
Abstract
Abstract
There is a lack of knowledge on the tissue-specific expression of miRNAs in response to dehydration stress in Brachypodium (Brachypodium distachyon (L.) Beauv), a model for temperate grass species. In this study, miRNA expression patterns of drought-tolerant Brachypodium were investigated using the miRNA microarray platform. A total of 205 miRNAs in control and 438 miRNAs in both drought-treated leaf and root tissues were expressed. Seven of the detected Brachypodium miRNAs were dehydration stress responsive. Expression levels of known drought-responsive miRNAs, miR896, and miR1867 were quantified by qRT-PCR in Brachypodium upon 4 h and 8 h dehydration stress applications. This was performed to compare drought responsiveness of miRNAs in closely related species. Target transcripts of selected drought responsive miRNAs, miR170, miR1850, miR896, miR406, miR528, miR390, were computationally predicted. Target transcript of miR896 was verified by retrieving a cleaved miR896 transcript from drought stress-treated leaf samples using a modified 5′ RLM-RACE. Brachypodium dehydration responsive miRNA were also detected in barley and wild emmer wheat. Hence, the outcomes highlighted the conserved features of miRNA upon dehydration stress in Triticeae.
Introduction
Micro-RNAs (miRNAs) are a class of small, noncoding RNAs that endogenously regulate gene expression via posttranscriptional or posttranslational mechanisms (Bartel, 2004; Kantar et al., 2011). Following the cloning of first plant miRNA in Arabidopsis thaliana (Llave et al., 2002), several other miRNAs from different plants have been discovered in vitro by direct cloning and in silico by computer-based methods (Kantar et al., 2010, 2011; Unver and Budak, 2009; Unver et al., 2009, 2010). The majority of the miRNAs identified to date have been isolated from Arabidopsis thaliana and Populus trichocarpa in dicots and from Orzya sativa and Sorghum bicholor in monocots. Several of these miRNAs have been shown to be well-conserved across species (Unver and Budak, 2009; Kantar et al., 2010). In plants, long stem–loop structure precursors (pre-miRNA) are processed into mature miRNAs by the action of an RNase III endonuclease family member, Dicer-like-1 nuclease. The mature miRNA is then incorporated into RNA-induced silencing complex (RISC) for subsequent target-specific degradation or inhibition of translation of transcripts (Unver et al., 2009). Strong conservation of pre-miRNA sequences among plants has enabled prediction of miRNAs in silico, which in turn enabled a faster, cost-effective and accurate way of identifying novel miRNAs, compared to direct cloning strategies (Unver and Budak, 2009).
Plant miRNAs are known to play a role in an array of biological processes, involving protein degradation and signal transduction (Archard et al., 2004; Guo et al., 2005; Zhang et al., 2006a), as well as developmental courses (Bao et al., 2004; Kim et al., 2005; Laufs et al., 2004; Mallory et al., 2005). Recently, plants miRNAs also found to be involved in stress responses against biotic and abiotic factors (Chen, 2005; Jones-Rhoades and Bartel, 2004; Kantar et al., 2010, 2011; Kasschau et al., 2003; Phillips et al., 2007; Unver et al., 2009). The first miRNA reported to be related to stress conditions was miR398, which is transcriptionally downregulated in response to oxidative stress. The targets of miR398 in Arabidopsis thaliana were revealed to be the transcripts of two Cu/Zn superoxide dismutases that are closely related (Kantar et al., 2011; Sunkar et al., 2006). Recently, a number of miRNAs have also been shown to govern responses to drought stress. For instance, miR393, miR319, and miR397 were found to be upregulated (Sunkar and Zhu, 2004), as well as miR167, miR168, miR171, miR396, and miR408, all of which were identified in array-based systems, in response to drought in Arabidopsis (Liu et al., 2008). Although several miRNAs have been implicated to be up- or downregulated in drought stress, the first and only plant miRNA that is fully characterized with its target is miR169 in rice as reported by Zhao et al. (2007). Targets of miR169 family in rice include several members of Nuclear Factor Y (NFY) gene family, particularly NFYA5 in Arabidopsis. NFAY5 is known to be upregulated in response to drought, which is partially dependent upon the downregulation of miR169 family members. Accordingly, miR169a and miR169c are dramatically downregulated in drought stress. In addition, both nfa5 knockout plants and miR169 overexpressing plants are shown to be drought sensitive. NFAY5 expression in stomatal cells is speculated to regulate stomatal aperture, which may help the plant to cope with the drought stress (Li et al., 2008). Despite the advances in miRNA prediction and identification, however, research on exact molecular functions of identified miRNAs is relatively in its infancy.
