
Abstract
Abstract
The analysis of plasma samples from HIV-1/HCV mono- and coinfected individuals by quantitative proteomics is an efficient strategy to investigate changes in protein abundances and to characterize the proteins that are the effectors of cellular functions involved in viral pathogenesis. In this study, the infected and healthy plasma samples (in triplicate) were treated with ProteoMiner beads to equalize protein concentrations and subjected to 4-plex iTRAQ labeling and liquid chromatography/mass spectrometry (LC-MS/MS) analysis. A total of 70 proteins were identified with high confidence in the triplicate analysis of plasma proteins and 65% of the proteins were found to be common among the three replicates. Apolipoproteins and complement proteins are the two major classes of proteins that exhibited differential regulation. The results of quantitative analysis revealed that APOA2, APOC2, APOE, C3, HRG proteins were upregulated in the plasma of all the three HIV-1 mono-, HCV mono-, and coinfected patient samples compared to healthy control samples. Ingenuity pathway analysis (IPA) of the upregulated proteins revealed that they are implicated in the hepatic lipid metabolism, inflammation, and acute-phase response signaling pathways. Thus, we identified several differentially regulated proteins in HIV-1/HCV mono and coinfected plasma samples that may be potential biomarkers for liver disease.
Introduction
The explosion of comparative proteomic technologies emphasize that analysis of differentially expressed proteins associated with HIV-1/HCV coinfection could potentially result in identification of biomarkers and drug targets (Liu et al., 1999; Luciano-Montalvo et al., 2008; Zhang et al., 2010). However, the vast number of biomarker discovery research is focused on cancer with limited proteomics data available for infectious diseases including HIV-1 and HCV. Although a few proteomics studies have focused on analysis of serum samples from HIV-1/HCV mono- and coinfected patients, fewer to no proteomic analysis of plasma have been reported thus far in this cohort of patients. Recently, SELDI-TOF-based proteomic analysis of sera from HIV-1-infected individuals with or without cognitive impairment, identified several biomarkers for HIV-1-associated dementia (Wiederin et al., 2009). These analyses demonstrated the value of mass spectrometry (MS) as a mainstay for the characterization of complex proteomes.
In the present study, we investigated the comprehensive proteome profile of plasma samples from HIV-1/HCV mono- and coinfected patients using a comparative proteomic analysis in an attempt to understand the systemic pathophysiology of liver disease in these patients. We used ProteoMiner equalization method (Guerrier et al., 2008) to reduce the dynamic range of the plasma proteome and isobaric tag for relative and absolute quantitation (iTRAQ) (Ross et al., 2004) technology in conjunction with MS. Our study is the first ever to investigate the proteome profiles of HIV-1/HCV mono- and coinfected patient's plasma samples. Herein we report that key proteins involved in various molecular mechanisms that are differentially and uniquely present in plasma samples from HIV-1/HCV mono- and coinfected patients as identified by iTRAQ quantitative proteomics and determined by literature reports and ingenuity pathway analysis.
Materials and Methods
Patient samples
The patient cohorts included in the study consists of three HIV mono-infected patients (two male, one female) with no prior treatment and with CD4 counts ranging from 300 to 600 with viral load 10,700 to 42,600. The three HCV mono-infected pateints (two male, one female) who failed prior antiviral treatment with viral load ranging from 500 to 3.5×106. The HIV/HCV coinfected patient cohort consists of two male and one female patient who have failed antiviral treatments with CD4 counts ranging from 220 to 630 and HIV viral load ranging from <75 to 10m,729 and HCV viral load from 0.11×106 to 0.85×106 (Rahman et al., 2011).
Plasma protein equalization with ProteoMiner treatment
Plasma samples were pooled from three HIV-1 mono-infected patients, three HCV mono-infected patients, three HCV+HIV coinfected patients, and three healthy individuals. Each of the pooled plasma samples were divided into three aliquotes and processed independently as biological triplicates to assess the reproducibility of the data. All the plasma samples were centrifuged at 3,000 rpm for 10 min prior to treatment with hexapeptide ligand library (ProteoMiner) beads. The plasma proteins were “equalized” using the ProteoMiner small capacity protein enrichment kit (BioRad Laboratories, Hercules, CA) according to the manufacturer's recommended protocol. Briefly, spin columns were washed with water 5 min (two times), and then by wash buffer 5 min (two times), and thereafter, 175 μL of the plasma was applied to each of the columns and incubated for 2 h at room temperature. The columns were then washed with wash buffer for 5 min (two times). After the buffers have been removed, 20 μL of rehydrated elution reagent (buffer containing urea, CHAPS, acetic acid) was added and incubated for 15 min (three times) at room temperature. All the three elution fractions were pooled and protein concentrations were estimated by Nanodrop (Nanodrop Technologies, Wilmington, DE) method.
In-solution digestion by Trypsin
The proteins from the plasma samples were prepared in 50 mM ammonium bicarbonate (AB) and reduced with dithiothretol (DTT) (5 μg/μL in 50 mM AB) by incubating the mixture at 65°C for 45 min and alkylated with iodoacetamide (15 μg/μL in 50 mM AB) by incubating the reaction mixture at the dark for 30 min. The alkylated proteins were digested by Trypsin (Promega, Madison, WI) (5 ng/μL in 50 mM AB) overnight at 37°C.
Purification of tryptic peptides by Sep-Pak C-18 columns
The Sep-Pak (C-18) columns (waters) were activated with 50% acetonitrile and equilibrated with 2% acetonitrile (ACN) and 0.1% trifluoroacetic acid (TFA) in water buffer. Then peptide mixture (dissolved in 2% ACN and 0.1% TFA in water) was slowly loaded into the C-18 columns. The columns were then washed thoroughly with 0.1% TFA to remove salts and buffers. Finally, the tryptic peptides were eluted in 1 mL of 80% acetonitrile and concentrated.
iTRAQ labeling
Peptide mixtures were labeled using 4-plex iTRAQ labeling kit (Applied Biosystems, Bedford, MA) following the vendors recommended protocol. Briefly, each vial of iTRAQ Reagent was allowed to reach room temperature and spun to bring the solution to the bottom of the tube. A total of 70 μL of ethanol was added to each room-temperature iTRAQ reagent vial. Then the solution in each vial was vortexed to mix and then spun. The contents of one iTRAQ reagent vial were transferred to one sample tube. (iTRAQ Reagent 114 vial to Normal serum protein digest; iTRAQ Reagent 115 vial to HCV serum protein digest; iTRAQ Reagent 116 vial to HIV-1 serum protein digest; iTRAQ reagent 117 vial to HCV+HIV serum protein digest). Each tube containing the reaction mixture was vortexed to mix and then spun. Finally, the reaction mixtures were incubated at room temperature for 1 hour. All the iTRAQ labeled tryptic peptide mixtures were combined for further purification and fractionation.
