
Abstract
Oligonucleotide- and polynucleotide-based gene modification strategies were developed as an alternative to transgene-based and classical gene targeting-based gene therapy approaches for treatment of genetic disorders. Unlike the transgene-based strategies, oligo/polynucleotide gene targeting approaches maintain gene integrity and the relationship between the protein coding and gene-specific regulatory sequences. Oligo/polynucleotide-based gene modification also has several advantages over classical vector-based homologous recombination approaches. These include essentially complete homology to the target sequence and the potential to rapidly engineer patient-specific oligo/polynucleotide gene modification reagents. Several oligo/polynucleotide-based approaches have been shown to successfully mediate sequence-specific modification of genomic DNA in mammalian cells. The strategies involve the use of polynucleotide small DNA fragments, triplex-forming oligonucleotides, and single-stranded oligodeoxynucleotides to mediate homologous exchange. The primary focus of this review will be on the mechanistic aspects of the small fragment homologous replacement, triplex-forming oligonucleotide-mediated, and single-stranded oligodeoxynucleotide-mediated gene modification strategies as it relates to their therapeutic potential.
Introduction
The ultimate goal of gene therapy for inherited diseases is the correction of genomic mutations responsible for disease pathology. The ideal approach would leave the endogenous gene structure intact and maintain the natural linkage between the coding sequences and native regulatory sequences. Classical gene targeting strategies have been used successfully for over 20 years to engineer mouse models of disease and, more recently, for genetically modifying genes in mammalian somatic cells (CAPECCHI, 1989, 1994, 2000). However, one significant limitation of classical gene targeting is the inefficiency of the process, with only 10−6 to 10−7 transfected cells facilitating homologous recombination (HR) between the gene targeting vector and the chromosomal target. This may, in part, be due to the presence of nonnative DNA, drug selection, and DNA sequences that undermine the stability of the homologous pairing process. Further, drug selection genes typically contained in classical targeting vectors require subsequent cellular manipulations for their removal and often leave behind genetic footprints of nonnative DNA sequences. The developments in oligo/polynucleotide gene targeting strategies and in the use of recombinant nucleases that introduce site-specific double-strand breaks (DSBs) to enhance the frequency of HR are the focus of this review (Fig. 1).

Sequence-specific gene modification. Modification of a specific chromosomal locus (star) in cells may be performed using DNA with the normal wild-type sequence to correct a mutation or mutant sequences to introduce a mutation. The modifying DNA may be oligonucleotides, polynucleotides, or classical gene targeting vectors transfected into mutant cells. Components of the cellular DNA replication and/or repair pathways catalyze the synapses of incoming exogenous DNA with the homologous chromosomal target gene to facilitate sequence-specific modification. The modifying DNA may also be engineered to contain silent mutations (circle) that would alter the target DNA sequence without changing the sequence of the resultant protein, thereby providing another marker for identifying loci that have undergone a homologous exchange. There is also the possibility that the incoming DNA gets randomly inserted either in its entirety or in a truncated form.
For over two decades there have been efforts to develop alternative gene therapy strategies that focus on gene targeting, that is, sequence-specific modification of endogenous mutant sequences without the limitations associated with classical gene targeting or transgene approaches (BOGGS, 1990; VEGA, 1991; WALDMAN, 1992; Porter and Dallman, 1997; GRUENERT, 1998, 1999; Yanez and Porter, 1998; Lai and Lien, 1999; Richardson et al., 2001; Vasquez et al., 2001b; Wu et al., 2001; Vasquez and Glazer, 2002; Gruenert et al., 2003; Igoucheva et al., 2004; Kuan and Glazer, 2004). The approaches discussed use oligonucleotide (25–200 nucleotides or bp in length) or polynucleotide (>200 nucleotides or bp in length) sequences to induce sequence-specific modifications of native genomic targets. Several of these strategies have shown promise in terms of both the cell-type versatility and the apparent efficiencies of gene modification that can be achieved. Each strategy can be defined in terms of the oligo- or polynucleotides used to affect the homologous exchange and include polynucleotide small DNA fragments (SDFs), triplex-forming oligonucleotides (TFOs), and single-stranded oligodeoxynucleotides (ssODNs/SSOs) (Table 1).
Classes of oligo/polynucleotide molecules used for gene targeting in mammalian cells are listed. Resistance to intracellular nuclease digestion and increased cellular half-life is conferred by base, backbone, 5′ and 3′ ends, and sugar modifications that can be introduced through direct chemical synthesis. Polynucleotides are typically generated by PCR amplification or from plasmids or phagemids.
dsDNA, double-stranded DNA; PCR, polymerase chain reaction; nt, nucleotide; SDF, small DNA fragment; ssDNA, single-stranded DNA; SSO, single-stranded oligonucleotide; TFO, triplex-forming oligonucleotide.
Chimeric RNA/DNA oligonucleotides (RDOs) are another class of oligonucleotides that have been used for gene targeting and have shown to achieve correction efficiencies up to 50% (Kren et al., 1999b). However, the reproducibility of the studies has been limited in mammalian systems (Zhang et al., 1998; de Semir et al., 2002; TAUBES, 2002a, 2002b). There are numerous speculations about the mechanism underlying RDO-mediated exchange (Kren et al., 1999a; Gamper et al., 2000a; Richardson et al., 2001; Andersen et al., 2002), and it has been suggested that the limited reproducibility of previous studies in multiple independent laboratories is related to the synthesis of the RDOs. Although this system may have applications for sequence-specific gene modification and repair, the difficulty in reproducibly achieving high levels of homologous exchange in multiple laboratories makes it less attractive as a potential therapy (Diaz-Font et al., 2003; Manzano et al., 2003) and will therefore not be discussed in detail.
