
Abstract
Aptamers are single-stranded DNA or RNA oligonucleotides that adopt specific three-dimensional structures binding with high affinity and specificity to their targets. These molecules are being currently used with detection and diagnosis purposes. Parasites of the genus Leishmania cause leishmaniosis in humans and animals. Interestingly, Leishmania do not condense their chromatin during mitosis, and histone genes could be responsible for this fact. Although histones are extremely conserved proteins, reflecting their apparent universality of function, sequence similarity of kinetoplastid core histones with that of higher eukaryotes is found predominantly in the globular region. However, high sequence divergences in the N-terminal and C-terminal domains are found that convert them into potential diagnostic and/or therapeutics targets. We have successfully isolated a pool of DNA aptamers, named SELH3, which binds to Leishmania infantum H3 with high affinity and specificity. Thus, it appears that this novel anti-H3 aptamer population may be of potential application as a diagnostic system for leishmaniosis.
Introduction
The selection process implies the incubation of a target with a library of nucleic acids, typically ranging from 1014 to 1018 different sequences. The target–nucleic acid complexes are isolated, and the nucleic acid molecule is amplified for a next round of selection. The process is repeated until the desired sample is enriched with sequences that display high affinity and specificity for the target of interest. In this way, it is often possible to identify aptamers with very high affinities (nanomolar or subnanomolar) that can be considered potential biorecognition molecules.
The causative agent of Leishmaniasis is a parasitic protozoa of the genus Leishmania that is transmitted to humans by sandflies. Most forms of leishmaniasis are originally infections of small mammals (“reservoir hosts”), which play a major role in the epidemiology of the disease. Sandflies become infected by ingesting blood from infected reservoir hosts or from infected people. Leishmania parasites possess a digenetic life cycle with 2 discrete morphological phases: the promastigote, which develops extracelullarly within the gut of the insect vector and the amastigote, which is specialized to survive within the macrophage's phagolysosome of the vertebrate host. These parasites, like other related kinetoplastid protozoa, are placed in the most primitive branch of the eukaryote evolution and possess very peculiar features of gene expression and organization. Among them, the organization of the nuclear genome differs from that of higher eukaryotes. The nuclear envelope persists during cell division, and chromosomes are not visualized at any phase of the cell cycle. Although chromatin is organized in nucleosomes, higher-order structures such as 30 nm fibers are not observed. Despite the fact that histones are among the most highly conserved proteins along the evolutionary scale, histones of trypanosomatids have accumulated substantial sequence differences, mainly at the N-terminal and C-terminal regions (Galanti et al., 1998). In fact, the specificity of the humoral response elicited against parasite histones during Leishmania infection is due to the localization of the main antigenic determinants in those divergent regions (Requena et al., 2000). This helped develop a recombinant multiepitope protein containing the antigenic determinants of the H3 histone that represent a valuable tool for the serodiagnosis of canine visceral leishmaniosis (Soto et al., 1998).
During the last years, we have dedicated a lot of effort to the selection of aptamers that specifically recognize Leishmania proteins to develop a detection system for Leishmaniasis. Recently, we published the selection and characterization of an aptamer population against LiH2A and now, we report the selection of an ssDNA aptamer population, designated as SELH3, that specifically recognizes the Leishmania infantum H3 antigen. This aptamer population can be used as a screening tool for detection and identification of the H3 antigen with detection limits as low as 50 ng of protein using optical detection. In this article, we detail the application of these aptamers in enzyme-linked oligonucleotide assay (ELONA), slot blot, and western blot assays. In addition, we have identified the regions in H3 that are recognized by SELH3 aptamer population to map the H3-aptamer interaction. In summary, these findings further support that aptamers represent a new class of reagents that might be used in the detection and diagnostic platforms that were previously dominated by antibodies.
Materials and Methods
Materials
Synthetic random DNA, nonlabeled and digoxigenin-labeled primers, and bovine serum albumin (BSA) were purchased from Sigma. Dulbecco's modified Eagle's medium (DMEM) and phosphate-buffered saline (PBS) were obtained from GibcoBRL.
