by the length of 10 nt from the 5′-end of the antisense strand (Elbashir et al., 2001c; SONTHEIMER, 2005). Any disturbance within the helical structure of the siRNA (eg, introduction of a wobble base pair into the central region of the duplex) results in a reduction of the silencing effect (Sipa et al., 2007; Nawrot and Sipa, 2006). Kim et al. reported that longer 27-mer (25 bp + 2 nt) dsRNAs effectively induce RNAi (Kim et al., 2005). The enhanced silencing potency of the longer duplexes is attributed to the fact that they are substrates for the Dicer endonuclease, directly linking the production of siRNAs with their incorporation into RISC where they function as conventional 21-mers (19 bp + 2 nt).
We and others have shown that even a slight shift in the siRNA target sequence can remarkably affect the extent of gene silencing (Reynolds et al., 2004; Sipa et al., 2007). Computational modeling and experimental data have demonstrated that the secondary structure of the target mRNA and its accessibility to the guide siRNA strand are important factors in siRNA silencing potential (Overhoff et al., 2005; Schubert et al., 2005; Ameres et al., 2007; Westerhout and Berkhout, 2007). Moreover, as we previously showed, thermodynamically stable siRNAs, in which all uridine (U) residues of the antisense strand were substituted with 2-thiouridine (s2U), are as active as their parent nonmodified precursors even if they are shortened to 17- or 15-bp duplexes. Importantly, the relative silencing activity of siRNAs decreases along with duplex length. Specifically, the ability of shortened 17- and 15-bp duplexes to silence green fluorescent protein (GFP) expression was reduced to ∼90% and 20%, respectively, of the activity exhibited by the 19-bp parent duplexes (Sipa et al., 2007).
Recently, Sun et al. investigated the minimal lengths of siRNA strands in mammalian cells and found that silencing requires at least 19-nt complementarity, confirming data from earlier studies on RNAi activity in Drosophila melanogaster lysate (Elbashir et al., 2001c; Sun et al., 2008). Asymmetric RNA duplexes consisting of a 19–21-nt antisense strand and a shorter 15–18-nt sense strand can mediate efficient gene silencing by recruiting RISC and initiating the RNAi pathway (Sano et al., 2008; Sun et al., 2008; Chang et al., 2009). Reversed asymmetric complexes with a 21-nt sense strand and an antisense strand of variable length showed no silencing activity.
Two years ago, Chu and Rana (2008) published their results suggesting that shorter 16-bp RNA duplexes are active RNAi triggers. They postulated the “16-bp rule,” which states that a minimal A-form helical dsRNA is required to assemble the catalytically active effector on the RISC complex and is able to induce a high level of RNAi. Moreover, the authors suggested that such short 16-bp siRNAs are more active in gene silencing than their 19-bp counterparts. Unfortunately, this statement with great importance for RNAi technology was in contradiction to our earlier conclusion that among the 19-, 17-, and 15-bp constructs screened, the longest duplexes possess the highest activity (Sipa et al., 2007). Therefore, to understand these discrepancies, we decided to verify some experiments on the dependence of the siRNA silencing activity on duplex length. We assumed that the discussed inconsistent results might originate from varying methods of siRNA duplex truncation.
Materials and Methods
Preparation of siRNA duplexes
Sequences of GFP-specific siRNA duplexes were designed as previously described (Sipa et al., 2007). CDK9-specific siRNAs were taken from Chu and Rana (2008). Sequences of all 15 siRNA duplexes are presented in Table 1. Synthesis of RNA oligonucleotides was performed according to routine phosphoramidite methodology (CARUTHERS, 1985) using LCA CPG glass supports and commercially available nucleoside phosphoramidites (Glen Research). Oligonucleotide synthesis was performed on an Applied Biosystems 394 instrument under the conditions recommended by the manufacturer. After synthesis, oligoribonucleotides were cleaved from the solid support, deprotected, and purified according to described procedures (Sipa et al., 2007). Assembly of siRNA duplexes was performed by mixing equimolar amounts of complementary sense and antisense oligoribonucleotides in phosphate-buffered saline (PBS) and heating at 96°C for 2 minutes, followed by slow cooling to room temperature (over 2 hours). The successful duplex formation was confirmed on a 4% agarose gel.