The oligo/polynucleotide gene targeting strategies using SDFs, TFOs, and SSOs have shown promise and may provide new paradigms for therapeutically manipulating the genome of individuals with inherited diseases or generating animal models for disease (Gruenert et al., 2003; Aarts et al., 2006, 2009; Goncz et al., 2006; Maurisse et al., 2006a; Andrieu-Soler et al., 2007; Murphy et al., 2007; Todaro et al., 2007; Sangiuolo et al., 2008; Zou et al., 2009). Thus, an understanding of the nuances of the application of these systems, their analysis and the potential mechanisms that underlie their efficacy is critical to their development as a viable therapy.
Oligo/Polynucleotide Gene Modification Strategies
Therapeutic oligo/polynucleotides generally carry a limited number (between 1 and 5) of modifying bases within a segment of DNA that is otherwise homologous to the target sequences. While more base mismatches may be used, their effect on efficacy needs to be tested empirically for each oligo/polynucleotide construct. The oligo/polynucleotide-mediated homologous exchange process relies on the endogenous cellular machinery to affect an exchange between the exogenous nucleotide sequences and the endogenous target sequences (Tables 2–4). This enzymatically driven “genetic surgery” both maintains the integrity of the gene and will minimize permanent regional chromatin remodeling that could change gene expression patterns (Cavazzana-Calvo et al., 2004; Cereseto and Giacca, 2004; Evans-Galea et al., 2007). The cellular machinery involved in oligo/polynucleotide homologous exchanges is not well defined for human cells. Extrapolating from what is know about classical HR pathways in bacteria and yeast, it is likely that DNA repair (particularly nucleotide excision repair, NER) and replication genes and pathways play a primary role the process of sequence-specific modification (Table 2 and Fig. 2). However, the actual mechanisms that underlie oligo/polynucleotide-mediated modification require further elucidation and are the subject of numerous studies (Swagemakers et al., 1998; HABER, 1999; Yanez and Porter, 1999, 2002; Taniguchi et al., 2002; Vasquez et al., 2002; GRUENERT, 2003; Gruenert et al., 2003; Villemure et al., 2003; Shivji and Venkitaraman, 2004; Yang et al., 2004; Igoucheva et al., 2006a; Sleeth et al., 2007; Zhang et al., 2007).
This list includes, but is not limited to, major DNA repair enzymes and pathways that are candidates for oligo/polynucleotide-mediated gene targeting. As there are continuous discoveries of new DNA repair enzymes and/or enzyme functions not previously recognized as having a role in DNA repair, there may be additional enzymes that could be candidates for mediating oligo/polynucleotide homologous exchange.
GGR, global genomic repair; TCR, transcription-coupled repair; HR, homologous recombination; NHEJ, nonhomologous end joining; SPR, short-patch repair; LPR, long-patch repair; BER, base excision repair; NER, nucleotide excision repair; DSBR, double-strand break repair; MMR, mismatch repair.
Details of gene targeting using unintegrated, extrachromosomal gene targets in mammalian cells are given. These experiments typically utilize mutant versions of the reporter genes transiently transfected into cells and rely on gene targeting by oligo/polynucleotides to convert into a functioning gene.
GFP, green fluorescent protein; Neo, neomycin resistance gene; HygB, hygromycin B resistance gene; Strep, streptomycin resistance gene; Zeo, zeocin resistance gene; βgal, β-galactocidase; luc, luciferase; supF, supF suppressor tRNA gene.
Details of gene targeting of transgenes at randomly integrated chromosomal sites are given. These experiments utilize mutant reporter genes (similar to those used in Table 3) randomly integrated into mammalian cell genomes and rely on homologous exchange between oligo/polynucleotides to restore function to the mutated genes. Gene abbreviations are as described in Table 3.
ES, embryonic stem.
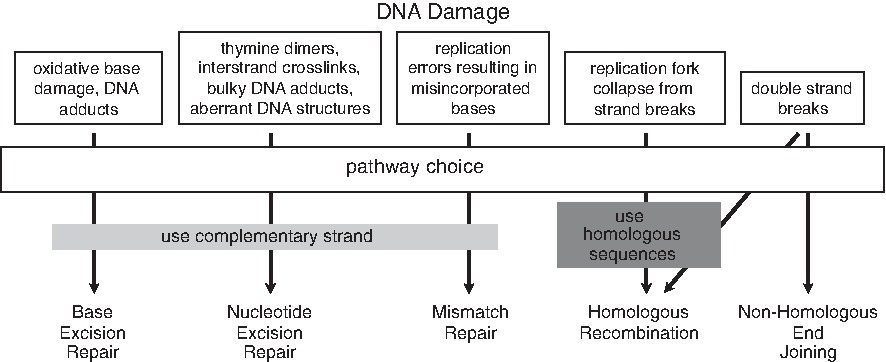
DNA repair pathways. There are 5 primary DNA repair processes that respond to sequence and/or structural changes in the DNA. Single-stranded DNA damage is generally repaired using the complementary strand as a template. Single-base damage generally engages the base excision repair pathway and involves the removal of the aberrant base from the deoxyribose-phosphate backbone DNA by a lesion-specific glycosylase. Subsequent apurinic/aprymidinic (AP) endonuclease cleavage of the phosphodiester bond in the backbone is followed by single-nucleotide gap filling with DNA-polymerase-β and ligation with DNA ligase III. Another repair pathway, nucleotide excision repair (NER), responds to helix-distorting damage such as that caused by ultraviolet light or bulky chemical DNA adducts. NER corrects disrupted base pairing with an endonuclease cleavage 3′ and 5′ of the damaged strand. The damaged strand is then removed through a 5′ → 3′ exonuclease digestion or a helicase that creates a gap of 24–32 nucleotides (nt). The gap is filled in by DNA polymerase δ or ɛ and the ends are ligated by DNA ligase III. A subset of NER, transcription-coupled repair, is specific for the repair of transcriptionally active genes and will displace the RNA polymerase complex to ensure transcriptional fidelity. Mismatch repair (MMR) comes into play during DNA replication or recombination to correct base mismatches (base transversions or transitions and insertions or deletions) that have been generated during these processes. The MMR pathway involves 4 sequential steps: (1) mismatch recognition, (2) recruitment of components and assembly of a MMR complex, (3) strand discrimination and degradation of the abnormal strand, and (4) strand resynthesis. Certain DNA lesions such as DNA double-strand breaks (DSBs) invoke 2 other DNA repair pathways, nonhomologous end-joining (NHEJ) and homologous recombination (HR). These pathways are multifactorial and involve enzymatic and structural elements of the DNA repair pathways mentioned above, factors involved in DNA replication, and factors that are unique to either NHEJ or HR. In response to DSBs, 2 kinases central to DNA repair and replication, ataxia telangiectasia mutated and ataxia telangiectasia related, phosphorylate components of the DNA replication complex to inhibit the cell cycle and enhance DNA repair. DSBs that result from collapsed replication forks are primarily repaired by HR, which is upregulated during late S and G2 phases when sister chromatids are available. NHEJ is the error-prone repair pathway where the ends DSBs are linked together without regard to homology.