Expression and purification of recombinant H3
L. infantum H3 protein (Mw = 11.2 kDa) was cloned in the pQe30 expression vector, and the recombinant H3 protein was purified by affinity chromatography on Ni-nitrilo-triacetic acid (NTA) resin columns as described (Iborra et al., 2004). Briefly, the cells expressing recombinant H3 were harvested and resuspended in sonication buffer (8 M urea, 0.1 M NaH2PO4, and 0.01 M Tris HCl, pH 8.0) at 2–5 volumes × g−1 of wet weight. After binding to a Ni-NTA column, the recombinant proteins were gradually refolded on the affinity column as described (Shi et al., 1997). Afterward, recombinant H3 was eluted with 0.3 M imidazol, dialyzed against PBS at 4°C, and lyophilized.
In vitro selection procedure
Selection of ssDNA aptamers for recombinant L. infantum H3 was performed as previously described by Ramos et al. (2007). In brief, ssDNA oligonucleotides contained a central randomized region of 40 nucleotides flanked by 2 conserved 18-nucleotide regions at each end (RND40, 5′ GCGGATGAAGACTGGTCT-40N-GTTGCTCGTATTTAGGGC 3′), were denatured at 90°C for 10 minutes, and then cooled on ice for 10 minutes. For the initial SELEX round, 50 μg of RND40 (Mw = 25 kDa) were mixed with 2 μg of recombinant His-tagged H3 in 200 μL of selection buffer [20 mM Tris-HCl (pH 7.4), 1 mM MgCl2, 150 mM NaCl, 5 mM KCl, and 0.2% BSA] and incubated at 37°C for 30 minutes. The bound aptamer-H3 complexes were purified by adding 20 μL of Ni-NTA superflow (Qiagen) for 1 hour at 4°C. After washing 3 times with 1 mL of selection buffer, the ssDNA–protein complexes were resuspended in 20 μL of distilled H2O and amplified by polymerase chain reaction (PCR) using the primers named F3 (5′ GCGGATGAAGACTGGTGT 3′) and R3 (5′ GTTGCTCGTATTTAGGGC 3′) under the following conditions: 1 μM each primer, 250 μM dNTPs, 2 mM MgCl2, and 2 U Taq DNA polymerase (Biotools) in a final volume of 200 μL. The mixture was thermally cycled 15 times through 95°C for 15 seconds, 56°C for 15 seconds, and 72°C for 30 seconds, followed by a 5 minute extension at 72°C. Double-stranded PCR product was ethanol-precipitated, and the new ssDNA population was then obtained by heating the dsDNA at 90°C for 10 minutes and cooling on ice for 10 minutes. After round 3, the pool, named SELH3, was amplified and labeled by PCR using digoxigenin-labeled primer F3 and nonlabeled primer R3.
Enzyme-linked oligonucleotide assay
ELONA was used to assess the enrichment of the selected population after 3 rounds. Recombinant L. infantum H3 protein was diluted to 1 μg/mL in selection buffer, and 200 μL of the solution were incubated in a 96-well microtiter plate (Nunc) overnight at 4°C and, then, washed 4 times in selection buffer. Afterward, digoxigenin-labeled SELH3 aptamer population or digoxigenin-labeled RND40 library were diluted in selection buffer at different concentrations (0–5 μg/mL), denatured for 10 minutes at 95°C, and cooled for 10 minutes on ice. Next, 200 μL of the solution was added to each well (0–1 μg/well; 0–40 pmol/well); the plate was incubated at 37°C for 30 minutes and then washed 4 times with selection buffer to remove unbound ssDNA. Afterward, 200 μL of a 1:1000 dilution of anti-digoxigenin antibody conjugated with horse-radish peroxidase (POD) (Roche) was added to the individual wells. After 30-minute incubation at 37°C on a shaking platform, the plates were washed 4 times and developed using z,z′-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid) (ABTS) solution (Boehringer–Mannheim) according to the manufacturer's instruction. OD405nm values were determined using a microplate reader from Tecan.