In this study, we aimed to identify the effect of dehydration stress on the expression and tissue specificity in Brachypodium miRNAs using a microarray platform composed of 853 miRNAs from 21 plant species and to find the possible targets of these miRNAs that may be involved in stress responses.
Materials and Methods
Plant materials and stress treatment
Seeds of BdTR2A Brachypodium inbred line (Filiz et al., 2009b) were surface sterilized in 4% hypochlorite for 5 min and pregerminated in Petri dishes with 5 mL milipore water at 25°C±1 at dark. Seedlings were transferred to continuously aerated Hoagland's solution. Growth condition was under defined parameters: 15 h photoperiod, temperature 23/21°C, relative humidity 60/70%, and photon flux density of 600–700 μmol m−2 s−1 (Kantar et al., 2011). Stress treatment was performed as outlined by Ergen et al. (2009). Briefly, 3 weeks after the transfer to Hoagland's solution, to apply dehydration stress, plants were removed from the tanks and placed on paper towels for 4 and 8 h under the same growth conditions, whereas control plants were left in the Hoagland's solution for the same period of time. Control and 4- and 8-h dehydration stress-treated root and leaf samples were collected (Ergen et al., 2009; Kantar et al., 2011) from four plants under each treatment that were pooled, frozen in liquid nitrogen, and stored at −80°C.
Total RNA isolation
Total RNA was isolated from leaves and roots of 4- and 8-h stress and control samples (Kantar et al., 2010, 2011; Unver and Budak, 2009). Total RNA was isolated using Trizol reagent (Invitrogen, Carlsbad, CA) according to manufacturer's instructions and the quality and quantity of isolated RNAs were measured using a Nanodrop ND-100 spectrophotometer (Nanodrop Technologies, Wilmington, DE). DNase treatment, the PCR reaction, and miRNA quantification were performed as outlined by Kantar et al. (2010).
MiRNA microarray
The miRNA chip (in situ synthesized by LC Sciences, Houston, TX: http://www.lcsciences.com/mirna.html) comprised 853 miRNA probes corresponding to miRNA transcripts published in Sanger miRBase release 12.0 (http://www.mirbase.org/) with a number of control probes (Kantar et al., 2011). These miRNAs were from Arabidopsis thaliana (158), Brassica napus (24), Glycine max (soybean) (63), Medicago truncatula (17), Gossypium hirsutum (Mexican cotton) (7), Populus trichocarpa (cottonwood) (114), Lycopersicon esculentum (tomato) (23), Vitis vinifera (grape) (79), Oryza sativa (rice) (216), Sorghum bicolor (39), Saccharum officinarum (sugarcane) (10), Triticum aestivum (wheat) (31), Zea mays (maize) (43), Pinus taeda (Loblolly pine) (34), Physcomitrella patens (187), S. moellendorffii (60), and 1 each from Brassica oleracea (wild cabbage), Brassica rapa, Carica papaya, Gossypium herbaceum (Levant cotton) and Gossypium raimondii.
A total of 5 μg of total RNA from each sample (either leaf or root; either control, 4-h stressed, or 8-h stressed) was used for this study (Cebeci and Budak, 2009; Ergen et al., 2009; Kantar et al., 2011).