GFP, green fluorescent protein; mRNA, messenger RNA; siRNA, short interfering RNA.
Culture and transfection of HeLa cells
HeLa cells were maintained at 37°C in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum, penicillin (100 units/mL), and streptomycin (100 mg/mL) (Invitrogen). Cells were regularly passaged at subconfluence and plated at 24 hours before transfection at 70% confluence. Lipofectamine 2000 (Invitrogen)-mediated transient co-transfection of plasmids and siRNAs was performed in 96-well plates. A transfection mixture containing 30 ng of pEGFP-C1, 15 ng of pDsRed2-N1 reporter plasmids (BD Biosciences), 1.0, 2.5, or 50 nM of each siRNA, and Lipofectamine 2000 (ratio, 2:1) in serum-reduced OPTI-MEM (Invitrogen) was added to each well. Cells were incubated for 5 hours and the medium with transfection mixture was then replaced with fresh culturing medium supplemented with antibiotics. After 48 hours of incubation at 37°C in an atmosphere of 5% CO2, the cells were washed 3 times with PBS and lysed overnight with a mixture of NP-40 buffer [150 mM NaCl, 1% IGEPAL, 50 mM Tris-HCl (pH 7.0), 1 mM PMSF, and PBS (ratio, 1:3)] at 37°C. Cell lysates were used to determine the level of enhanced GFP (EGFP) expression by measuring its fluorescence. Cells for mock experiments were treated under the same conditions with control siRNA.
Dual fluorescence assay
Fluorescence values of EGFP and red fluorescent protein (RFP) were measured using a Synergy HT reader (BIO-TEK); data quantification was performed using KC4 software. Excitation and emission wavelengths were as follows: GFP λEx 485/20 nm and λEm 528/20 nm; RFP λEx 530/25 nm and λEm 590/30 nm. The siRNA activity was calculated as the ratio of GFP to RFP fluorescence values, averaged 8 repetitions. The relative level of fluorescence (GFP/RFP) in control cells (transfected with pEGFP-C1, pDsRed2-N1, and control siRNA) was defined as the reference (100%). Each experiment was repeated 4 to 6 times.
Reverse transcription–polymerase chain reaction (RT-PCR) analysis
HeLa cells on a 6-well plate were transfected with plasmid pEGFP-C1 and siRNA: G1(19), G1(16), G3(19), and G3(16) (100 nM) for GFP mRNA level determination or CDK9-1(19), CDK9-1(16), CDK9-2(19), and CDK9-2(16) (100 nM) for CDK9 mRNA level determination using Lipofectamine 2000 according to the manufacturer's protocol. The cells were collected at 48 hours after transfection and lysed with TriPure Isolation Reagent (Roche Applied Science), and total RNA was extracted and analyzed by RT–PCR. For a single reaction, 1 μg of total RNA was used as template. Sequences of primers (synthesized in-house) were as follows: for GFP, forward 5′-ACA AGC AGA AGA ACG GCA TC-3′ and reverse 5′-ACT GGG TGC TCA GGT AGT GG-3′; for CDK9, forward 5′-GCC TTG CGG GAG ATC AAG-3′ and reverse 5′-CAG CCC AGC AAG GTC ATG-3′; for glyceraldehyde 3-phosphate dehydrogenase (GAPDH), forward 5′-GAG TCA ACG GAT TTG GTC GT-3′ and reverse 5′-TTG ATT TTG GAG GGA TCT CG-3′. Reverse transcription was performed using ImProm-II Reverse Transcription System kit (Promega) according to the manufacturer's protocol. PCR-amplified samples were analyzed by 2% agarose gel electrophoresis and quantified using the G-BOX system (Syngene). The level of GAPDH mRNA was used as a reference.
Results and Discussion
Design of truncated siRNA duplexes for GFP mRNA
We used two siRNA duplexes, G1 and G2, designed against the mRNA of EGFP. The G1 duplex was previously described by Chiu and Rana (2002) and the G2 duplex was studied previously in our laboratory (Sipa et al., 2007). In the latter paper, we demonstrated that these two duplexes (G2 is shifted by 7 nt upstream of G1) exhibit quite different silencing activities. The 5′-end of G1 (with respect to the polarity of the guide strand) is located at position 258 and that of G2 is at position 251, where these correspond to the cleavage sites at phosphate groups located between nt 248/249 and 241/242, respectively (Fig. 1 and Table 2).