Purification of iTRAQ-labeled peptides by strong cation exchange (SCX) chromatography
The concentrations of buffer salts and organics in iTRAQ labeled peptide solution were reduced by diluting the sample mixture by 10-fold with cation exchange buffer-load [10 mM potassium phosphate (KH2PO4) in 25% ACN at pH 3.0]. The sample mixture was reconstituted with 1-mL cation exchange buffer-load and vortexed to mix. An aliquot was removed and the pH was adjusted by adding 1 M HCl to bring it down to pH ∼3. To condition the cartridge, 1 mL followed by 2 mL of the cation exchange buffer-clean (10 mM KH2PO4 in 25% ACN/1 M KCl at pH 3.0) was injected and diverted to waste. The diluted sample mixture was slowly injected (≈1 drop/s) onto the cation-exchange cartridge and the flow-through was collected in a sample tube. A total of 1 mL of the cation exchange buffer-load was injected to wash the Tris(2-carboxyethyl)phosphine (TCEP), SDS, calcium chloride, and excess iTRAQ reagents from the cartridge. To elute the peptides, 500 μL of the cation exchange buffer-elute [10 mM KH2PO4 in 25% acetonitrile/350 mM potassium chloride (KCl) at pH 3.0] was slowly injected (∼1 drop/s) and the eluate was captured in a fresh tube.
Fractionation of peptides by C-18 chromatography
Prior to the fractionation, the peptides were purified using waters Sep-Pak C-18 columns as described above. The fractions were collected as a part of the elution step of the peptides from C-18 columns using three different (20, 50, and 80%) acetonitrile in water elution buffers. These peptide fractions were eluted in 100 μL (two times) of elution buffers and concentrated for subsequent analysis.
MS experiments
The purified iTRAQ labeled tryptic peptides were analyzed by nano LC-MS/MS experiments on a QSTAR-Elite hybrid mass spectrometer (ABSCIEX™) interfaced with a nanoscale reversed-phase high-pressure liquid chromatography (Tempo) with a 10 cm-180 i.d. glass capillary packed with 5-μm C18 Zorbax™ beads (Agilent, Placerville, CA). The buffer compositions were as follows: Buffer A was composed of 98% H2O, 2% ACN, 0.2% formic acid, and 0.005% TFA; buffer B was composed of 100% ACN, 0.2% formic acid, and 0.005% TFA. Peptides were eluted from the C-18 column into the mass spectrometer using a linear gradient of 10–80% Buffer B over 60 min at 400 μL/min (10-40% buffer B for 50 min, followed by 5 min at 80% B, followed by 5 min 10% B). LC-MS/MS data were acquired in a data-dependent fashion by selecting the six most intense peaks with charge state of 2 to 4 that exceeds 20 counts, with exclusion of former target ions set to “120 s” and the mass tolerance for exclusion set to 100 ppm. Time-of-flight MS were acquired at m/z 400 to 2,000 Da for 0.5 s with 12 time bins to sum. MS/MS data were acquired from m/z 50 to 2,000 Da by using “enhance all” function and 24 time bins to sum, dynamic background subtract, automatic collision energy, and automatic MS/MS accumulation with the fragment intensity multiplier set to 6 and maximum accumulation set to 2 s before returning to the survey scan (in iTRAQ acquisition mode).
Identification and quantification of proteins
The raw data obtained from the LC-MS/MS experiments was processed and searched by ProteinPilot 3.0 software using ProGroup search engine to identify proteins. Carbamidomethylation was selected as the fixed cysteine modification, trypsin as the digestion enzyme, “biological modifications” were selected as the “ID focus” and a “Thorough ID Search Effort” was selected. The SwissProt human database (released on 03-23-2010 with 20,280 human entries) was used for all searches with either a confidence interval of 80 or 95%. A decoy database search strategy was also employed to estimate the false discovery rate (FDR) and it was calculated searching the spectra against the SwissProt decoy database. The total number of proteins reported in each set were based on ≥80% confidence for protein identification as determined by ProteinPilot (protein score ≥0.7). The iTRAQ labeled peptides were quantified by ProteinPilot software and to determine the differential expression of proteins in the HCV, HIV, and HCV+HIV, the average ratio of identified peptides was calculated based on the iTRAQ reporter ion intensities. The results were then exported into excel for manual data interpretation. Although relative quantification and statistical analysis were provided by the ProteinPilot 3.0 software, an additional 1.5-fold change cutoff for all iTRAQ ratios (ratio >1.5 or <0.6) was selected to categorize proteins as up- or downregulated. Proteins with iTRAQ ratios below the low range (0.6) were considered to be downregulated, whereas those above the high range (1.5) were considered upregulated.
Ingenuity network and pathway analysis
Differentially regulated proteins identified in this analysis were further investigated using IPA (Ingenuity Systems, Mountain View, CA; http://www.ingenuity.com). IPA was used to infer upregulated proteins in terms of an interaction network and predominant canonical pathways. A protein interaction network was generated as follows: a dataset containing the upregulated proteins (focus proteins) from the infected patients plasma samples, was uploaded into the IPA. By running the core analysis, these proteins were overlaid onto a global molecular network developed from the information in the ingenuity knowledge base (IKB), a regularly updated and curated database that consists of interactions between different proteins gathered from scientific literature. Networks of these focus proteins were then algorithmically generated by including as many primary proteins as possible and other nonfocus proteins from the IKB that are needed to generate the network based on connectivity. Canonical pathways were identified from the IPA library based on their significance to the dataset. The significance of the association between the upregulated protein dataset and the canonical pathway was measured as described in the literature (Kulkarni et al., 2010).