It has been difficult to gain insight into similarities and differences in the mechanisms underlying the various oligo/polynucleotide gene targeting approaches, because of the variability in the exogenous DNA, the genetic target, the cell systems employed, and the approaches used to analyze the efficiency of homologous exchange. Since there are multiple DNA repair and replication pathways (Fig. 2 and Table 2) that depend not only on the type of DNA modification, but also on the cell type and the stage of the cell cycle (eg, HR is likely involved in S-phase repair), it is difficult to generalize about the involvement of specific mechanisms in the homologous exchange process for an individual strategy. The situation becomes even more complicated when the exogenous modifying/therapeutic DNA (referred to as “therapeutic DNA” from this point forward) is either single-stranded DNA (ssDNA) or double-stranded DNA, is of varying lengths, and/or contains a variable number of mismatches, modifications of the bases, sugars, 5′ or 3′ ends, or the backbone; placed in different regions and sequence contexts within the therapeutic DNA (Jain et al., 2008). A number of studies have begun to investigate potential pathways for specific gene targeting approaches and have shed some light on the involvement of general pathway and enzymatic features of a given approach (Kucherlapati et al., 1985; KUCHERLAPATI, 1987; GRUENERT, 1998, 2003; Yanez and Porter, 1999; Datta et al., 2001; Vasquez et al., 2001b; Igoucheva et al., 2004, 2006a; Terunuma et al., 2004; Ferrara and Kmiec, 2006; Goncz et al., 2006; Knauert et al., 2006; Radecke et al., 2006b) (Table 2). Understanding the role that these pathways play in modulating oligo/polynucleotide-based sequence modification will be critical for development and optimization of therapeutic efficacy and assessing potential adverse outcomes.
ssODN/SSO targeting
ssODN/SSOs (for the purposes of this review, SSOs will be used) are <200 bp in length. SSOs are comprised of a single mismatch to the target sequence that is generally in the middle of the molecule (Fig. 3). SSOs were initially evaluated using plasmids carrying reporter genes (Campbell et al., 1989) or in the targeting of the HPRT1 gene in lymphoblasts (Hunger-Bertling et al., 1990; Kenner et al., 2002, 2004; Hegele et al., 2008; Wuepping et al., 2009). Although SSOs have been synthesized with phosphorothioate backbones (Gamper et al., 2000b), 2′-O-methyl uracil (Igoucheva et al., 2001) or with 5′ or 3′ thymidine clamps (Hegele et al., 2008) to inhibit degradation, they have also been used without modification to facilitate homologous exchange.
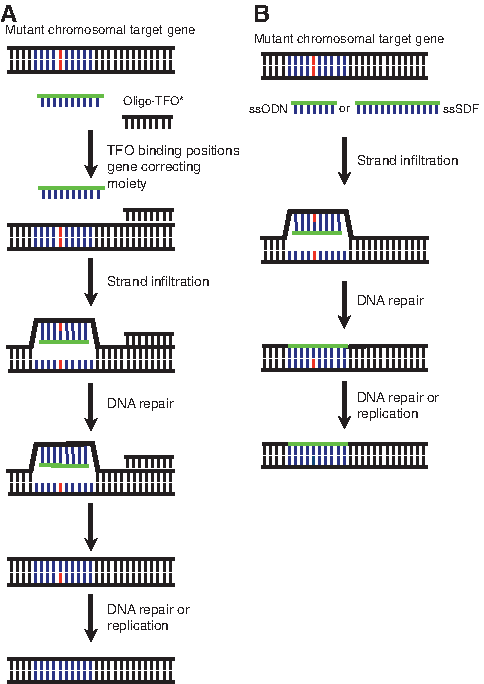
Oligo/polynucleotide homologous exchange.
Analysis of cellular responses after transfection with either SSOs or plasmids indicated that a greater number of genes involved in DNA repair, cell cycle arrest, and apoptosis were upregulated after transfection with a plasmid when compared with transfection of SSOs (Igoucheva et al., 2006a). Thus, the frequency of genomic correction was ∼5-fold higher when the SSOs were cotransfected with an unrelated plasmid when compared with transfection with the correcting SSO alone. These studies clearly suggest that cellular DNA repair processes influence SSO-mediated, sequence-specific modification of genomic DNA.
One repair pathway, mismatch repair (MMR), appears to suppress SSO-mediated targeting of genomic DNA, but not episomal gene targeting through MSH2 (Dekker et al., 2003; Aarts et al., 2006). Studies evaluating MMR showed that SSOs were effective in correcting mutations in reporter genes that had been introduced into msh2 −/− cells, but not in wild-type cells, unless the expression of MSH2 was transiently inhibited by RNA interference. However, when the target was episomal, SSO-mediated modification was observed in both the wild-type and msh2 −/− mouse embryo fibroblasts, whereas it was reduced in CHO cells defective in NER (Igoucheva et al., 2006a).