In other set of experiments, recombinant H3 protein was diluted to the required concentrations (0–1.25 μg/mL), and the experiment was performed as described above. To assess the specificity of the aptamers, similar experiments were performed coating the wells with 200 μL of other recombinant Leishmania proteins (H2A, H2B, LiP0, LiP2a, and LiP2b) diluted to 1 μg/mL in selection buffer. Next, proteins were incubated with 200 μL of digoxigenin-labeled SELH3 aptamer population diluted in selection buffer at 125 ng/mL concentration and the ELONA developed as described above.
Slot blot analysis with aptamers
Various amounts of recombinant L. infantum H3 protein (0–500 ng) (0–45 pmol) and 500 ng (7.5 pmol) of BSA, as negative control, were transferred onto nitrocellulose membranes under vacuum. Filters were washed 3 times in PBS-T for 10 minutes and then blocked with 5% milk in PBS-T for 1 hour at room temperature. Afterward, membranes were incubated with 1 or 2.5 μg/mL (40 or 100 nM) of digoxigenin-labeled SELH3 aptamer population in selection buffer for 1 hour at room temperature. Next, membranes were washed 3 times with selection buffer and probed with anti-digoxigenin-POD antibody diluted 1/1000, for 1 hour at room temperature. Excess enzyme was removed by 3 subsequent washes with selection buffer. Finally, the membranes were developed with enhanced chemiluminescence's kits (GE Healthcare) and exposed to hyperfilm.
Immunoblotting
Promastigotes from L. infantum were grown in DMEM containing 10% of fetal bovine serum at 26°C. Nuclear and cytosolic fractions were obtained as previously described by Ramos et al. (2007). Thirty micrograms of cytoplasmic and nuclear proteins from L. infantum, and 1 μg of recombinant H3 were separated on 15% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride (PVDF) membranes. Afterward, membranes were incubated with digoxigenin-labeled SELH3 aptamer population at 5 μg/mL in selection buffer for 1 hour at room temperature with gentle rocking, then washed with selection buffer 3 times, and finally probed with anti-digoxigenin-POD antibody diluted 1/1000, for 1 hour at room temperature. Finally, after 3 subsequent washes with selection buffer, the membranes were developed as described above.
Mapping of the H3-SELH3 interaction
Twelve peptides corresponding to overlapped sequences of L. infantum H3 protein (2 μg/well) were incubated in a 96-well microtiter plate (Nunc) overnight at 4°C and, then, washed 4 times. Afterward, 200 μL of digoxigenin-labeled SELH3 aptamer population at 0.5 μg/mL, prepared as indicated above, were added to each well. Next, the plate was incubated at 37°C for 30 minutes and washed 4 times; and anti-digoxigenin-POD antibody was added, incubated at 37°C for 30 minutes, and developed using ABTS solution (Boehringer–Mannheim). Peptides were synthesized by the simultaneous multiple solid-phase synthetic method using a polyamine resin and FMOC chemistry (HOUGHTEN, 1985). Purity was checked by amino acid analysis and high-performance liquid chromatography.
Statistical analysis
Data are presented as an average value ± standard error of the mean from 3 to 6 independent measurements in separate experiments and analyzed using GraphPad Prism v4. The statistical significance was performed by analysis of variance followed by Tukey's test.
Results and Discussion
Histones are among the most highly conserved proteins along the evolutionary scale. However, histones of trypanosomatids have accumulated substantial sequence differences and, in fact, the specificity of the humoral response elicited against parasite histones during Leishmania infection is due to the main antigenic determinants being located in those divergent regions (Requena et al., 2000). This information suggests that histones could represent a valuable tool for the serodiagnosis of canine visceral leishmaniosis (Soto et al., 1998). Additionally, it has been shown that aptamers are able to discriminate closely related molecules and also exhibit chiral discrimination of their targets (Jenison et al., 1994). It is reasonable to assume that DNA aptamers without further modifications could perform better in detection and diagnostic assays in which the aptamers may come in contact with different biological samples for a brief period of time. Although other types of detection and diagnostic platforms utilizing peptides and antibodies are available, DNA-based technology may be more suitable for the detection of Leishmania in environmental conditions in which this parasite is endemic.