Results
The strategy used in the current study is depicted in Figure 1. The plasma samples from HIV-1/HCV mono- and coinfected individuals and the controls were processed by ProteoMiner kit to reduce the dynamic range of the plasma protein concentration. Approximately 30 μg of the eluted protein was digested with trypsin and the resulted peptide mixtures were labeled with iTRAQ reagents and fractionated by an off-line HPLC as described in the Method section. A representative image of the HPLC chromatograms of the iTRAQ labeled peptide mixtures resulted in the triplicate analysis of the plasma samples is shown in Figure 2. All the labeled peptides were analyzed by LC-MS/MS experiments and the resulting raw data were searched in SwissProt human database to identify the proteins. The quantitation of differentially expressed proteins and FDR analysis were performed by ProteinPilot 3.0 software.

Strategy used for the identification of differentially expressed proteins in HIV-1/HCV mono- and coinfected patient's plasma.
Partial HPLC chromatograms of the fractionation of iTRAQ labeled tryptic peptides by C-18 reversed-phase chromatography.
In total, 70 proteins were identified in the triplicate analysis of Itraq-labeled tryptic peptides (Supplementary Tables S1 and S2). The results of protein identification by ProteinPilot software obtained at different confidence levels are summarized in Table 1. A total of 57 proteins in Replicate 1, 51 in Replicate 2, and 48 in Replicate 3 were identified with over 80% confidence. The Venn diagram in Figure 3 shows that 34 of these proteins were common to three replicates. There were high correlation rates between three biological replicates because 60% of proteins identified in Replicate 1 were detected in Replicates 2 and C, 66% of proteins identified in Replicate 2 were detected in Replicates 1 and 3, and similarly, 71% of proteins identified in Replicate 3 were detected in Replicates 1 and 2. These results were supported by the analogous HPLC chromatographic profiles observed for the iTRAQ-labeled peptide mixtures in three replicates (Fig. 2). FDR analysis was performed using ProteinPilot software and the results are given in Supplementary Table S3. It is evident from the data that FDR results are less informative as this analysis in ProteinPilot software (or in any other software program) requires a larger data set (more than 100 proteins). Nonetheless, we meticulously verified manually all the raw tandem mass spectrometry data to gain high confidence in protein identification and made sure that there are no false positive hits in the final protein list.
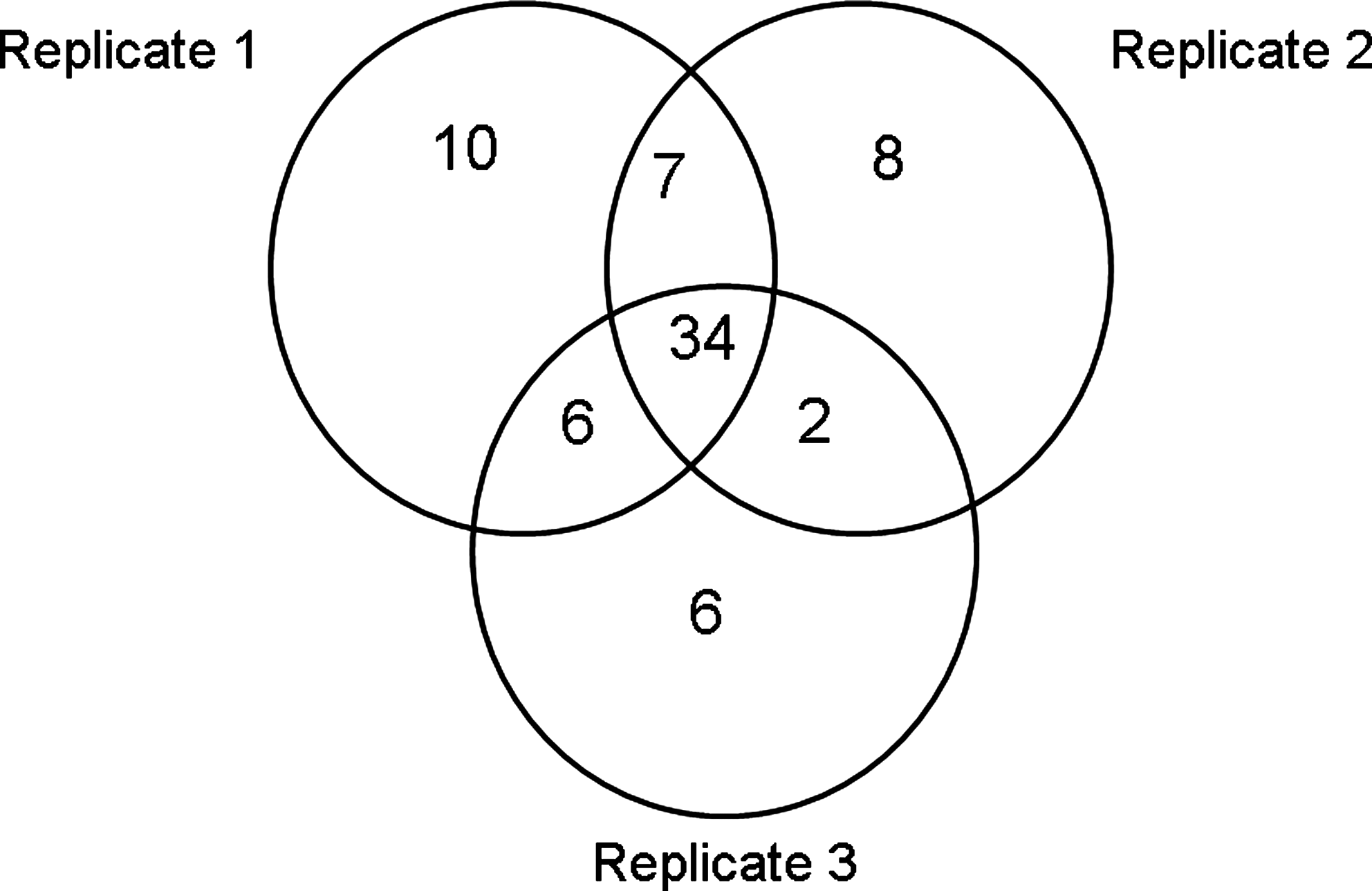
Ven diagram showing the overlap of proteins identified in three replicates.