Studies targeting different reporter genes generally indicate that there is a higher efficiency of correction with the antisense, nontranscribed SSO target (Igoucheva et al., 2001; Nickerson and Colledge, 2003; Pierce et al., 2003; Olsen et al., 2005b; Yin et al., 2005). When comparing the efficiency of correction in CHO cells carrying a single integrated lacZ, the clone with higher lacZ mRNA expression had a relatively greater gene targeting efficiency for the transcribed sense than the untranscribed antisense SSOs compared with a clone that had lower level of expression (Igoucheva et al., 2003). This study suggests that transcription may be a factor in the correction process. However, one might also view this result in the context of chromatin accessibility. As the lacZ transgenes in the individual clones are likely integrated in different regions of the genome, their chromosomal environment is different in terms of gene expression. There are data to suggest that the antisense SSOs are more effective at facilitating homologous exchange than their sense counterparts; however, there is also evidence that there are no strand-associated differences in SSO-mediated targeting (Dekker et al., 2003). Whether these contradictory findings reflect differences in the cells used in the targeting studies requires further analysis.
In addition to the implications that DNA repair pathways are involved in modulating SSO-mediated gene modification, higher efficiencies of modification were also observed for cells stalled in S-phase (Brachman and Kmiec, 2005; Olsen et al., 2005a; Wu et al., 2005), indicating that DNA replication and/or the chromatin structure may also be involved. In this context, there is a possibility that an SSO can be incorporated into the genome as Okazaki fragment during replication (GRUENERT, 1998; Wu et al., 2005; Radecke et al., 2006b). Several studies showed physical incorporation of an SSO into the genome (Radecke et al., 2006b; Hegele et al., 2008) and also indicated that in the presence of DSBs, SSOs appear to act as a template and are not incorporated (Radecke et al., 2006a). This suggests that the mechanism for incorporation of the SSO into the genome may not involve the HR or the nonhomologous end joining (NHEJ) pathways that are activated by DSBs (Thompson and Schild, 1999).
Small fragment homologous replacement/SDF
Small fragment homologous replacement (SFHR) modification is distinct from SSO-mediated modification in that the SDF polynucleotides used in SFHR (generally between 200 and 2000 bp/nt in length) are larger than the SSOs and are either individual ssDNA, complementary ssDNA, or double-stranded DNA. The term “polynucleotides,” rather than oligonucleotides, will be used to designate SDFs.
SFHR effectively relies on (1) an SDF, carrying a single or multiple base alterations, finding its sequence homolog (genomic or episomal), and (2) the cellular enzymatic pathways that facilitate homologous exchange between target sequences and the SDF. Although there has been speculation about the mechanism(s) that underlie SFHR (GRUENERT, 1998, 1999, 2003; Gruenert et al., 2003), there is still only limited knowledge of the cellular factors that influence SFHR (Fig. 3).
SFHR has been shown to occur in a variety of cell types both in vitro and in vivo, targeting a number of different genes related to inherited diseases (Table 3). Previous studies have demonstrated SFHR-mediated modification of genes associated with sickle cell disease (Goncz et al., 2006) and β-thalassemia (Colosimo et al., 2007) (β-globin), cystic fibrosis (CFTR) (Kunzelmann et al., 1996; Goncz et al., 1998, 2001, 2002; Sangiuolo et al., 2002, 2008), Duchene's muscular dystrophy (dystrophin) (Kapsa et al., 2001, 2002; Todaro et al., 2007), severe combined immune deficiency (DNA-dependent kinase catalytic subunit) (Zayed et al., 2006), alpha-1 antitrypsin deficiency (alpha-1 antitrypsin) (McNab et al., 2007), spinal muscular atrophy (survival motor neuron-1) (Sangiuolo et al., 2005), and Lesch-Nyhan syndrome (hypoxanthine-guanine phosphoribosyl transferase, HPRT) (Bedayat et al., 2010).
Although most corrections are single base substitutions, studies with the CFTR concentrated on the most common mutation, a 3-bp deletion in human CFTR exon 10 resulting in a phenylalanine deletion at codon 508 (ΔF508) (CONSORTIUM, 1990; Tsui et al., 2010). Approximately 1% of immortalized CF airway epithelial cells homozygous for the ΔF508 mutation have been functionally and/or genotypically corrected following transfection with an SDF that reinserts the 3 deleted bases (Kunzelmann et al., 1996; Bruscia et al., 2002; Sangiuolo et al., 2002). A ΔF508, 3-bp deletion mutation was also introduced into primary human airway epithelial cells in vitro (Goncz et al., 1998), mouse airway epithelium in vivo (Goncz et al., 2001), and in mouse embryonic stem cells (Sangiuolo et al., 2008). Studies using the SDF-transfected mouse embryonic stem cells indicated that at least 12% of the CFTR mRNA transcript was ΔF508 and that there was a significant reduction in cAMP-dependent chloride efflux.
Thus far there have been only a limited number of studies that have explored the mechanisms that underlie SFHR-mediated modification. One study, using an episomal target, evaluated how components of different DNA repair pathways influence SDF-mediated episomal repair (Fig. 2 and Table 2) (Morrison and Wagner, 1996). This study indicated that cells defective in genes encoding for Ku80 (NHEJ), Mlh-1 (MMR), xeroderma pigmentosum complementation group C (NER), ataxia telangiectasia mutated (DNA damage response/repair, cell cycle regulation), or ADP-ribosyl transferase (base excision repair) show no differences in their ability to undergo homologous replacement when compared with normal controls. The study evaluated correction of an episomal target within 48 hours of transfection, thereby suggesting that episomal homologous exchange mediated by a double-strand SDF is not affected by each of these pathways independently. Although this study and that evaluating SFHR in a DNA-dependent kinase catalytic subunit knockout mouse cells (Zayed et al., 2006) suggest that SFHR is independent of the NHEJ pathway, the role of the HR repair pathway needs to be confirmed. In addition, the possibility that components from both the NHEJ and HR repair pathways, acting in concert, may influence episomal homologous exchange cannot be ruled out.
