
Abstract
Abstract
Many plant genomes are already known, and new ones are being sequenced every year. The next step for researchers is to identify all of the functional elements in these genomes, including the important class of functional elements known as microRNAs (miRNAs), which are involved in posttranscriptional regulatory pathways. However, computational tools for predicting new plant miRNAs are limited, and there is a particular need for tools that can be used easily by laboratory researchers. We present semirna, a new tool for predicting miRNAs in plant genomes, available as a Web server. This tool takes a putative target sequence such as a messenger RNA (mRNA) as input, and allows users to search for miRNAs that target this sequence. It can also be used to determine whether small RNA sequences from massive sequencing analysis represent true miRNAs and to search for miRNAs in new genomes using homology. Semirna has shown a high level of accuracy using various test sets, and gives users the ability to search for miRNAs with several different adjustable parameters. Semirna, a user-friendly and intuitive Web server for predicting miRNA sequences, can be reached at http://www.bioinfocabd.upo.es/semirna/. It is useful for researchers searching for miRNAs involved in particular pathways, as well as those searching for miRNAs in newly sequenced genomes.
Background
A large number of miRNAs have been experimentally identified, both in plants (Pilcher et al., 2007; Sunkar et al., 2005; Sunkar and Zhu, 2004) and animals (Chen et al., 2009; Lim et al., 2003; Wienholds et al., 2005). However, laboratory methods are laborious, and computational methods have been developed to help predict the sequences of miRNAs and to aid in the discovery of all miRNAs that are expressed in known genomes (for a recent review, see Mendes et al., 2009). In silico miRNA discovery in plants is complex due to the heterogeneity of precursors, but it is facilitated by the high complementarity between mature miRNAs and their targets. However, there are currently only a limited number of publicly available plant miRNA prediction tools (Adai et al., 2005; Bonnet et al., 2004; Dezulian et al., 2006; Jones-Rhoades and Bartel, 2004; Lindow and Krogh, 2005; Moxon et al., 2008), which are specific for different prediction tasks, and to the best of our knowledge, nothing so far exists for the ab initio prediction of miRNAs starting from their putative targets.
At present, 232 miRNAs have been identified for the genome of the model plant Arabidopsis thaliana, most of which have been validated by experimental methods. More and more new plant miRNAs are being discovered, and a number of computational studies have proposed that thousands of different miRNAs may be expressed in this plant (Lindow and Krogh, 2005). Therefore, new, more sensitive methods are needed to bridge this gap. These methods should predict miRNA sequence accurately, in a manner similar to coding gene prediction tools, in order to help researchers to discover miRNAs that regulate molecules and mechanisms of interest.
We have developed a computational algorithm to predict de novo miRNA sequences, starting with both a plant genome and a target sequence. Our method searches throughout the genomic sequence, looking for fragments that fulfill specific criteria that enable them to act as a miRNA: length, folding, free energy, and complementarity against the target sequence.
The algorithm has been implemented as a Web server (semirna), which allows miRNA prediction to be performed on all completed plant genomes. This tool can return predictions for any target sequence submitted by the user, such as mRNA from the same plant, or foreign RNA, and the data returned is presented as a set of putative miRNAs, together with detailed information about the predictions.
Semirna has been trained with known miRNAs and tested on plant genomes with low numbers of known miRNAs. Furthermore, the tool has been tested in other scenarios (such as searching for precursor sequences for mature miRNAs and searching for miRNAs by homology), where it has provided accurate results. Taken as a whole, this testing shows semirna to be a versatile tool, useful for obtaining new knowledge in the field of small plant RNA sequences.
Materials and Methods
Web server programs and libraries
The miRNA algorithm (semirna) was written in the Perl programming language and has been implemented as a Web server using the CGI Perl library. The algorithm uses Blastn for the initial miRNA candidate search. To predict RNA secondary structure and calculate the free energy of these structures, several programs from the Vienna RNA package are used (Gruber et al., 2008): RNAfold, RNAplot, and RNAcofold. The program spends 10 to 120 s on average to annotate a sequence of 100 to 10,000 nt in length, respectively, using a dual-processor Intel Pentium 4 CPU 3.00 GHz with 1 Gb of RAM.