During the last years, we have selected aptamers that specifically recognize Leishmania proteins to develop a detection system for leishmaniosis. Recently, we published the selection and characterization of an aptamer population against LiH2A (Ramos et al., 2007) and, in this study, L. infantum H3 antigen was selected as a target due to its potential as an excellent candidate for Leishmania diagnosis (Soto et al., 1992, 1995) and therapy (Iborra et al., 2004).
DNA aptamers specific for L. infantum H3 protein were selected from an initial library that theoretically contained 1014–1016 different sequences. The number of PCR cycles was optimized to avoid overamplification. ssDNA sequences exhibiting affinity to the target were captured by agarose-conjugated Ni beads. The process was stopped after 3 rounds of selection, and the selected population, named SELH3, was amplified by PCR using digoxigenin-labeled primer F3 and nonlabeled primer R3.
The H3-binding properties of the starting RND40 and final SELH3 pools were compared in an ELONA experiment, which provides a rapid assessment of the relative binding capabilities of the 2 populations. As shown in Fig. 1, the SELH3 population displayed increased binding to H3 protein compared with the starting pool, suggesting that SELH3 is enriched in ssDNA sequences that recognize H3 protein relative to RND40. Data were analyzed using nonlinear regression showing that they respond to a 1-site binding curve with an equation y = (x × Bmax)/(x + KD), where Bmax is the maximal binding and KD is the concentration of