Studies with hRad51, a key element in HR, indicate that hRad51 overexpression will slightly enhance (up to 3-fold higher) homologous replacement in an episomal target (Yanez and Porter, 1999; Thorpe et al., 2002), but will not significantly improve the frequency of homologous exchange in genomic DNA (Yanez and Porter, 2002). Evaluating an episomal target provides some insight into the process of polynucleotide-mediated homologous exchange, whereas episomal gene targeting and genomic gene targeting appear to be mechanistically distinct. This is consistent with the findings of studies using SSOs indicating that there are differences between episomal targeting and targeting in chromatin (Igoucheva et al., 2001, 2006a, 2008; Olsen et al., 2005a; Ferrara and Kmiec, 2006; Radecke et al., 2006a, 2006b; Murphy et al., 2007). Whether there are elements of each type of homologous exchange that overlap is clearly an area open to further investigation.
Another study evaluating genomic homologous exchange mediated by an SDF used a severe combined immune-deficient mouse defective in the DNA-dependent kinase catalytic subunit (DNA-PKCS), a component of the NHEJ repair pathway. This study indicated that SDFs could mediate homologous exchange at the DNA-PKCS locus (Zayed et al., 2006). Taken further, one would expect an increase in homologous replacement as the cells are forced to rely on the HR machinery because of the defect in the NHEJ pathway. It therefore appears that the NHEJ pathways do not play a substantive role in the SFHR/polynucleotide-mediated homologous exchange process.
TFO-mediated targeting
TFOs are ssODNs/SSOs, generally 10–40 nt in length, that bind to specific regions in duplex DNA as a third strand to form a triple helix. These triple helices, originally described >50 years ago (Felsenfeld and Rich, 1957), were formed at polypurine or polypyrimidine regions of DNA bound via Hoogsteen hydrogen bonds to TFOs (Kallenbach et al., 1976). Because of their high binding affinity, TFOs have been used to manipulate genes and gene function, for example, for sequence-specific induced mutagenesis in plasmids (Wang and Glazer, 1995) or mice (Vasquez et al., 2001a) or for enhanced intrachromosomal recombination (Luo et al., 2000; Datta and Glazer, 2001) (Fig. 3). TFOs have also been used successfully for gene targeting by tethering an SDF to a TFO directed to the proximity of the sequence to be modified. These studies showed correction of an episomal reporter gene that had been transfected into mammalian cells (Chan et al., 1999; Maurisse et al., 2002). In addition, TFO-mediated correction of adenosine deaminase-deficient human lymphocytes and p53 mutant human glioblastoma cells has been demonstrated (Culver et al., 1999).
Studies evaluating the enzymatic pathways that may underlie the TFO-mediated homologous exchange indicate that NER is involved (Wang et al., 1996; Duval-Valentin et al., 1998; Christensen et al., 2008) (Fig. 2). These studies indicated that sequence-specific mutagenesis or intramolecular episomal HR was not detectable in cells deficient in the xeroderma pigmentosum group A (XPA) gene, whereas XPA cells complemented with the wild-type XPA cDNA or naive wild-type cells were able to facilitate the HR (Faruqi et al., 1996; Wang et al., 1996; Datta et al., 2001; Vasquez et al., 2002; Simon et al., 2008; Lonkar et al., 2009). TFO-induced mutagenesis was also not detected in cells deficient in the Cockayne's syndrome group B gene that is involved in the transcription-coupled NER pathway (Wang et al., 1996). In addition, there appear to be other repair pathways involved in TFO-mediated homologous exchange that can be enhanced in the presence of DSBs (Vasquez and Glazer, 2002; Villemure et al., 2003; Chin et al., 2007; Zhang et al., 2007; Chin and Glazer, 2009).
As sequences rich in polypurine and polypyrimidine often occur in the human genome (Goni et al., 2004), this sequence specificity also limits the ability of TFOs to target any DNA lesion. One approach to overcome this limitation has been to develop TFOs that have modified nucleotides such as locked nucleic acid or peptide nucleic acid residues to increase the affinity of the TFO for specific target sequences (Simon et al., 2008). Another approach has been linkage of the TFO to psoralens that bind to DNA upon UV light irradiation and both anchor the TFO to its target site and also stimulate DNA repair (Gruenert and Cleaver, 1985; Luo et al., 2000; Vasquez et al., 2001a; Broitman et al., 2003; Varganov et al., 2007; Liu et al., 2009b). Although the use of psoralens might be useful in a laboratory setting, it may not meet safety standards because of its ability to enhance DNA damage and potentiate carcinogenesis (Pathak et al., 1959; Pathak and Joshi, 1984; Gruenert et al., 1985; Tamaro et al., 1986). The UVA irradiation requirement for psoralens to form covalent linkage to the DNA is only feasible in skin or ex vivo through the use of a 2-photon laser (Vasquez and Legerski, 2010). Further, the UVA irradiation component of psoralen addition presents additional problems because of the production of reactive oxygen species (YOUNG, 1986; Carraro and Pathak, 1988; Moller et al., 1995; Viola et al., 2008).