MiRNA microarray data analysis
Data was collected via Axon Gene Pix 4000B Microarray Scanner and analyzed using ArrayProTM image analysis software (Media Cybernetics, Silver Spring, MD). Two conditions were to be met to accept a miRNA signal as detectable: signal intensity higher than three times background standard deviation, and spot CV<0.5: (CV=signal standard deviation/signal intensity). Signals from four technical replicates each of RNA derived from stressed and control plants were compared using paired, two-tailed Student's t-test; only signals with p values<0.05 and greaterh than threefold increased or decreased differential expression were considered as significant (Cebeci and Budak, 2009).
Stem–loop reverse transcription
Stem–loop reverse transcription and RT-PCR were performed for miR896 and miR1867, which were previously shown to be present and responsive to dehydration in Triticeae (Kantar et al., 2010, 2011; Unver et al., 2009). Quantification of these miRNAs in Brachypodium was performed using the same RNA samples used in the microarray study. Stem–loop RT primers for miR896 and miR1867 were designed as described by Varkonyi-Gasic et al. (2007) (Supplementary Table 1). miRNA specific RT reactions were performed using Superscript III First-Strand Synthesis System for RT-PCR from Invitrogen, Cat no: 18080-51. The miRNA stem–loop reverse transcription was performed as outlined by Kantar et al. (2011).
miRNA quantification using qRT-PCR
Expression level differences of miR896 and miR1867 in leaf and root tissues upon 4 and 8 h of dehydration were quantified via qRT-PCR with Brilliant II SYBR Green QPCR Master Mix from Stratagene, Cat no: 600028 (La Jolla, CA) on an Icycler Multicolor Real-Time PCR Detection Systems (Bio-Rad Laboratories, Hercules, CA) (Ergen and Budak, 2009; Ergen et al., 2009; Kantar et al., 2010). Briefly, 3 μL RT stem–loop cDNA products was used in qRT-PCR reactions and PCR reactions were performed as 10 μL 2×Master mix, 0.6 μL forward (300 nM), 0.6 μL reverse (300 nM) primers, 0.3 μL (30 nM) reference dye, and 5.5 μL nuclease free water. The forward primers were designed for each individual miRNA, whereas the universal reverse primer, described by Varkonyi-Gasic et al. (2007), was used as a reverse primer. For normalization, 18s rRNA (GenBank ID: AF147501, forward primer: GTGACGGGTGACGGAGAATT/reverse primer: GACACTAATGCGCCCGGTAT) was used (Kantar et al., 2010). In qRT-PCR, the samples were heated to 95°C for 10 min, followed by 40 cycles of 95°C for 30 s, 58°C for 1 min, and 72°C for 30 s, followed by 72°C for 10 min. The melting curves were generated using the following program: PCR products were denatured at 95°C and cooled to 65°C. The fluorescence signals were collected continuously from 65 to 95°C as the temperature increased at 0.2°C per second. All reactions were repeated for three times and noRNA and noRT primer controls were used for each sample (Kantar et al., 2010).
In silico target mRNA identification
Sequences of differentially expressed miRNA probes from the Brachypodium microarray and other Triticeae miRNA sequences previously reported to be drought responsive were used to interrogate sequences for target sites on the psRNAtarget web server (http://biocomp5.noble.org/psRNATarget/). The target transcripts containing complementary sequences of miRNAs were determined with previously established criteria (Kantar et al., 2010, 2011; Unver and Budak, 2009; Yin et al., 2008; Zhang et al., 2006a, 2006b). miRNAs targets were exposed to homology search against the protein-coding (mRNA) database among all plant species.
Target mRNA validation with qRT-PCR and 5′ RLM-RACE
As noted above, target ESTs of several miRNAs were predicted using psRNAtarget, a plant miRNA target finder software (http://biocomp5.noble.org/psRNATarget/) including the computationally proposed target EST (GenBank ID: TC5463) of miR896. To validate this target, qRT-PCR and RACE reactions were applied.