ligand required to reach half-maximal binding. Results indicate that SELH3 aptamers are able to detect recombinant H3 protein in a concentration-dependent manner with a KD = 0.94 ± 0.19 nM, significantly lower than that for RND40 (KD = 1.80 ± 0.52 nM). In view of these results, we decided to characterize different aspects of the SELH3 population to determinate whether these aptamers could be used as biorecognition molecules.
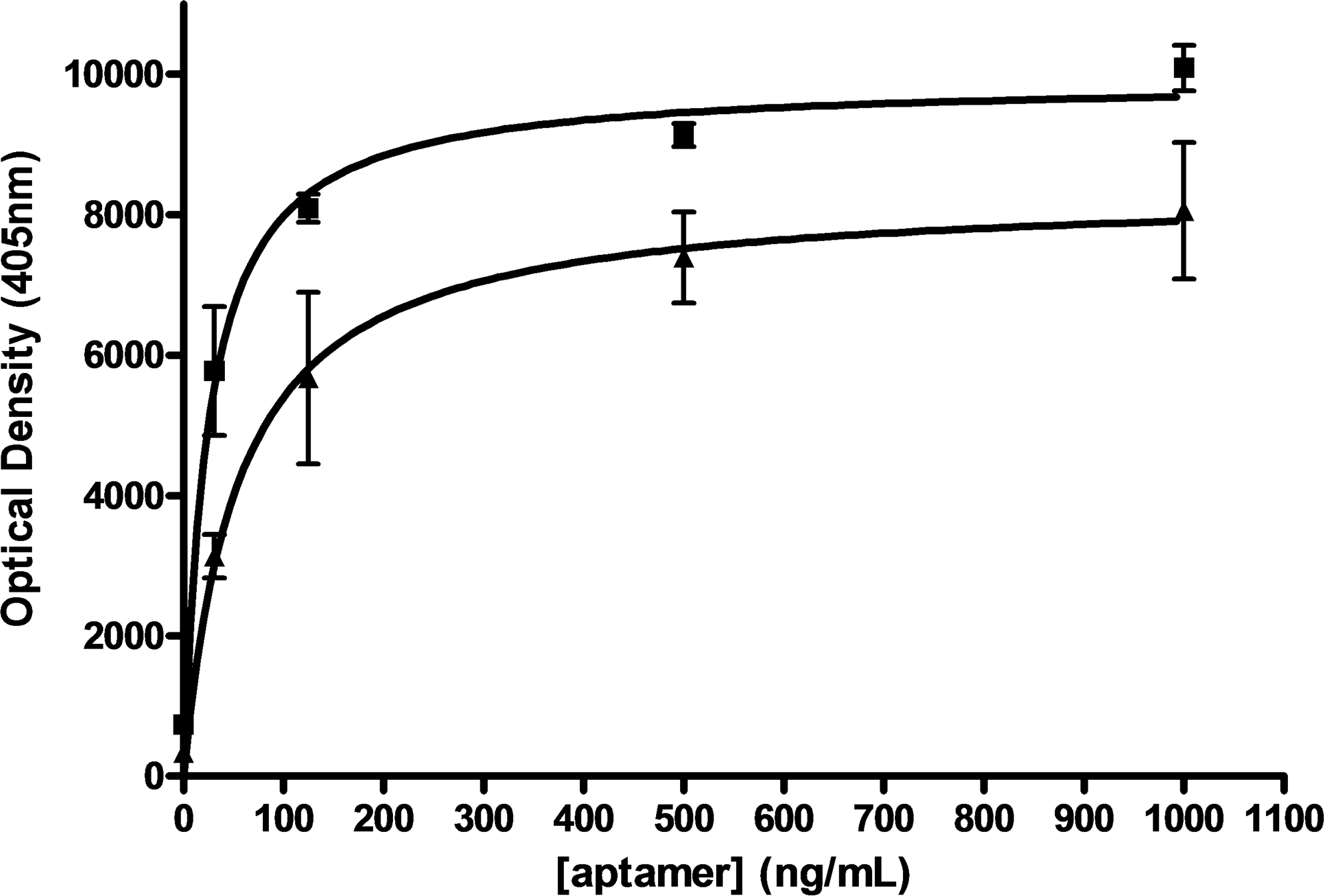
The stringency of the systematic evolution of ligands by exponential enrichment (SELEX) procedure was monitored using enzyme-linked oligonucleotide assay as described in Materials and Methods section. SELEX pool from round 3 (SELH3) and the starting round (RND40) were assayed for binding to Leishmania H3 protein. Squares, SELH3 aptamers; triangles, RND40.
Binding affinity of SELH3 aptamer population to L. infantum H3 was studied using slot blot and ELONAs. Thus, in a first approach, several amounts of recombinant H3 protein and BSA were transferred onto nitrocellulose membranes, and the immobilized proteins were probed with 2 different concentrations of digoxigenin-labeled SELH3 aptamer population as described in Materials and Methods section. As shown in Fig. 2A, SELH3 population showed strong affinity to recombinant H3 protein but did not show any affinity to negative control BSA, thereby indicating their specificity toward the H3 antigen. In these type of experiments, as few as 25 ng of H3 were detected after incubation of the nitrocellulose membrane with only 1 μg/mL (40 nM) SELH3.