Optimizing Gene Targeting
Optimization of the efficiency of oligo/polynucleotide-driven homologous exchange is a critical component for achieving therapeutic efficacy. As most of the oligo/polynucleotide-based approaches appear to involve elements of DNA repair and/or DNA replication (Biggerstaff et al., 1993; KUNKEL, 1995; GRUENERT, 1998, 2003; JIRICNY, 1998; Yanez and Porter, 1998; HABER, 1999; Ng and Baker, 1999; Bode et al., 2000; CAPECCHI, 2000; Ray and Langer, 2002; Saintigny and Lopez, 2002; Thompson and Schild, 2002; HELLEDAY, 2003; Wang and James Shen, 2004; Durant and Nickoloff, 2005; Wu et al., 2005; Bugreev et al., 2007; Cuozzo et al., 2007; Sleeth et al., 2007; Li and Heyer, 2008; Waldman et al., 2008), there are specific enzymatic mechanisms that can be investigated. It will be important to develop assay systems that can be readily used to quantify homologous exchange in the context of particular repair or replication mechanisms. There are a number of gene and cell systems that have already been developed, including the HPRT gene (Doetschman et al., 1987; Hunger-Bertling et al., 1990; Valancius and Smithies, 1991; Kenner et al., 2002; Hendrie et al., 2003; Ohbayashi et al., 2005; Pierce and Jasin, 2005; Hinz et al., 2006; Bedayat et al., 2010), green fluorescent protein (GFP) (Thorpe et al., 2002; Radecke et al., 2004; Olsen et al., 2005a; Tsuchiya et al., 2005c; Vasileva et al., 2006; Sakamoto et al., 2007; Kamiya et al., 2008), neomycin resistance (G418) (Song et al., 1985; Campbell et al., 1989; Manivasakam et al., 2001; Hendrie et al., 2003; Murphy et al., 2007), zeocin resistance (Colosimo et al., 2001), and β-galactocidase (Nickerson and Colledge, 2003). Although the reporter/selectable marker gene systems are often used, the HPRT system represents a gene in its natural genomic context and may more accurately reflect homologous exchange at other genomic loci. However, if these analytical endpoints are to be instructive with regard to specific DNA repair and replication mechanisms, it will be necessary to evaluate these endpoints under conditions where specific enzymatic components of a given pathway are incapacitated or activated. This can be either done in cell systems that are defective in specific DNA repair and/or replication enzymes, in cells where specific enzyme components are repressed, or in systems where specific pathways are activated.
Although some of these approaches have been pursued in particular cell systems where there is a genetic defect in an aspect of DNA repair or replication, there is still a great deal of ambiguity about which pathways predominate for any given oligo/polynucleotide base approach. Studies with TFOs have already demonstrated that there is a correlation between the XPA gene and the TFO targeting (Datta et al., 2001; Vasquez et al., 2002; Simon et al., 2008; Lonkar et al., 2009), and studies with SSOs suggest that MMR is involved (Whitehouse et al., 1996; Yin et al., 2005; Liu et al., 2009a; Olsen et al., 2009). On the other hand, SDFs appear to be DNA replication dependent and do not involve the NHEJ pathways (Morrison and Wagner, 1996; GRUENERT, 1998, 2003; Goncz et al., 2006; Zayed et al., 2006). There is still the issue of episomal vs. chromatin targeting that needs to be resolved, before there can be any accurate association between a specific pathway and a specific oligonucleotide strategy.
One potential avenue for optimizing the efficiency of homologous exchange is through the introduction of DSBs (Thompson and Schild, 2002; Golding et al., 2004; Nickoloff and Brenneman, 2004; Radecke et al., 2006a; Engels et al., 2007; Zhang et al., 2007). Although DSBs can be cytotoxic and mutagenic, cells have the capacity to repair such lesions through either NHEJ or HR (THOMPSON, 1991; JEGGO, 1998; Johnson and Jasin, 2001; Sonoda et al., 2001; Tamulevicius et al., 2007). DSBs can be induced through physical or chemical DNA damaging agents that either directly or indirectly generate lesions in the DNA. The random nature of physically or chemically induced DSBs make these approaches more challenging when developing a therapeutic regimen to correct pathological mutations (Li and Heyer, 2008). To circumvent the issue of random DSBs throughout the genome, strategies have been developed using rare cutting, sequence-specific endonucleases (Rouet et al., 1994; Choulika et al., 1995; Smih et al., 1995; Cohen-Tannoudji et al., 1998; Elliott et al., 1998; Johnson and Jasin, 2001; Cabaniols and Paques, 2008; Grizot et al., 2009, 2010), chimeric endonucleases with sequence-specific zinc finger motifs (Kim and Chandrasegaran, 1994; Kim et al., 1997; Chandrasegaran and SMITH, 1999; Smith et al., 1999; Bibikova et al., 2001, 2003; Porteus and Baltimore, 2003), and TFOs conjugated with125I (Mezhevaya et al., 1999; Sedelnikova et al., 2000) or pyrene (Benfield et al., 2008) to introduce DSBs at defined genomic loci.
The most studied rare-cutting endonuclease, I-SceI, is derived from yeast and has an 18-bp recognition site. As this 18-bp recognition sequence is not found in mammalian cells, the target DNA (whether episomal or genomic) must be modified to contain the unique recognition sequence before DSBs can be introduced (Choulika et al., 1994, 1995; Rouet et al., 1994; Smih et al., 1995; Sargent et al., 1997; Cohen-Tannoudji et al., 1998; Kim et al., 2001; Willers et al., 2001; Akyuz et al., 2002; Nickoloff and Brenneman, 2004; Saleh-Gohari and Helleday, 2004; Puttini et al., 2005; Radecke et al., 2006a). Numerous studies have clearly demonstrated that, when compared with uncleaved DNA or DNA that does not contain the recognition sequence, the frequency of homologous exchange is significantly increased when the target DNA containing the I-SceI recognition sequence is cleaved (Smih et al., 1995; Liang et al., 1998; Moynahan et al., 1999; Johnson and Jasin, 2001). A primary drawback in using I-SceI cleavage for generating DSBs in mammalian DNA is that there are no endogenous I-SceI recognition sequences in mammalian cells. The 18-bp recognition sequence must always be introduced into the cells near the target site to have a maximum effect. There is also a relationship between the cut site and the distance from the target site that shows a significant decrease as the distance from the target sequence increases, falling to 0% conversion after ∼500 bp in one model system (Elliott et al., 1998).