5′ RLM-RACE was performed for the confirmation of miRNA896 target transcript as previously performed (Arenas-Huertero et al., 2009; Wei et al., 2009). cDNA was produced using the RLM-RACE kit (Ambion, Austin, TX; Cat no: 600028) following the manufacturer's directions, except that the alkaline phosphatase and acid pyrophosphatase steps were omitted. PCR amplification of cDNA fragments was achieved using primers designed based on the sequence information. PCR fragment was cloned and independent clones were sequenced to identify the 5′-end of the amplified fragments. Specific primers for RLM-RACE are included in Supplementary Table 1.
Results
miRNAs identified in leaf and root tissue by miRNA microarray analysis
Dehydration stress was used as the stress condition to profile the majority of dehydration responsive miRNA genes (Ergen et al., 2009). The microarray was probed with pooled Brachypodium RNAs isolated from four seedlings under each condition. In addition to 56 hybridization controls, for each miRNA probe four technical replicas were tested for the control condition and four replicas for each stress condition (Kantar et al., 2011).
The cumulative number of miRNAs listed in miRBase (http://www.mirbase.org/, release 16) along with the ones found in a recent study performed by our group is 35 Brachypodium miRNAs and experimental validation is present for seven of them. (Unver and Budak, 2009). Additionally, deep sequencing of Brachypodium small RNAs by two groups has revealed several miRNAs either conserved among plants or that are Brachypodium specific. One of the studies revealed 94 conserved and 12 novel miRNAs and the other group has shown the presence of 27 conserved and 129 novel miRNAs (Wei et al., 2009; Zhang et al., 2009). Our findings in this study are also parallel with the findings cited above.
Microarray analysis in this study revealed hybridization between 443 probes and Brachypodium miRNAs (of 853 total), which are also shown to hybridize to Triticum dicoccoides miRNAs in our previous study (Kantar et al., 2011), pointing out to a high degree of conservation between Brachypodium and T. dicoccoides. In general, a greater number of miRNAs were observed to be induced under drought stress conditions compared to control conditions, which were 438 and 205 miRNAs, which have been found to be upregulated in this study, under drought and control conditions, respectively.
Stress-responsive miRNAs identified by miRNA microarray analysis
Differential expression of miRNAs in response to drought stress were analyzed for all miRNAs detected in both control and drought conditions and more than threefold up- or downregulation was demonstrated at a p value<0.05. Those results are summarized in Figure 1.

Venn diagram indicating common and unique differentially expressed miRNAs in root and leaf tissues under two 4-h and 8-h drought treatments.
Among the dehydration-responsive Brachypodium miRNAs exhibited the most significant differential expression profiles in this microarray study; four are previously reported: miR1850, miR528, miR390, and miR170 (Unver and Budak, 2009; Wei et al., 2009; Zhang et al., 2009). Our study uncovers three previously unreported Brachypodium miRNAs, namely, miR1450, miR406, and miR1881 (Fig. 1). Of the seven miRNAs that were found to be differentially regulated, five of them were observed to be upregulated in response to drought. These miRNAs are miR1850, miR390, miR170, miR1450, and miR1881 (Fig. 1).
In leaf tissue, miR1850 was found to be upregulated under both drought stress conditions. Moreover in leaf tissue, a probe for miR528 showed downregulation only after 8 h of stress. Roots that had been drought-stressed for 4 h and 8 h showed the most changes in miRNA levels: miR1450 was induced and miR406 was suppressed only upon 4-h stress; miR1881 and miR170 showed upregulation only upon 8-h stress, and miR390 was responsive to stress in both stress conditions in roots. miR390 was also induced upon 8-h stress in leaf tissue. The distributions of differentially expressed miRNAs by tissue and drought condition are displayed in Figure 1 by Venn diagram.