Note: The numbers highlighted in gray are the mean ratios of differentially expressed proteins.
This protein is included in no change in concentration list as its STDEV numbers are very high especially for coinfected plasma.
For quantitation purpose, we selected only 47 proteins that were identified in two and three replicates with reasonable standard deviations. Table 1 shows the iTRAQ quantitative mean ratios and standard deviations of a list of differentially expressed proteins identified in the current analysis. Ratios for each iTRAQ label were obtained by means of iTRAQ reporter ion 114 (control) as the denominator. The quantitative analysis revealed that 28 proteins were differentially regulated in HIV-1/HCV mono- and coinfected patients plasma compared to healthy controls. A subset of these proteins, apolipoprotein A-II (APOA2), apolipoprotein C-II (APOC2), apolipoprotein E (APOE), Complement C3 (C3), histidine-rich glycoprotein (HRG), and a few immunoglobulins were upregulated in all the HIV-1/HCV mono- and coinfected plasma samples. The quantitative ratios of these proteins and their standard deviations shown in Figure 5a offer a closer examination of their expression in different patient's plasma. APOC3 and POTEE were upregulated in only HIV-1 or HCV mono-infected samples. In HIV-1 mono-infected plasma, only transthyretin (Prealbumin/TTR) was upregulated other than immunoglobulin proteins. Apolipoprotein B-100 (APOB) was found to be upregulated only in HCV mono-infected