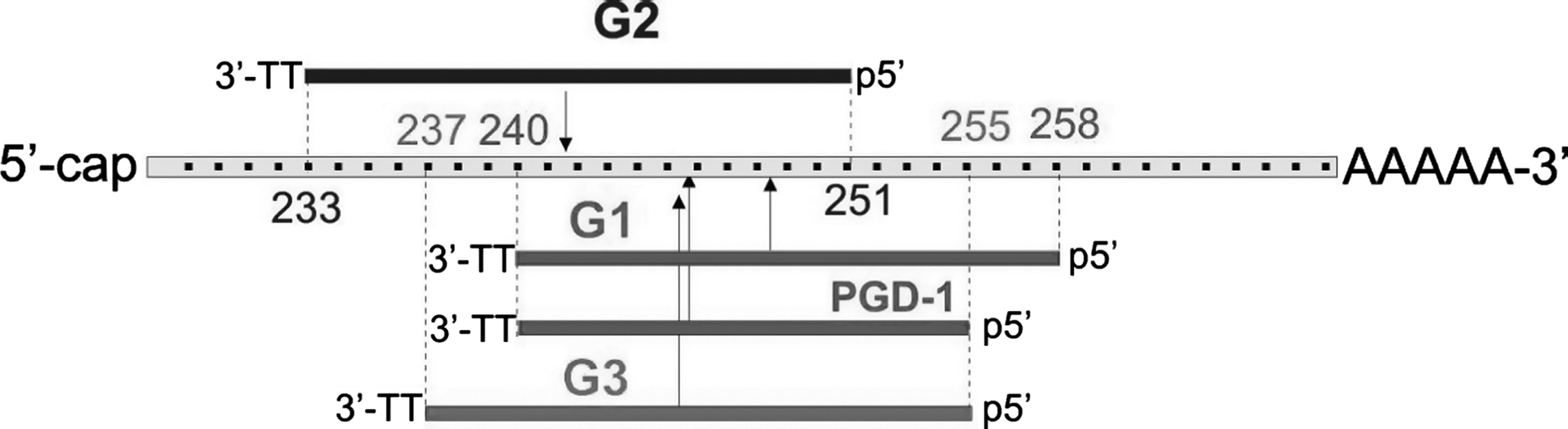
Schematic representation of the target sites of enhanced green fluorescent protein mRNA complementary to antisense strands of G1, G2, PGD-1 and G3 siRNAs. The numbering of the mRNA begins at the start codon. The sequence of G1 duplex was originally designed by Chiu and Rana (2002) and we designed G2 duplex (Sipa et al., 2007). PGD is the siRNA molecule named and tested by Chu & Rana (2008). The target sites of G3 and G2 are shifted upstream of G1 by 3 and 7 nt, respectively. Vertical arrows indicate mRNA cleavage sites guided by the indicated siRNAs. mRNA, messenger RNA; siRNA, short interfering RNA.
GFP, green fluorescent protein.
As it was shown in a study by Sipa et al. (2007), at a 1.0 nM concentration, G1- and G2-assisted RNAi decreased GFP expression by ∼18% and 90%, respectively (mock GFP expression was defined as 100%). Thus, shifting the mRNA target site upstream of the G1 target site caused elevation of the silencing activity of G2. The reason for this difference was not investigated, but it might originate, in part, from higher G2 duplex thermodynamic asymmetry (A-U bp at the 5′-end) compared with G1 (C-G bp at the 5′-end with respect to guide strand polarity) (Freier et al., 1986). Also, it might be partially attributed to the secondary structure of GFP mRNA, that is, the target sequence strand of GFP mRNA might be better suited for binding to the antisense strand of G2 rather than G1.
Chu and Rana (2008) have reported that the G1 duplex truncated by removal of 3 nt from its 5′-end (originally designed by the authors as “PGD-1”) is more active than the G1 duplex itself. We recognize that the cleavage site guided by the shorter PGD-1 duplex is positioned closer to the cleavage site guided by the more active G2 duplex (Fig. 1).