The development of chimeric zinc finger nucleases (ZFNs) has provided an alternative to the I-SceI endonuclease to generate sequence-specific DSBs (Kim and Chandrasegaran, 1994; Kim et al., 1996; Porteus and Baltimore, 2003; CARROLL, 2004). These chimeric endonucleases are comprised of a customized sequence-specific zinc-finger domain linked to nonspecific nucleases for targeted cleavage (Kim et al., 1997; Chandrasegaran and SMITH, 1999; Bibikova et al., 2001; Porteus and Carroll, 2005). ZFNs have been successfully used to enhance targeted gene replacement by a donor plasmid in Drosophila (Bibikova et al., 2002, 2003; Carroll et al., 2008; Bozas et al., 2009) and human cells (Porteus and Baltimore, 2003; Alwin et al., 2005; Urnov et al., 2005; Potts et al., 2006; Lombardo et al., 2007; Pruett-Miller et al., 2008; Mittelman et al., 2009; Zou et al., 2009). The donor plasmid contains sequences essentially homologous to the endogenous target, with the exception of the bases to be modified in a bacterial plasmid backbone. One study has demonstrated that a ZFN/donor plasmid-mediated approach could correct a one-base frameshift mutation in the IL2RG gene in human erythroleukemia K562 cells and also demonstrated the expression of IL2RG mRNA and protein after correction (Urnov et al., 2005). In addition, ZFNs have been shown to enhance the correction of a mutant EGFP integrated in HEK293 cells by SSOs (Olsen et al., 2009) and SDFs (H. Parsi and D.C. Gruenert, unpublished data).
Although the endonuclease and ZFN strategies have a certain appeal for enhancing homologous exchange, they are similar to other cDNA-based gene therapies, in that they involve introduction of a therapeutic cDNA into cells to facilitate gene modification. There is the potential of eliciting an immune response to the ZFN protein because it is a foreign protein; however, this may be somewhat mitigated ex vivo. Moreover, it is possible that the ZFNs will cut at sites other than the ZF-directed site (Porteus and Carroll, 2005; CARROLL, 2008; Radecke et al., 2010). This nonspecific offsite cutting would generate multiple DSBs that could affect the karyotypic and genetic stability of the target cell. Clearly, these limitations will affect the therapeutic potential of any approach that relies on the introduction of DSBs. It will therefore be paramount to evaluate these aspects of enzymatically induced DSBs in cell systems that are directly relevant to the cells targeted by any given therapeutic strategy rather than surrogate cell lines.
Considerations
The enhancement of oligonucleotide-mediated modification requires evaluation of multiple parameters that might influence efficacy or the assessment of efficacy. These include, but are not limited to, the genetic target to be modified, the degree of homology associated with the size and character of the therapeutic DNA, the cell type, the mode of transfection, the enzymatic pathways involved, and the method of assessing homologous exchange. Several studies have shown that even when the transfection efficiency between different cell types is similar, the efficiency of targeting can be quite different. One study indicated that, although the efficiency of transfecting a plasmid or oligonucleotide into CHO-K1 and HEK239 cells was the same (∼80%), the efficiency of episomal gene correction varies from 29% for CHO-K1 cells to <2 × 10−3 for HEK293 cells (Igoucheva et al., 2006b). Another study also evaluating episomal targeting for the same pair of cells and the same target gene (lacZ) indicated an efficiency of 0.44% for the CHO-K1 cells and 2.6% for the HEK293T cells (Nickerson and Colledge, 2003). Although on the surface it appears that the studies are similar, there are significant differences that may provide some insight into factors that are significant for a particular gene modification method. The above studies were carried out with SSOs that were of different sizes, 45 nt (Igoucheva et al., 2006b) vs. 35 nt (Nickerson and Colledge, 2003). Although this 10 nt difference is small, it represents a significant fraction of the SSO length and may reflect the importance of length in SSO targeting. Alternatively, this difference may be a function of the proportion of cells in S-phase or the transfection method. Further, an examination of the sequences, that is, base composition, could be important in establishing reproducibility. Along those lines, one study indicated that a TA repeat at the 5′ end of an SSO is more effective at facilitating homologous exchange (Wuepping et al., 2009). Sequence is particularly important for TFO recognition and sequence-specific modification (Fox and Brown, 2005).
Other approaches using TFOs or SDFs also require close scrutiny of the transfection conditions for oligo/polynucleotide introduction, cell type, composition, and target. There appears to be a relationship between the SDF size and the targeting efficiency. However, there is only limited data in this regard. Preliminary studies in HEK293 cells indicate that smaller SDFs, within a range from ∼200 to 500 bases, are more effective at targeting an integrated GFP (H. Parsi and D.C. Gruenert, unpublished observations). As indicated for the SSOs, there is also a distinct difference between the modification of a genomic or an episomal target. The reasons for this difference are not well defined; however, a previous study evaluating the induction of genes by a plasmid vs. an oligonucleotide suggests that the quantity of repair and replication genes transcribed after the introduction of a plasmid is significantly greater than those induced by an oligonucleotide alone (Igoucheva et al., 2006b). Therefore, the plasmid introduced during a study evaluating episomal targeting would likely activate multiple pathways relevant to the oligo/polynucleotide modification of the plasmid and thereby facilitate episomal targeting. Moreover, the episomal copy number (especially with replicating plasmids) is generally greater than that of the genomic target and further enhances the potential of detecting a targeting event.
Another consideration involves the assessment of the homologous exchange (Fig. 4). In the event that a phenotypic readout is not possible, as with correction of an allele where there is no chemical or physical means to facilitate enrichment of the modified cells, it becomes necessary to devise screening protocols that can identify modifications in the DNA and RNA. The most obvious assessment strategy is polymerase chain reaction (PCR) and reverse transcription–PCR analysis. Although, on the surface, PCR appears straightforward, there are important experimental design features that must be implemented to avoid aberrant or misleading results (Zhang et al., 1998; De Semir and Aran, 2003, 2006; Gruenert et al., 2004; Maurisse et al., 2006a). In particular, assessment of genomic DNA modification requires that the DNA be gel purified, especially at early time points following the transfection, to eliminate spurious amplification due to the presence of unincorporated oligo/polynucleotide (Maurisse et al., 2006b). The assessment of modification at the transcriptional level requires that the RNA samples be treated with DNase to avoid any contribution to the reverse transcription–PCR by residual oligo/polynucleotide (Goncz et al., 1998, 2001; Maurisse et al., 2006b). Analysis of the genomic DNA is generally more of a screening, whereas analysis of the RNA can be confirmatory under the appropriate conditions, for example, DNase treatment and a unique silent mutation present in the modifying oligo/polynucleotide that results in introduction of an restriction fragment length polymorphism. Clearly, a phenotypic functional readout is the most definitive indication that a homologous exchange has occurred.