plasma. However, no protein was upregulated that is unique to coinfected plasma. The list of downregulated proteins comprises complement proteins, fibrinogen proteins, and apolipoproteins. There was no change in the levels of other 19 proteins as listed in Table 1. Representative tandem mass spectrometry data of an upregulated C3 and a downregulated complement C1s subcomponent (C1S) are given in Figure 4.
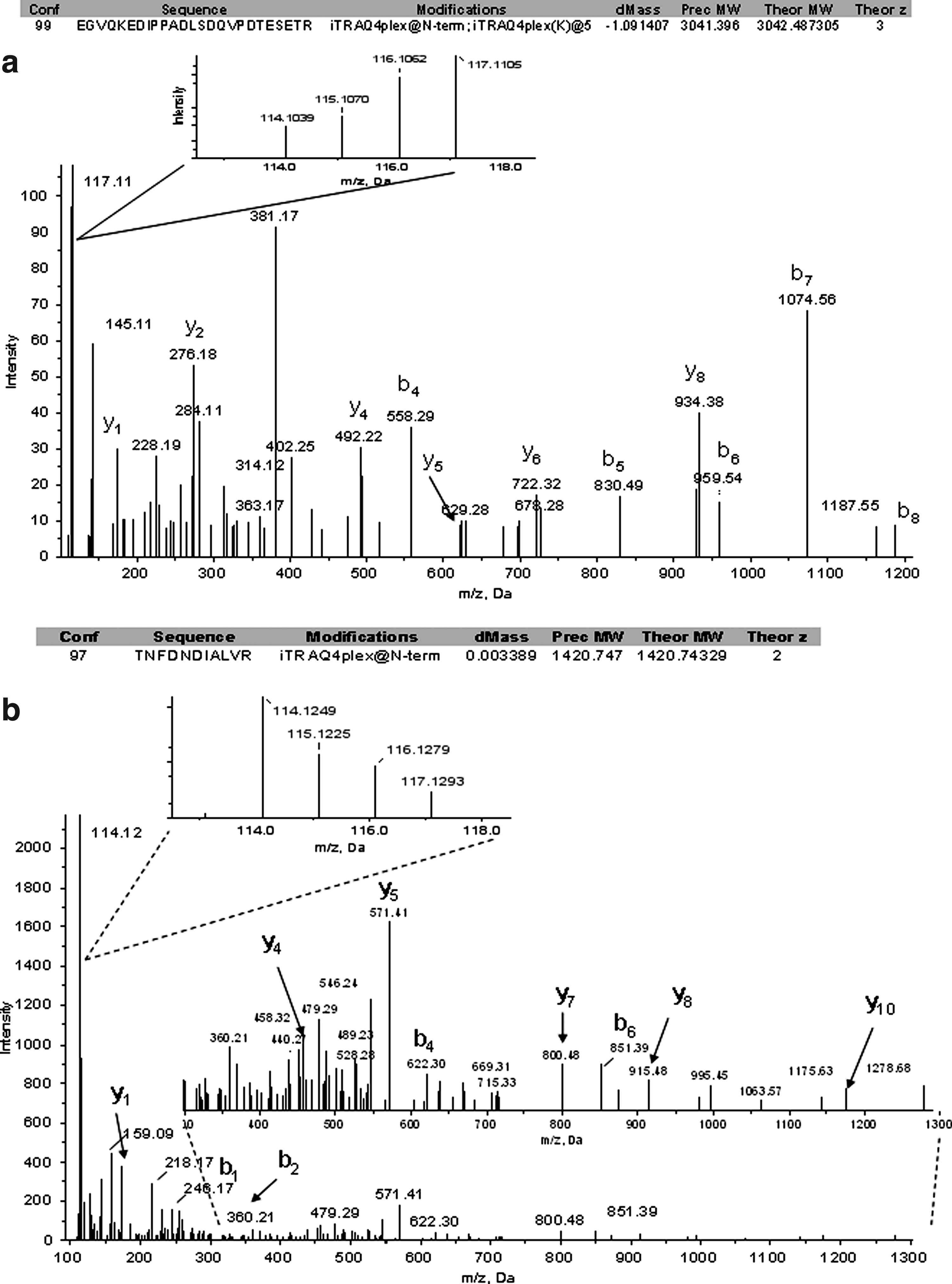
LC-MS/MS spectra of (
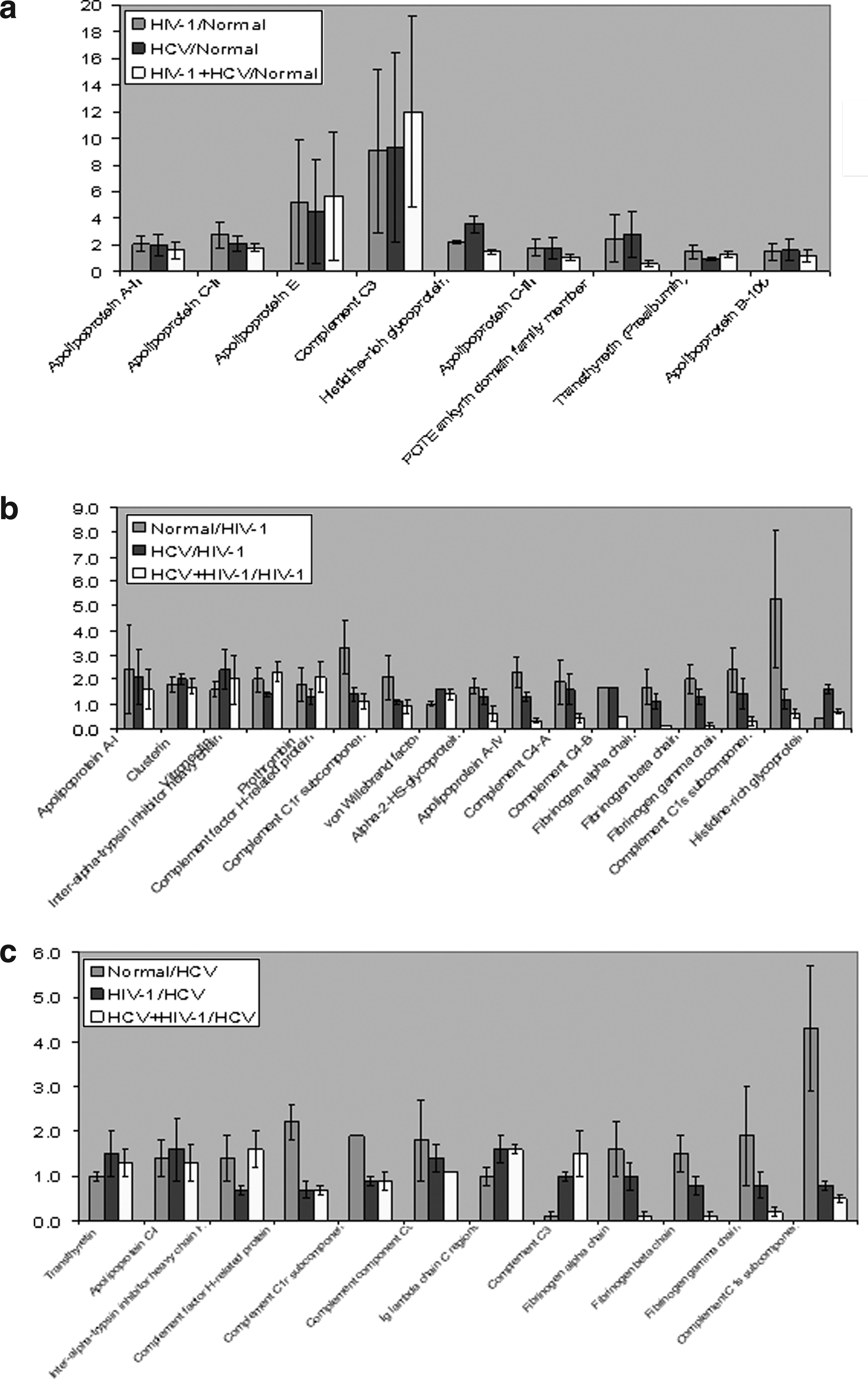
Upregulated proteins and their standard deviations in healthy and plasma proteome analysis to (
Among quantitated proteins in infected patients' plasma, majority of them were upregulated in HCV mono-infected plasma compared to other two infected plasma samples (Table 1). Also, if HCV was considered as a denominator to calculate the quantitative ratios, the differences were less pronounced (Table 1). The data processed with the values from HIV-1 and HCV mono-infected samples as denominators to delineate the differential expression of proteins are shown in Figures 5b and 5c, respectively. It is evident in the data that majority of the proteins are downregulated in infected plasma compared to normal plasma. However, HRG is the only protein that was upregulated in HIV-1 mono-infected plasma compared to HCV mono-infected plasma and normal plasma (Fig. 5b). Similarly, C3 was upregulated in HIV-1 mono-infected and coinfected plasma compared to HCV mono-infected plasma and normal plasma (Fig. 5c).