Therefore, the higher activity of the PGD-1 duplex compared with G1 might originate from the fact that the antisense strand PGD-1 siRNA has greater access to its target site. Interestingly, we noticed that PGD-1 has a different “identity” than the G1 duplex as the 5′-end of PGD-1 is located at position 255, which results in an altered cleavage site (the phosphate group is positioned between nt 245 and 246) (Table 2).
Therefore, we designed corresponding siRNA duplex (G3) with the target site, which is located between nt 255 and 237, starting from the start codon of GFP mRNA. The 5′-end, seed region, and the cleavage site guided by G3 are identical to those of PGD-1. All 3 screened duplexes, G1, G2, and G3, were prepared as a series of 3′-end truncated constructs to assure their “recognition identity,” containing 19- to 15-bp duplexes (Fig. 2 and Table 1) with 3′-terminal TT overhangs. We aimed to (i) determine the RNAi effect caused by the G1, G2, and G3 duplexes in their long (19-bp) and shortened versions (18-, 17-, 16-, and 15-bp); (ii) compare the RNAi activity of G1(19) and G3(16) [note that G3(16) is identical to the PGD-1 duplex]; and (iii) verify the “16-bp rule.”
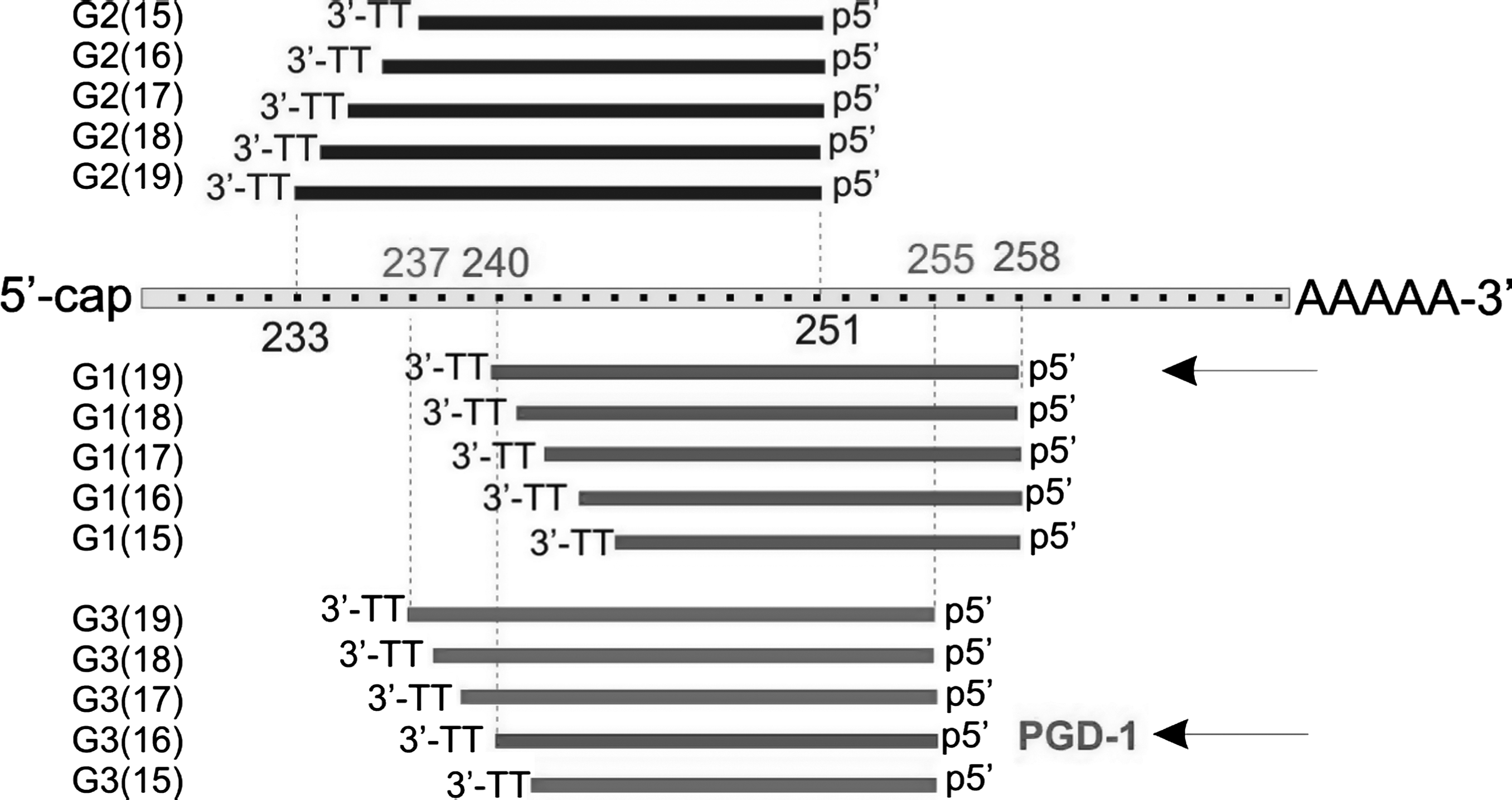