qRT-PCR validation and measurement of miRNAs in response to dehydration stress
We quantified differential miRNA expression under drought stress with qRT-PCR using the SYBR Green I assay (Ergen et al., 2007; Kantar et al., 2010, 2011; Unver and Budak, 2009). Two additional miRNAs that were shown to be differentially expressed in response to drought in T. dicoccoides samples in a previous microarray study (Kantar et al., 2011) were validated by qRT-PCR quantification. This was performed to check the conservation of these miRNAs through Triticeae and observe the similar or different trends in the drought induced or suppressed expression of miRNAs in closely related species. qRT-PCR was performed using leaf and root tissues of 4-h and 8-h dehydrated samples (Fig. 3A). In Brachypodium, miRNA896 expression was shown to be upregulated in 4- and 8-h stress treatments in root tissue; miRNA1867 was shown to be upregulated in 8-h stressed leaf and 4-h stressed root. The trend in expression level change of miRNA1867 in leaf tissue upon 8-h dehydration was similar to that observed in T. dicoccoides. In Triticum samples, differential expressions of these two miRNAs were as follows: miR1867 was upregulated in both stress treatments in leaf; miR896 was transiently downregulated at 4-h stress, but reinduced after 8 h of dehydration in leaf (Kantar et al., 2011). Hence, this result indicates that drought stress miRNAs are conserved in T. dicoccoides and Brachypodium.
Prediction of Brachypodium miRNA targets using bioinformatics tools
miRNAs are shown to regulate gene expression post-transcriptionally by binding to the complementary sites in their target mRNAs. Binding of miRNA leads to either the cleavage or inhibition of translation of the corresponding mRNA (Bartel, 2004). Such targeting of mRNAs by miRNAs is dependent on the complementarity and more than four mismatches are not allowed (Bartel, 2004; Rhoades et al., 2002; Shwab et al., 2005).
We predicted the potential mRNA targets of potential or verified Brachypodium miRNAs using psRNATarget Web server (http://biocomp5.noble.org/psRNATarget/). Target sequences were predicted using probe sequences of miRNAs shown to be differentially expressed in the dehydration stressed Brachypodium and T. dicoccoides microarrays (Kantar et al., 2010). Criteria were applied as previously mentioned and potential target ESTs were identified in Brachypodium for the following miRNA queries: miR170, miR406, miR1850, miR528, miR390 from the Brachypodium microarray, and miR896 from the T.dicoccoides microarray. The targets for all miRNAs are listed in Table 1. Brachypodium miRNA target genes are found to be diverse in sequence and function, and most of them are assigned to functional proteins in plant metabolism, structure maintenance, development, cell–cell signaling, cell cycle and senescence (Table 1).
Some of the targets predicted for miR1850 and miR528 are proteins involved in ubiquitination for proteasomal degradation. In silico targets of miR896, miR406, miR528, and miR390 include proteins related to several abiotic and biotic environmental stress response including cyanogenesis and UV-B response. These results are similar to previous findings (Bonnet et al., 2004; Rhoades et al., 2002; Zhang et al., 2006a).
Target mRNA validation with qRT-PCR and 5′ RLM-RACE
A computationally predicted target EST (GenBank ID: TC5463) of miRNA896 was monitored with qRT-PCR. Expression level of the target gene of miR896 was quantified relative to controls in 4-h and 8-h dehydration stressed Brachypodium root tissue. miR896 target was found to be suppressed in 4-h and 8-h stress-applied root samples. So an inverse correlation between levels of miRNA and its corresponding target mRNA was observed supporting miRNA896 action upon drought stress. Figure 3B demonstrates a representative expression pattern of miR896 target (GenBank ID: TC5463) in leaf and root tissues upon 8-h dehydration stress quantified by qRT-PCR.
Experimental validation of target mRNAs in plants takes advantage of the preferred mode of action of plant miRNAs. In this work a modified 5′ RLM RACE experiment was performed with total RNA extracted from root tissues. The predicted target EST of miR896 was verified with 5′ RLM-RACE (Fig. 2).

RLM-RACE of miR896. (
Discussion
Plant genes involved in responses to abiotic stresses such as drought may be regulated at the posttranscriptional level, and miRNAs have a major role in posttranscriptional regulation (Kantar et al., 2010, 2011). miRNAs are involved in the regulation of various cellular events (reviewed by Jones-Rhoades et al., 2006). Several elements related to miRNA metabolism have been shown to be involved in abscisic acid (ABA) signaling (Nishimura et al., 2005; Xiong et al., 2001), which is an important component of the plant response to abiotic stresses (Ergen and Budak, 2009). “Omics” technology have been used to identify systematically drought-responsive miRNAs in Hordeum vulgare (Kantar et al., 2010) and Triticum dicoccoides (Kantar et al., 2011).