Discussion
Combining equalizer beads and multiplex iTRAQ proteomics technologies, we identified 70 proteins including a set of differentially regulated proteins in HIV-1/HCV mono- and coinfected plasma samples. In order to ascertain protein identification as well as their differential regulation, the results were reproduced in three biological replicates (Ye et al., 2010). Although we identified several plasma proteins that are reported in the literature, the identification of relatively a low total number of proteins in our analysis may be due to the lack of multidimensional fractionation approach (SCX fractionation followed by reversed-phase) (Ye et al., 2010). Furthermore, we used the equalizer bead protein enrichment strategy to reduce the complexity of the plasma proteome of HIV-1/HCV mono- and coinfected patients (Supplementary Table S1). Equalizer bead (ProteoMiner) technology is an established method to equalize/enrich medium to low abundant proteins in various biological samples (Guerrier et al., 2008; Ye et al., 2010). This technology has been applied successfully for the investigation of low abundant plasma and serum proteome (Ye et al., 2010). ProteoMiner equalization was also proved to be a complementary method for quantitative proteomics analysis (Dwivedi et al., 2010) and it was successfully applied to plasma biomarker discovery (Ernoult et al., 2010) and clinical proteomics (Hartwig et al., 2009). By combining the proteominer enrichment and quantitative proteomics, we identified a number of medium to low abundant proteins in addition to several classical plasma proteins, in the analysis of plasma samples from uninfected and HIV-1/HCV-infected patients. Examples of medium abundant proteins are various complement factors, plasminogen, and ceruloplasmin. Histone H2B type 1-L and myoglobin are tissue leakage proteins and are the examples of low abundant proteins (Keidel et al., 2010) (Supplementary Table 1S). However, no differential regulation was observed for these medium and low abundant proteins in our analysis. We also observed high standard deviations for some of the proteins shown in Table 1, which could be explained by the multiple mechanisms attributed to the ProteoMiner bead methodology including (1) selective binding of proteins to highly diverse but specific peptide binding sites, (2) a general hydrophobic interaction (Keidel et al., 2010) proposed for the action of hexapeptide ligand library beads that has been described to contribute and increase variations up to twofold (Dwivedi et al., 2010) in the relative efficiencies of capture of some proteins by equalizer beads in ProteoMiner kit. In this study, we pooled the samples from each cohort prior to the triplicate analysis. Although pooling normalizes the variations between the individual sample it may also be possible that high level of a protein in a single patient may skew the overall data. Validation studies with individual patient samples may further resolve the true differences between the patient cohorts.
It is interesting to note that significant changes (up- or downregulation of various proteins) were observed in HIV mono-infected patients, who were treatment naive when compared to HCV mono- or coinfected patients, who have failed prior antiviral treatments. In the present study, the more effective proteins in HIV-1/HCV pathogenesis appear to be the apolipoprotein family and the complement system proteins. In addition to these proteins, the remaining up- or downregulated proteins identified in infected patient's plasma samples are further discussed here in relevance to HIV-1/HCV infection.
Upregulated proteins
A set of glycoproteins, APOA2, APOC2, APOE, C3, HRG were upregulated in all the HIV-1/HCV mono- and coinfected patients' plasma samples compared to healthy controls. However, other than Ig-gamma-1 chain C region, the expression of APOE and C3 were higher compared to other upregulated proteins in all the infected plasma samples although the standard deviations are relatively higher in case of these two proteins. Kim et al. (2007) showed that APOA1 present heterologous posttranslational expression and different isoforms of APOA1 were found to be downregulated in HIV-1-infected patients plasma compared to uninfected control plasma. APOC2 was found to be differentially expressed in a proteomic analysis of HCV cirrhosis and HCV-induced HCC and it was downregulated in early HCV-HCC plasma samples (Mas et al., 2009). Using SELDI-TOF proteomics approach, complement C3a which is a fragment of C3, was discovered as a candidate biomarker in patients with chronic hepatitis C and HCV-related HCC (Lee et al., 2006). C3 was also found to be one of the HIV-modulated proteins associated with essential steps during angiogenesis (Rasheed et al., 2009). In a cerebrospinal fluid proteome profiling study of HIV-1-infected patients with cognitive impairment, C3 was found as a marker protein as it was downregulated in HAD compared to ND (Rozek et al., 2007). This protein was also differentially expressed between HIV-1-resistant women and control groups and downregulated by threefold in cervical mucosa of HIV-1-resistant women (Burgener et al., 2008). Gangadharan et al. (2007) showed that both C3 and TTR were downregulated in the sera of hepatitis C patients with liver fibrosis.
The family of apolipoproteins has been shown to play a major role in HIV-1 and HCV. One example is APOE, a protein that binds to receptors on both liver and peripheral cells. Although APOE plays a protective role against HCV progression (Kuhlmann et al., 2010), the protein causes harmful effects for HIV patients, promoting the fusion of the virus to host cell membranes (Kuhlmann et al., 2010). Other apolipoproteins play a more specialized role with either HIV or HCV; one example is APOA1, a protein found in blood plasma, which has a role in lipid metabolism. By interacting with gp41, an HIV peptide, which is essential to fusion between the virus and a host cell, APOA1 has been shown to inhibit HIV pathogenesis (Martin et al., 1992). In comparison, both APOC1 and APOC2 play roles in the HCV infection process. APOC1 has been shown to be essential to the HCV replication cycle (Meunier et al., 2008), whereas levels of APOC2 decrease in the presence of chronic HCV (Koike, 2005). Other proteins in the apolipoprotein family, such as APOC3, play less significant role in HIV and HCV infection. The role of APOC3 is to inhibit lipoprotein lipase and hepatic lipase in order to decrease the general uptake by the hepatic cells. Studies have shown that HIV-1 patients possess increased levels of APOC3 (Bard et al., 2006), whereas other reports have found that APOC3 responds to stimuli in obese patients who have been affected by diseases such as hepatitis C (Harvey et al., 2009). APOB is another protein related to HCV progression. APOB is a protein that is part of the lipoprotein family and one that largely moves cholesterol around the body. A study found that proteins involved in the virus' progression inhibit the secretion of the APOB protein; the significance of the finding is that it provides evidence that HCV alters the lipid structure throughout the body (Domitrovich et al., 2005).
The complement system, a group of proteins with a role in the immune system, naturally is important to both HIV/HCV pathogenesis and treatment. One particular complement protein, C3, is related to both the HIV and HCV infections. The main purpose of C3 is to activate the complement system; this also directly relates to its role as a part of the innate immune response. The HIV virus was found to upregulate the levels of C3 in astrocytes and neurons (Bruder et al., 2004), which is an example of how the infection increases the C3 protein expression in certain immune cells. In a separate study, Heckmann et al. (1999) examined three HCV patients of ages 55, 62, and 71, and concluded that C3 levels were diminished as a result of the HCV infection.
HRG protein is found in human plasma and platelets and is shown to act as an extracellular adaptor protein. Although our studies found that the levels of this protein were slightly up regulated in plasma of HIV-1 mono-infected patients, other data contradicted these assumptions. According to a recent report, the levels of HRG protein dropped in AIDS patients (Toulon, 1998). This detail suggests that this glycoprotein may decrease at later stages during the progression of HIV infection. Another upregulated protein in relation to HIV-1/HCV infection is TTR. This protein is found in the cerebrospinal fluid and has been linked to diseases characterized by neurodegeneration and organ failure. It has been previously suggested that levels of TTR may correlate with the progression to later stages of HIV-1 infection (Baeten et al., 2006).