Structures of antisense strands from short interfering RNA duplexes G1, G2, and G3 evaluated in this study. For each 19-bp duplex, a series of shorter duplexes was designed by shortening the 19-bp duplex at its 3′-end by 1–4 bp. The 3′-ends consist of a TT dimer and the 5′-ends are intact within each series. In this way, each series maintains its “recognition identity” and guides the same target cleavage site. Duplexes compared by Chu & Rana (2008) are indicated by arrows.
Screening of the silencing activity of G1, G2, and G3 in a dual fluorescence assay
Silencing activity of all duplexes was measured by a dual fluorescence assay, in which reduction in GFP expression in the presence of duplex was measured. Expression of RFP was measured as an internal control. Screening was conducted at several siRNA concentrations ranging from 0.5 to 50 nM. At higher concentrations, the silencing activity of siRNA duplexes was too high to highlight any differences between the compounds in each series. Therefore, we decided to screen the silencing activity at a 2.5 nM concentration. Figure 3 presents relative GFP silencing in the presence of 19- to 15-bp duplexes for G1–G3. The GFP-to-RFP fluorescence ratio in cells transfected with control siRNA was used as a mock.
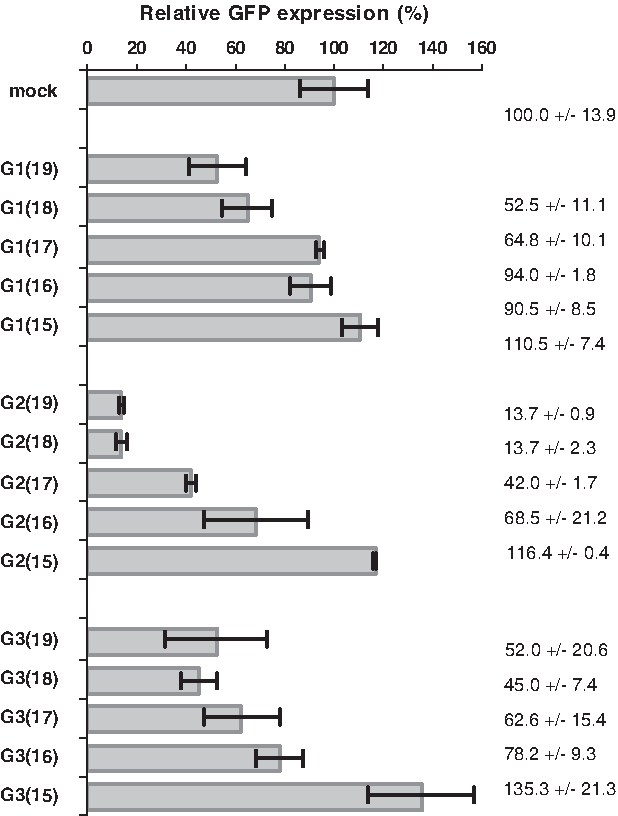
Relative GFP expression in HeLa cells transfected with pEGFP-C1 plasmid and with the 19- to 15-bp duplexes G1–G3 (1 nM concentration) determined in a dual fluorescence (GFP/RFP) assay. The cells transfected with control short interfering RNA were used as a mock. GFP, green fluorescent protein.