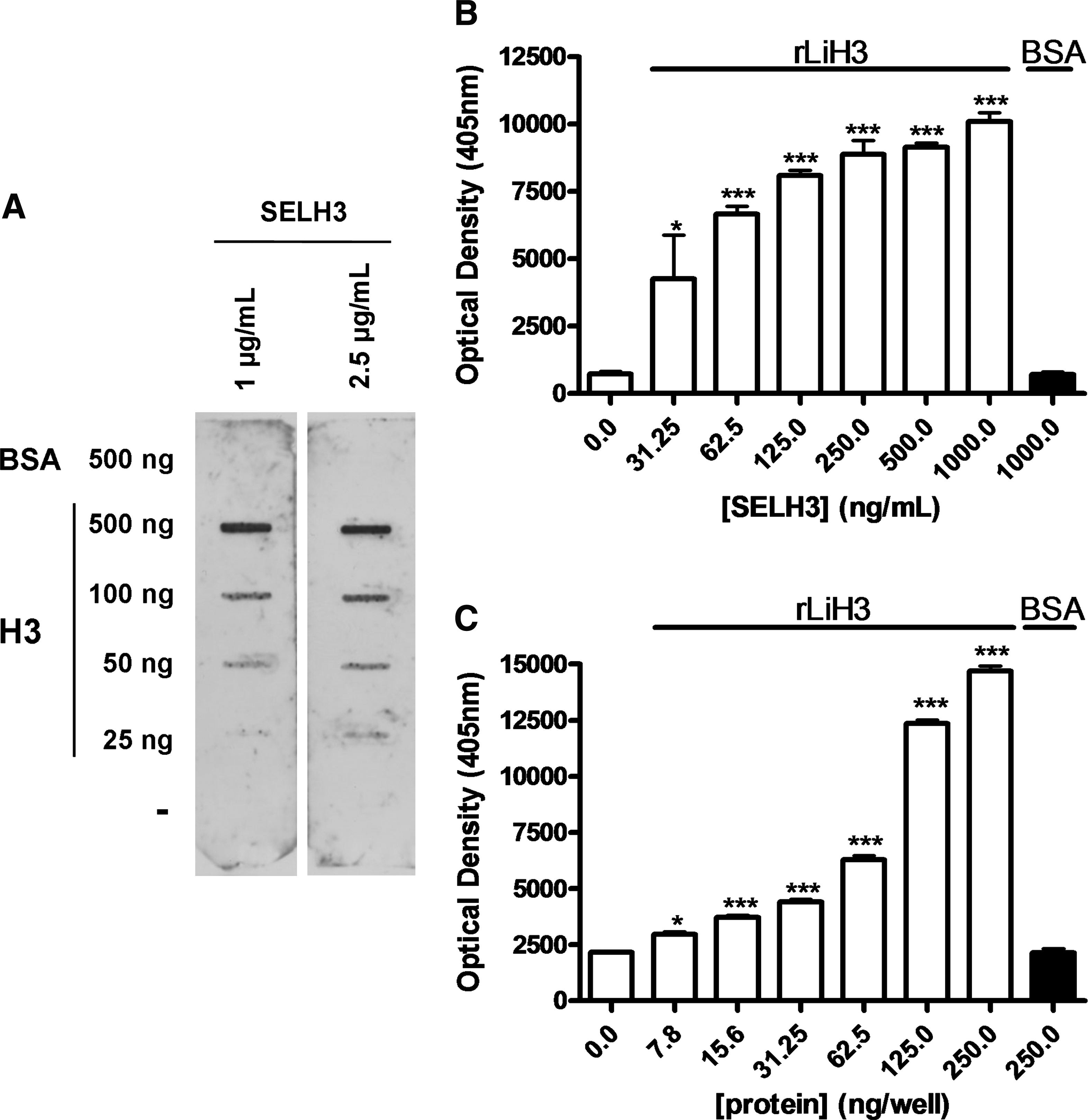
Binding capability of the SELH3 aptamer population for Leishmania infantum H3 proteins. The assays were performed as described in Materials and Methods section. (
In the ELONAs, the wells were coated with recombinant H3 protein and different concentrations of digoxigenin-labeled SELH3 were tested as described in Materials and Methods section. An exhaustive analysis of the data represented in Fig. 2B showed that concentrations as low as 31.25 ng/mL of SELH3 were able to detect from 200 ng (18 nmol) of recombinant H3 protein in a significant manner (P < 0.05). To determine the sensitivity of the selected aptamer population SELH3, we performed an analysis in which wells were coated with quantities ranging from 0 to 250 ng of recombinant H3 or 250 ng of BSA protein and incubated in the presence of digoxigenin-labeled SELH3 as described in Materials and Methods section. The results showed that SELH3 population is able to detect recombinant H3 protein in a concentration-dependent manner. As seen in Fig. 2C, it is further confirmed that the protein affinity to the aptamer population is directly proportional to the quantity of H3. A more detailed analysis of the data indicates that 50 ng/well of SELH3 aptamer population is able to significantly detect from 15.6 ng (1.4 pmol) of H3 protein in a very significant manner (P < 0.001). All the above results confirm that SELH3 recognizes recombinant H3 protein with high affinity. Enzyme-linked assays have provided a means of quickly evaluating the binding affinity. Drolet et al. (1996) reported the first enzyme-linked aptamer assay (ELAA) consisting of a mixed ELISA/ELAA sandwich to detect human vascular endothelial growth factor on a microtiter plate. Since then, similar assays have been used with either labeled or immobilized nucleic acid molecules as capture or detecting agents. Aptamer-conjugated immunoassays using other platforms to detect specific proteins were reported earlier (Rye and Nustad, 2001; Yang et al., 2003). The most remarkable applications were the demonstration of a fiber-optic microarray biosensor using aptamers (Lee and Walt, 2000) and an aptamer-based quartz crystal biosensor (Liss et al., 2002). None of these reports utilized nucleic acid-based enzymatic assays as described by us (Ramos et al., 2007), consisting of a digoxigenin-labeled aptamer assay within a 96-well microtiter plate format. Very recently, in a displacement ELLA using thrombin, Baldrich et al. (2005) or MUC1, Ferreira et al. (2008) have also demonstrated the superior performance of aptamers when compared with antibodies.