Downregulated proteins
The complement proteins that were downregulated in the range of 2.5- to fivefold in HIV-1/HCV mono- and coinfected patients plasma are Complement factor H-related protein 1 (CFHR1), complement C1s subcomponent (C1S), complement C1r subcomponent (C1R), and complement C4-A (C4A). Whereas, APOA4 was downregulated in HIV-1 mono-infected plasma (2.5-fold) and coinfected plasma (10-fold). The downregulated proteins that are unique to coinfected plasma are AHSG, fibronectin (FN1), fibrinogen proteins (FGA and FGB), and C4A.
Complement factor H is a glycoprotein found in human plasma with the main goal of directing the complement system toward pathogens rather than host cells. The protein interacts with the HIV glycoprotein 120 during the early stages of infection (Pinter et al., 1995); this finding is important because the protein could possibly prevent HIV binding to host cell membrane through means of the gp120 peptide. Another study on the C-dependent cell infection of HIV-1 proposes the idea that complement proteins C1r and C1s are important in the HIV-1 pathway (Tacnet-Delorme et al., 1999). However, complement proteins have also been studied for their possible influence during HIV/HCV treatments, such as complement component C4. This protein is a widely used biomarker, which is expressed when antibodies bind specific target molecules. C4 levels have been investigated in HCV patients in order to better understand the correlation between the stages of viral infection and C4 activity (Dumestre-Perard et al., 2002).
FN1 is involved in HIV-1 cycle; it is a peptide that binds to cell receptor proteins and plays an important role in wound healing. Data shows that patients with HIV-infected lymphocytes have elevated levels of FN1 in the cells themselves; this correlation is supported by the idea that FN1 plays a critical role in stabilizing the HIV infection process (Pal and Schnapp, 2004). Although the quantity of blood cell proteins has been up regulated, the amount of FN1 will be downregulated in plasma to compensate for the increased cellular levels. FN1 also played a role in differentiating HCV patients with liver fibrosis with those who did not have fibrosis (Attallah et al., 2007). Serum tests found a 75% efficiency rate in using FN1 to determine the presence of fibrosis in these HCV patients (Attallah et al., 2007). AHSG was also proved to be an important protein that is downregulated in the presence of HCV. AHSG is a serum protein that is involved in brain development and formation of bone tissues. A study on hepatitis C patients demonstrated downregulation of AHSG expression in serum samples and concluded that this protein may offer a new biomarker in determining fibrosis progression in HCV patients (Cheung et al., 2009).
Ingenuity pathway analysis
In order to delineate the potential role of the upregulated proteins in HIV-1/HCV mono- and coinfection, we performed ingenuity pathway analysis by grouping the proteins that shared similar functions. A network was generated by uploading the upregulated protein dataset and mapping to corresponding gene objects in IKB as shown in Figure 6. A total of 35 molecules per network and 25 networks per analysis were selected in this pathway analysis. The resulted network exhibited a score of 23 containing 8 focus genes and 28 nonfocus genes. HRG and APOE are directly interacting with heparin which is an anticoagulant and plays a vital role in cell with regard to binding, proliferation, adhesion, chemotaxis, activation, growth, G1/S phase transition, migration, replication, and maturation of cells. APOB and APOA2 are directly connected to lipid proteins, phospholipid transfer protein (PLTP), and glycosylphosphatidylinositol specific phospholipase D1 (GPLD1). All these proteins in this network, although connected indirectly, interacting with Akt (serine/threonine protein kinase) which is involved in apoptosis, proliferation, and cell death and implicated in a variety of cancer indications. Similarly, C3 is indirectly interacting with IL1 and IL6 that are implicated in several inflammatory diseases. The results of IPA revealed that all of the upregulated proteins predominately associated directly with lipid metabolism, immunological disease, metabolic diseases, and cell death signifies the role of these proteins in HIV and HCV infection. The complete association of the functional pathways of these proteins is shown in Supplementary Table S4.
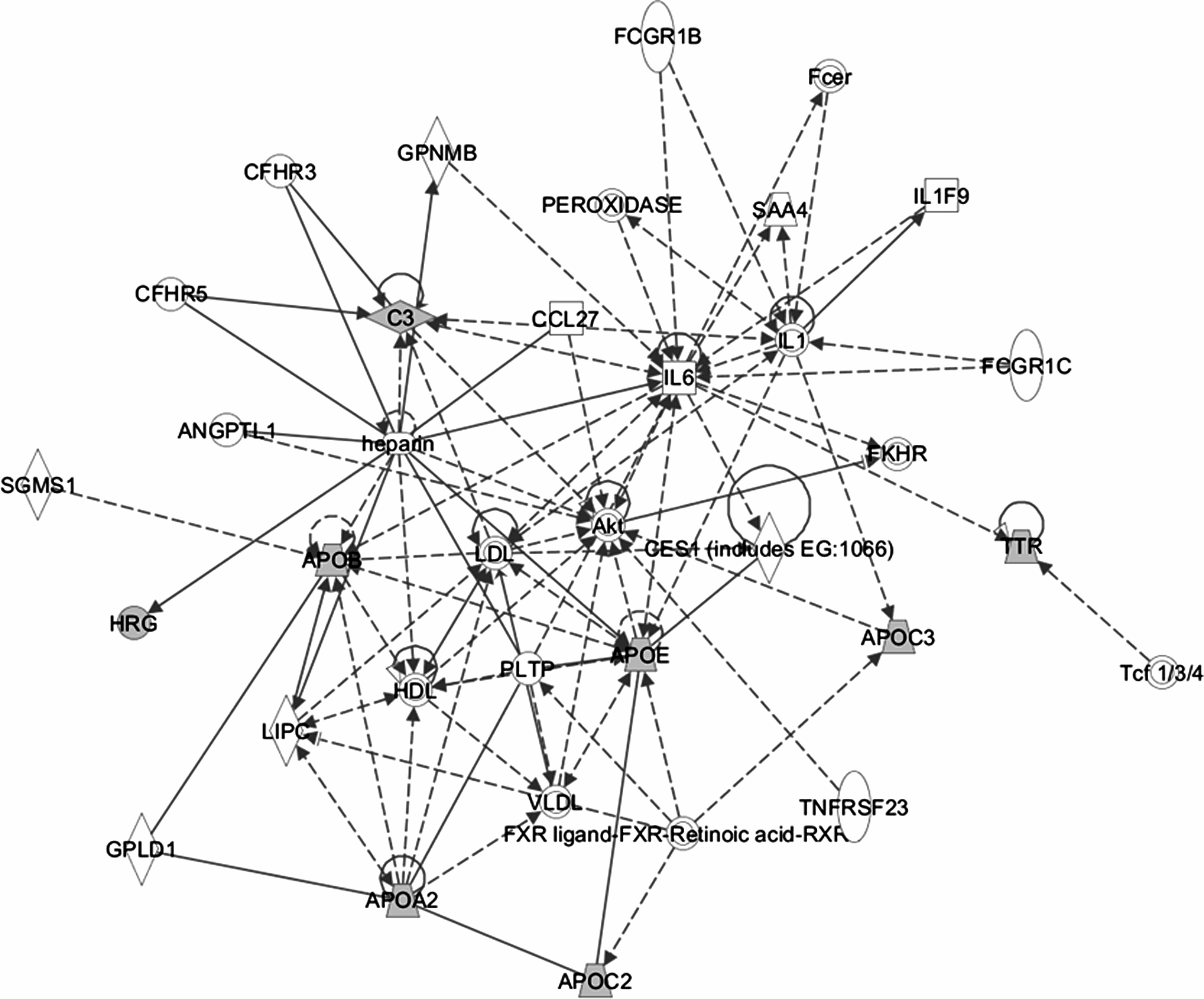