Here we performed the first systematic identification of miRNAs in Brachypodium, the wild temperate grass species, under normal growth and dehydration stress conditions. In our previous studies in wild emmer wheat, dehydration stress was demonstrated to bring about detectable changes in the expression patterns of a higher proportion of drought-responsive genes than other type of drought treatments (Ergen and Budak, 2009; Ergen et al., 2009). However, it is highly likely that other genes are also involved in long-term drought acclimation that can be detected using a soil-drying type experiment (Kantar et al., 2010).
In a previous study of our group, Brachypodium miRNAs were computationally identified and some of them were validated by qRT-PCR quantification (Unver and Budak, 2009). In this study, we performed a large-scale study to detect new Brachypodium miRNAs and analyze their dehydration stress responsiveness to better understand the role of plant miRNAs in stress adaptation. Currently, there is a limited number of Brachypodium miRNAs and targets either computationally identified or experimentally confirmed. Using a cross-species miRNA microarray enabled us to identify miRNAs expressed in a different species. The disadvantage of this approach is that the small RNA binding to each probe may differ by one or more nucleotides from the miRNA it was designed against, so only the family, not the exact sequence of the putative miRNA, can be deduced. Thomson et al. (2004) also noted that miRNAs hybridizing to more than one probe may well also hybridize to multiple target sequences in vivo.
By the microarray approach, several previously unidentified Brachypodium microRNAs were revealed. Among the seven stress responsive miRNAs that exhibited the most significant expression differences in response to dehydration stress treatment, four were previously identified, which are miR1850, miR390, miR170, and miR528 (Unver and Budak, 2009; Wei et al., 2009; Zhang et al., 2009). Newly identified Brachypodium miRNAs include miR1450, miR406, and miR1881 (Fig. 1). We have also detected that most of the Brachypodium miRNAs are homologues to Oryza sativa (osa-miRNA), Arabidopsis thaliana (ath-miRNA), Populus trichocarpa (ptc-miRNA), and Physcomitrella patens (ppt-miRNA). Because these are not close relatives of Brachypodium, we can conclude that this may result from extensive miRNA sequence conservation in plants. Additionally, microarray approach can result in a high number of false positives. To reduce the risk of selecting false positives, in the analysis of stress related differential expression of miRNAs, we only examined drought-sensitive miRNAs that were detected in both stressed and control plants. However, for the newly detected drought-specific miRNAs, which are particularly interesting as potential Brachypodium miRNAs, further experimental verification by an independent technique is required.
From the microarray data, it was also notable that although both control and stressed leaves contained a larger variety of miRNAs than control roots, the majority of the drought stress-regulated miRNAs were specific to root tissue. From the miRNAs that show differential regulation, four were root-specific and two were leaf-specific. This is understandable as the root tissue would be the first to be affected by drought conditions. We have shown that miR1850 and miR528 are responsive to drought specifically in Brachypodium leaves, whereas miR1450, miR406, miR1881, and miR170 are drought responsive in root tissue (Fig. 1). It would be interesting in further studies to find out whether a larger number of miRNAs are up- or downregulated in leaves after prolonged drought.
Over half of the probes (443 out of 853) on the microarray hybridized to miRNAs present in T. dicoccoides, as expected from the high conservation of miRNAs between plant species. Furthermore, more than twice as many miRNAs were detected in plants undergoing drought stress than in controls (438 compared to 205).This suggests that miRNAs are generally upregulated and have an important role in abiotic stress responses. By acting at the posttranscriptional level, they enable the plant rapidly to shut down nonessential gene expression without waiting for new proteins to be produced. The majority (five of seven) of differentially regulated miRNAs were upregulated in drought. These included miR1850, miR 390, miR170, miR1450, and miR1881 (Fig. 1).