To study the specificity of SELH3 population to other proteins, we tested the binding of these aptamers against the histones H2A, H2B, and H4 and the ribosomal proteins LiP0, LiP2a, and LiP2b by ELONA (Fig. 3A). Data showed that the signal output for H3 is almost 2-fold higher than that for H2A and 3-fold higher than that for H4, indicating that although SELH3 recognized several histones (H3, H2A, and H4), H3 was recognized with a significant higher signal than for H2A and H4 (P < 0.001). Additionally, western blot assays were also performed in which cytoplasmic and nuclear fractions from promastigotes of L. infantum and recombinant H3 protein were blotted on polyvinylidene fluoride membranes and then probed with digoxigenin-labeled SELH3 aptamer population. As shown in Fig. 3B, the selected aptamers specifically recognized endogenous H3 in the nuclear fraction from L. infantum promastigotes, but this band does not appear in cytoplamic fraction. Other bands with a lower molecular weight also appeared probably corresponding to H2A. In the same experiments, we included recombinant H3 protein as a positive control. The position of this band can be used as a reference.

Specificity of the SELH3 aptamer. (
To determine the H3 regions that are recognized with higher affinity by SELH3 population, we performed ELONAs using as antigen 12 peptides corresponding to overlapped sequences of H3 protein. As shown in Fig. 3C, SELH3 mainly recognized peptide 10 with the highest affinity (P < 0.01), although peptides 5, 4, and 8 are also highly recognized but with a significantly lower affinity (P < 0.001) than peptide 10.
In summary, we have generated a population of ssDNA aptamers that bind with high specificity to L. infantum H3 antigen and do not bind to other proteins of the parasite. This article demonstrates that aptamers can be used for detection of specific targets in an ELONA format. It is important to point out that aptamers have an unlimited potential to circumvent limitations associated with antibodies. Although the antibody detection system is the gold standard for protein detection and identification, the data presented in this study strongly support the feasibility of the aptamer-based diagnostic system. In conclusion, it is reasonable to expect that the ELONA platform and western blot generated by aptamers will pave the way for future detection systems in which antibodies have been previously used.
Footnotes
Acknowledgments
E. R. is supported by a contract from the Fondo de Investigaciones Sanitarias (FIS), and V.M.G. and M.E.M. are researchers from FIBio-HRC supported by Consejeria de Sanidad (CAM). This work was supported by Grant FIS05/0453 and FIS08/0801 from the FIS of the Ministerio de Ciencia e Innovación (Spain). The authors gratefully acknowledge Dr. M. Salinas for her help during all these years as head of laboratory.
Author Disclosure Statement
No competing financial interests exist.