Potential pathways for dsSDF-mediated homologous exchange: SDF repair of chromosomal DSBs. DSBs [eg, nuclease-induced (yellow) as well as replication anomalies and damage caused by physical or chemical agents] can activate intracellular exonucleases to produce 3′ single-strand DNA ends. Cellular proteins involved with HR and repair of DNA ends include BRCA2, RPA, and Rad gene family members (Rad51/Rad52/Rad54). One potential HR mechanism is diagrammed for DSB repair with an SDF as a template for repair synthesis. The color scheme for the bases and DNA backbone is the same as in Fig. 3.
If oligo/polynucleotide-based gene modification strategies are to have therapeutic applications, the degree of random integration must be measured. The finding that the random integration of recombinant retrovirus led to adverse affects such as leukemia in the cDNA-based severe combined immune deficient-X1 clinical trials is an indication that insertional mutagenesis can compromise therapeutic efficacy (Cavazzana-Calvo et al., 2004; Williams and Baum, 2004; Evans-Galea et al., 2007). Given this observation, it appears that random integration of cDNA with its associated regulatory elements can significantly affect the surrounding chromatin and gene expression profile. Although it is possible that a randomly integrated oligo/polynucleotide may result in detrimental insertional mutagenesis, the potential will be minimized because of the lack of regulatory elements directly associated with the therapeutic DNA. In studies evaluating a clonal population of cells modified by HPRT-specific SDFs, random integration was not apparent (Bedayat et al., 2010). However, the potential for random integration needs to be thoroughly evaluated. This can be achieved through direct sequencing, inverse PCR, assessment of adverse effects in animal models, and/or transfection of sequences not found in the genomic DNA that are of similar size. Ultimately, before any oligo/polynucleotide-based approach can be used clinically, the extent of random integration needs to be assessed in terms of risks vs. potential benefit.
Prospects
The prospect of a genetic therapy that will “surgically” correct a disease-causing mutation in an endogenous gene is not only appealing but also represents the ultimate goal for gene therapy. The possibility of treating the myriad of genetic disorders by correcting specific genomic sequences and maintaining the integrity of the genetic structure is clearly very enticing. However, evaluation of the therapeutic potential of oligo/polynucleotide-based gene modification remains in its infancy and additional mechanistic studies are required to gain insight into the weaknesses and strengths of the different approaches, as well as how they might be optimized to maximize therapeutic efficacy without adverse effects.
It will be critical to determine whether the introduction of large quantities of DNA will lead to apoptosis (GRUENERT, 2003; Gruenert et al., 2003; Liu et al., 2009a; Olsen et al., 2009). There are dosage and threshold issues that need to be clarified to determine whether the dose of the oligo/polynucleotide required to achieve maximal modification induces significant apoptosis undermining therapeutic efficacy by decreasing the number of corrected cells.
The safe clinical application of the cDNA/donor plasmid-based ZFN (CARROLL, 2004; Alwin et al., 2005; Porteus and Carroll, 2005; Urnov et al., 2005) or the meganuclease (Cabaniols and Paques, 2008; Grizot et al., 2009, 2010) gene targeting approaches also requires careful assessment of offsite DSBs (CARROLL, 2008; Bozas et al., 2009). Minimizing the number of extraneous DSBs will be critical to ensure that genomic integrity is maintained and does not lead to karyotypic instability and/or neoplasia (Griffin and Thacker, 2004; Acilan et al., 2007; Mittelman et al., 2009). Recent studies using reengineered FokI nucleases may be one means to address this limitation (Kandavelou et al., 2009; Ramalingam et al., 2010).
As is the case with any new therapy, it will be necessary to appropriately evaluate underlying mechanisms and how they relate to efficiency, efficacy, and safety. The potential candidate pathways and enzymes and the studies investigating aspects of individual approaches outlined in Tables 2–5 represent a partial list of an ongoing effort to gain a better understanding of the mechanisms involved and the therapeutic potential of individual approaches. Although Tables 3–5 are extensive, they are not comprehensive and are intended to highlight representative studies that have provided insight into both the phenomenology and the mechanisms involved and lay the foundation for future studies that will help determine how a given oligo/polynucleotide-based strategy can be most effectively applied. It may be that there is not one approach that will predominate, but rather that each approach has specific applications where it will be the most effective and/or that a given gene targeting strategy complements another therapeutic strategy. Whatever the case, the oligo/polynucleotide-based strategies offer the potential for exciting alternatives that have limited other genetic therapy approaches.
Details of gene targeting to modify native endogenous genes in mammalian cells existing gene mutations or introduce DNA sequence changes are given.
Gene nomenclature: α1-AT, alpha 1 antitrypsin, β-globin, beta globin gene; CFTR, cystic fibrosis transmembrane conductance regulator; HPRT, hypoxanthine-guanine phosphoribosyltransferase; SMN2, survival of motor neuron 2; PRKDC, protein kinase, DNA-activated, catalytic polypeptide (DNA-dependent protein kinase catalytic subunit, DNA-PKcs); γ-globin, gamma globin; DMD, Duchene's muscular dystrophy (dystrophin); K-17, keratin 17; Rb, retinoblastoma; ADA, adenosine deaminase; TP53, p53 gene; Kit, c-kit; Hoxa7, Hox 1.1 (homeobox 1.1).
HSPCs, hematopoietic stem/progenitor cells.
Footnotes
Acknowledgments
The authors thank Drs. Hooman Parsi, Hamid Emamekhoo, Rosalie Maurisse, and David DeSemir for their input and for sharing their data, as well as Drs. Karen Vasquez and James Cleaver for their reading of the manuscript and constructive comments. This overview has been supported by NIH grants (GM075111 and GM075111-04S1) and Pennsylvania Cystic Fibrosis, Inc.
Author Disclosure Statement
No competing financial interests exist.