In this study, qRT-PCR experiments on miR896 and miR1867, which were previously shown to be drought responsive in other species belonging to Triticeae, were performed (Kantar et al., 2010, 2011). The presence of miR1867 and miR896 in Brachypodium was verified, emphasizing the conservation of these miRNAs in closely related species. Another aim of this study was to track the similarities and differences in the mode of differential miRNA expression in stress conditions between different species. miR1867 was found to be upregulated in 8-h stressed leaf both previously in T. dicoccoides and in this study in Brachypodium (Fig. 3) (Kantar et al., 2011). Hence, the trend of differential regulation is similar for both species only for miR1867 in 8-h stressed leaf in comparison to control. However, a general similarity in the trend of expression level changes in response to drought could not be observed among different Triticeae species. This finding highlights the possibility that the same miRNA may be acting on different targets in different species under drought conditions. Target predictions for the miRNAs covered in this study, suggest the regulation of an array of processes, including basic developmental and metabolic processes, by these miRNAs. Presence of a large variety of potential miRNA targets offers an explanation for the different trends in miRNA expression upon drought in different tissues and species (Table 1).
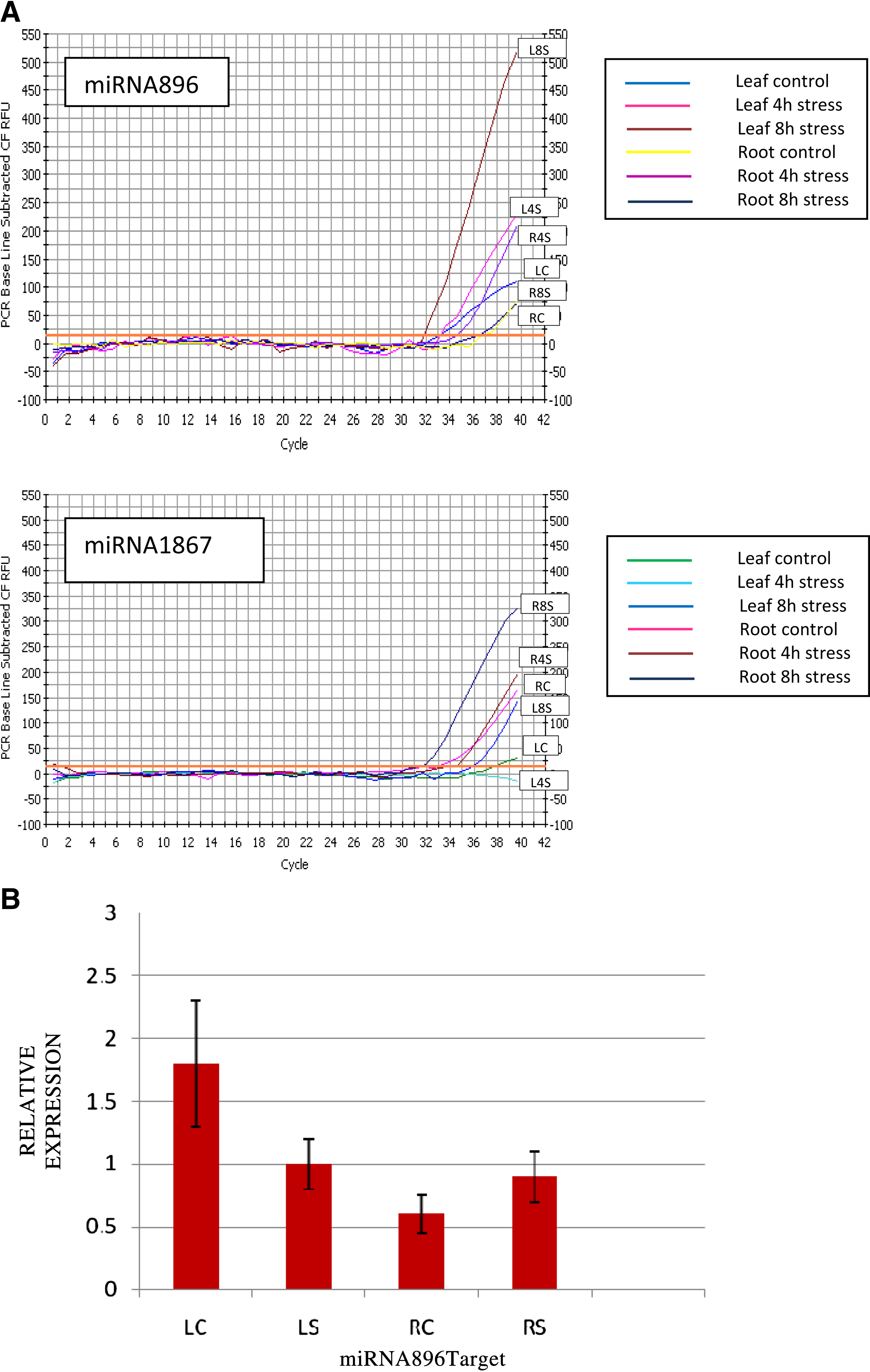
Quantitative real-time PCR results. (
Brachypodium miR896 was shown to be induced in 4-h and 8-h stressed roots in this study (Fig. 3). However, Tdi-miR896 was previously shown to be suppressed in 4-h stressed leaf and reinduced after 8 h of stress (Kantar et al., 2011). To relate miRNA action on the target, a putative target EST of Brachypodium miR896 was computationally predicted and quantified (Table 1). Target expression decreased in 4- and 8-h stressed root. An inverse correlation between levels of miRNA and its corresponding target mRNA was observed. The putative target EST of miR896 was verified by 5′ RLM-RACE (Fig. 2).