Analysis of upregulated proteins using a manually curated structured network tool (ingenuity pathway analysis). The shapes are indicative of the protein family. Proteins highlighted in grey shading were upregulated and identified in the LC-MS/MS analysis. Proteins highlighted in clear were not identified in the LC-MS/MS analysis but were linked to the identified protein by examination of the ingenuity pathway analysis curated database describing protein:protein interactions. Lines connecting the molecules indicate molecular relationships. There are two line styles: dashed lines indicate indirect interactions and solid lines indicate direct interactions.
The upregulated dataset was also analyzed in canonical pathways of IPA library in order to investigate the relationship between the upregulated proteins and various biological pathways. The canonical pathways that are identified from the IPA library based on their significance to the dataset are shown in Figure 7. Two major canonical biological pathways were identified for the upregulated protein dataset. The first pathway is Farnesoid X receptor (FXR)/retinoid X receptor (RXR) activation (p-value=8.57×108/threshold=0.039) in which all apolipoprotein genes are involved. FXR is a key player in the control of numerous metabolic pathways and along with RXR, FXR plays a crucial role in linking bile acid regulation with lipoprotein, lipid, and glucose metabolism. The other significant canonical pathway of four upregulated HRG, APOA2, C3, and TTR proteins is an acute phase response signaling pathway (p-value=7.85×107/threshold=0.02). The acute phase response is a rapid inflammatory response that provides protection against microorganisms using innate defense mechanisms. During this response in an infected individual, an increase in inflammatory factors and a change in concentration of several plasma proteins occur because of an altered hepatic metabolism. This ultimately leads to a change in the concentration levels of acute phase plasma proteins. Thus, based on the results obtained from ingenuity network and canonical pathway analysis, it is likely that all the upregulated proteins in infected plasma samples are involved in acute phase signaling pathway and are strongly implicated in the HIV-1 and HCV infections. Consequently, they may function in opsonization and trapping of microorganisms, complement activation, neutralizing enzymes, and modulating the immune response.

Predominant canonical pathways of upregulated proteins in infected plasma as identified by ingenuity pathway analysis (IPA).
Although in this study, we observed significant changes between the groups, one of the limitations of the study is lack of validation of the findings using large sample set. Future experiments will focus on validation of the key targets with individual patient samples. However, the significance of the findings are well supported by the triplicate analysis and the reproducibility of the data on the differential expression of proteins (in particular, the upregulated proteins in all the infected patients plasma samples). The results of the Ingenuity network and pathway analysis further confirm the vital role of these proteins in hepatic lipid metabolism, inflammation, and acute-phase response signaling pathways.
Conclusions
We have investigated for the first time, the plasma proteome profile of HIV-1, HCV mono-, and coinfected patients, by employing ProteoMiner bead protein equalization, iTRAQ technolongy, and ingenuity pathway analysis. We found that majority of the identified and differentially regulated proteins belong to apolipoprotein family and complement system. All the differentially regulated proteins identified in this study have been shown to be strongly implicated previously in HIV-1 and HCV infections. Furthermore, the triplicate experiments conducted in this study resulted in a set of plasma proteins (APOA2, APOC2, APOE, C3, HRG) that are consistently upregulated in all the HIV-1/HCV mono- and coinfected plasma samples. We also identified a few downregulated proteins (AHSG, FN1, FGA/FGB, C1r, and C1s) that are unique to only coinfected patient samples. Further analysis of upregulated proteins by IPA software revealed that they play a vital role in many important functional pathways including lipid metabolism and inflammation. The results of canonical pathway analysis also supported the function of these upregulated proteins in hepatic lipid metabolism and their critical role in acute-phase response signaling pathway. These results contribute to the understanding of proteome changes upon HIV-1/HCV mono- and coinfection and provide valuable information for further research to investigate them as possible plasma biomarkers.
Supporting Information
Supplementary information is available on-line at http://www.liebertonline.com/omi
Footnotes
Acknowledgments
These studies were supported by the corporate funding provided to the Immunotope, Inc. and also by the departmental funds provided to Dr. Pooja Jain. We thank Ingenuity systems for providing us a copy of the trial version of the IPA software.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.