First, we compared activities within the G1 series. The most active were siRNA duplexes G1(19) (ca. 50% silencing) and G1(18) (ca. 35% silencing), whereas the shorter versions of G1 [G1(17)–G1(15)] were inactive. In contrast, G2 duplexes were remarkably more active than G1, as reported previously (Sipa et al., 2007). Compounds G2(19) and G2(18) had >85% silencing activity (GFP expression level was lowered to ca. 15%), whereas G2(17) reduced GFP expression by about 60% and G2(16) by about 30%. The 15-bp G2 [G2(15)] was inactive under these conditions. Silencing activity of G3 was low in comparison to G2(19) but similar to that of G1(19). We found a similar silencing effect for G3(19), G3(18), and G3(17) duplexes (ca. 40%–55% GFP silencing). The shorter siRNA duplex G3(16) reduced GFP expression by about 20%, whereas the G3(15) duplex was inactive.
The above results lead to the following conclusions: (i) in each series of siRNAs, the most active is the longest 19-bp duplex; (ii) the duplexes shortened by 1 bp (18-bp) are equally (or only slightly less) active; (iii) further, 3′ truncation of the siRNA duplexes leads to a partial or entire loss of silencing activity.
Interestingly, in our experiments performed at 2.5 nM siRNA concentrations, the relative GFP silencing activity of G1(19) was higher (ca. 50%) than that of G3(16) (ca. 20%). At higher concentrations, the silencing effect was also elevated; therefore, one may assume that the activity of these two silencers might be similar in other conditions as shown by Chu and Rana (measurements done at 50 nM concentrations).
Screening of the silencing activity of G1 and G3 (19-bp and 16-bp) by RT-PCR
The above experiments suggested that 19-bp siRNA duplexes are better silencers than their shorter 16-bp versions. To further support these data, we measured the level of GFP mRNA in HeLa cells expressing EGFP from the exogenously delivered pEGFP-C1 plasmid. The cells were transfected with pEGFP-C1 plasmid and with siRNAs G1(19), G1(16), G3(19), or G3(16) as well as control siRNA (nonhomologous to the target gene). After 48 hours of incubation, total RNA was isolated from cell lysates. The level of GFP mRNA was determined by reverse transcription and PCR amplification. As shown at Fig. 4, the shorter versions of siRNAs were again less active as RNAi triggers than their parent (19-bp) precursors. In this case, duplex G3(19) was a more potent RNAi trigger than G1(19), as the duplexes decreased EGFP mRNA levels to 43% and 67%, respectively. However, both shorter 16-bp duplexes, G1(16) and G3(16), were inactive under these conditions.

RT–PCR analysis of the relative GFP (GFP/GAPDH) expression in HeLa cells expressing the pEGFP-C1 plasmid and transfected with G1(19) and G3(19) siRNA duplexes or with their 16-bp versions (100 nM duplex concentration). The GFP messenger RNA amplicons were analyzed by 2% agarose gel electrophoresis. “Mock” denotes the sample treated with non-silencing control siRNA. RT-PCR, reverse transcription-polymerase chain reaction.
These results confirm that in each series of siRNAs, the longer duplexes (19-bp) are more effective in triggering RNAi than their 16-bp shorter versions. It is also evident that G1(19) is much more active than G3(16). We were unable to confirm the results by Chu and Rana, in which in similar conditions duplex G3(16) triggered more pronounced RNAi than G1(19). We do not have any rational explanation for the observed discrepancies because the only difference between experiments conducted by Chu and Rana and ours is the concentration of siRNA used (50 vs. 2.5 nM, respectively).