Some of the stress responsive miRNAs we have detected in Brachypodium were also previously shown to be expressed in either biotic or abiotic stress treated P. trichocarpa tissues, and these are miR170, miR1450, miR406, and miR390 (Lu et al., 2008). None of the dehydration responsive miRNAs identified in this study were detected to be responsive to salt, mannitol, or cold treatments in Arabidopsis thaliana (Liu et al., 2008). miR169, which was shown to be drought responsive in Oryza sativa, was not detected to be dehydration responsive in Brachypodium (Zhao et al., 2007) miR1450, miR1881, miR528 are the common stress-responsive miRNAs identified both in T. diccocoides and Brachypodium microarrays (Kantar et al., 2011). Further, seven miRNA families were reported to respond to drought in various plant species (reviewed by Covarrubias and Reyes, 2010). These discrepancies in various studies could be explained by the different types of stress conditions applied, or to differences in miRNA regulation between species.
At least one putative target for each of the identified 26 miRNAs including miR170 and miR1850 were computationally proposed in a previous study of our group (Unver and Budak, 2009). In this study, target ESTs were also predicted for miR896, miR406, miR528, and miR390 (Table 1). Computationally identified Bdi-miRNA targets were consistent with previous findings (Zhang et al., 2006a, 2006b). The targets include proteins related to disease resistance, stress response, chromosome condensation, integrity of the cell wall and transport. A considerable number of in silico target ESTs were involved in ubiquitination for proteasomal degradation and several of the targets were proteins involved in abiotic and biotic environmental stress response (Kantar et al., 2010, 2011; Liu et al., 2008; Lu et al., 2008; Zhao et al., 2007).
In conclusion, Brachypodium miRNAs responsive to initial and ongoing drought stress were identified and quantified with the miRNA microarray. Furthermore, additional miRNA targets were predicted computationally. A large number of miRNAs were induced or suppressed upon drought treatment, showing that miRNAs have important roles in abiotic stress responses. To our knowledge, this is the first systematic identification of drought-responsive miRNAs in Brachypodium. As a model organism, Brachypodium is important in the drought resistance-related studies of crops, and this study can be a valuable source identify differences in miRNA expression that correlate with drought resistance and developing drought-resistant crops. Also, those miRNAs that were downregulated indicate targets that are likely to have positive effects on the drought response.
Footnotes
Author Disclosure Statement
The authors declare that no competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.