Silencing activity of siRNA duplexes against endogenous CDK9
In their paper, Chu and Rana (2008) demonstrated the significance of the “16-bp rule” with the example of the CDK9 gene. The authors screened the duplex CDK9(19) [named here CDK9-1(19)] and its shorter 5′-end truncated version CDK9(16). Through multiple experiments, the authors demonstrated higher RNAi activity of the shorter duplex CDK9(16) in comparison to the longer one CDK9(19). We were curious of the origin of this phenomenon, so we designed two sets of duplexes: the duplex CDK9-1 of 19 bp [CDK9-1(19)] and its shorter 3′-truncated version CDK9-1(16) as well as siRNA duplex CDK9-2(19) and its 3′-truncated version CDK9-2(16), which is identical to the CDK9(16) duplex cited by the authors (antisense strands of those duplexes are shown in Fig. 5A). Both 19-bp duplexes CDK9-1 and CDK9-2 differ by 3 nt, and the latter duplex target site is located upstream of the target site for the CDK9-1. The siRNA duplexes were transfected into HeLa cells, and after 48 hours of incubation, total RNA was isolated from cell lysates. The level of CDK9 mRNA was determined by RT-PCR using the specific primers used by Chu and Rana. Figure 5B shows the agarose gel analysis of the amplification products of mRNA isolated from HeLa cells.

RT–PCR analysis of the level of endogenous CDK9 mRNA in HeLa cells transfected with screened siRNA duplexes CDK9-1(19) and CDK9-2(19) or their 16-bp versions CDK9-1(16) and CDK9-2(16), respectively. (
For both screened duplexes, CDK9-1 and CDK9-2, their longer (19-bp) versions were more potent RNA triggers (silencing the target gene to 42% and 53%, respectively) than their shorter (16-bp) versions (ca. 80% GFP expression). However, the silencing potency of CDK9-1(19) and CDK9-2(16) was significantly different, opposite to the results of cited authors. Interestingly, in our hands, shorter siRNA duplexes exhibited lower potencies for the induction of target gene silencing than their long precursors, when shortening was performed by truncating the 3′-end of the parent duplexes. Moreover, shorter versions of siRNA duplexes led to cleavage of target mRNA at identical sites. Considering our previous data, we might assume that the CDK9-1 duplex would be more potent than CDK9-2 because of its greater thermodynamic asymmetry (Sipa et al., 2007). The slightly higher silencing potency of the previous duplex may be attributed either to other siRNA structural features or to CDK9 mRNA secondary structure and availability of the target site for siRNA (guide strand) binding.
Conclusions
In the series of experiments performed with siRNA duplexes of various lengths (from 19 to 15 bp) designed to silence either overexpressed EGFP or endogenously expressed CDK9, we confirmed the generally accepted opinion that 19-bp siRNAs are more efficient RNAi triggers than shorter versions of the duplexes with intact 5′-ends. These results contradict the conclusion that 16-bp siRNA duplexes are more potent RNAi triggers than their 19-bp precursors. This conclusion was drawn from results obtained by screening 19- and 16-bp duplexes, with the latter generated by the 5′-end shortening (by 3 nt) of the 19-bp duplex. One must note that shortening siRNA duplexes from their 5′-ends results in duplexes of different “identity,” that is, duplexes that guide RISC to mRNA sequences shifted upstream of the targets of the parental 19-bp siRNA duplexes. In consequence, mRNA cleavages occur at sites displaced by 3 nt. In this study, we used duplexes that retained intact 5′-ends but shortened at their 3′-ends and confirmed our previous results demonstrating that, within a series of siRNAs, the most active silencers are those with 19-bp duplexes, whereas shorter duplexes are less active. Any criteria formulated by comparing duplexes of varying 5′-ends should be carefully crafted. Even if such 16-bp duplexes are more active than 19-bp duplexes, their 5′-end-intact precursors should be more active still. Although we did not perform detailed experiments verifying the “16-bp rule,” in our opinion this rule may be valid for duplexes in which the guide strand remains intact while the passenger strand is truncated. This conclusion is in agreement with recent results from Sun et al. (2008), Sano et al. (2008), and Chang et al. (2009), who demonstrated noticeable silencing activity of shorter siRNA duplexes containing a truncated passenger (15–18 nt) strand and an intact guide strand (19–21 nt). Such asymmetric RNA duplexes can effectively trigger the RNAi pathway, confirming that optimization of siRNA structure is not a closed chapter.
Footnotes
Acknowledgment
This work was supported by PBZ-MNiSW-07/I/2007 for years 2008–2010.
Disclosure Statement
No competing financial interests exist.