Training sequences
To train the miRNA search algorithm, we used the 232 A. thaliana sequences taken from the miRBase database, release 14.0 (http://microrna.sanger.ac.uk/cgi-bin/sequences/mirna_summary.pl?org=ath). This database stores all known miRNA sequences from already sequenced or ongoing genomes and contains sequences from many species, ranging from viruses to humans (Griffiths-Jones et al., 2008).
Several target mRNAs for miRNAs stored in miRBase have been discovered by wetlab experimentation. We took the nucleotide sequences of these target mRNAs to use as query sequences in the training set. The miRNAs (or miRNA families) taken from miRBase with known target mRNA sequences were: miR156–miR171 (Rhoades et al., 2002); miR162 (Xie et al., 2003); miR163 (Allen et al., 2004); miR172 (Kasschau et al., 2003); miR173 (Allen et al., 2005); miR393 and miR394, miR395, miR396, and miR397 (Jones-Rhoades and Bartel, 2004); and miR400–miR408 (Sunkar and Zhu, 2004) (see Table 1).
(1) miRBase miRNAs, number of members from each family in miRBase; (2) predicted miRNAs, predicted miRNAs using default values (10 and 80 nt as flanking lengths); (3) semirna miRNAs by default parameters, known miRNAs found using default values; (4) semirna miRNAs by specific parameters, additional known miRNAs found using specific values (pre-miRNA known lengths), when they are not previously found; (5) other miRNAs, known miRNAs different to the expected ones; (6) gene, miRNAs falling within protein coding genes; (7) novel miRNAs, novel putative miRNAs predicted by semirna.
A very similar sequence to the true positives were also found.
Used genomes
The miRNA search starts with a nucleotide query sequence (a gene, mRNA, CDS, or multitranscriptional fragment, such as a virus genome or a BAC whole sequence) and a genome in which to search for potential miRNAs. Currently, all sequenced plant genomes can be selected: A. thaliana (chromosome collection from Genbank database), Oryza sativa (chromosome collection from DDBJ database), Medicago truncatula (collection of BACs MtBACsPh3 gathered from http://www.medicago.org, 02/02/2009), Populus trichocarpa (BACs collection gathered from DoE Joint Genome Institute and Poplar Genome Consortium, ftp://ftp.jgi-psf.org/pub/JGI_data/Poplar/assembly/v1.0/), Shorgum bicolor (BACs gathered from DOE-JGI Community Sequencing Program, ftp://ftp.jgi-psf.org/pub/JGI_data/phytozome/v4.0/Sbicolor/assembly/Sbi1/), Vitis vinifera (chromosome collection from NCBI, April 7, 2009), and Zea mays (collection of BACs gathered from ftp://ftp.tigr.org/pub/data/MAIZE/, 06/08/2005). We will include each newly sequenced plant genome as they are uncovered.
The genome sequences have been filtered to mask low-complexity regions. Di- and tri-nucleotide simple repeats are removed.
Searching for alignments between target and genome sequences
This initial step of semirna is performed by a highly close-fitting identity search using Blastn, with extra parameters, allowing a short alignment between both query and genome sequences (–q –1 –W 7; q=–1 for penalizing mismatches, and W=7 for searches with short-length windows). The blast threshold e-value is dynamically calculated by multiplying the number of query sequence nucleotides by 0.32. The initial alignments can be extended when weaker G-U matches are found at the ends.
Rule for filtering miRNA-target alignments
To evaluate the quality of the miRNA-target pairings, a rule was established based on the characteristics of the training set containing the 187 known miRNAs from A. thaliana. We determined both the number of mismatches and the positions where they appear. The results of this test were (in the 5′–3′ direction in the miRNA): in the first third (approximately seven nucleotides), only a maximum of one mismatch can appear; in the second third, a maximum of two can appear; and in the last third, again a maximum of two can appear. In addition, in the entire alignment, there should not be more than three mismatches in total. This rule must be fulfilled in sequences with at least 20 nucleotides, so if it is not fulfilled, the miRNA candidate is refused.
Precursor structure
The RNAfold program from Vienna Package is used both to create a secondary structure model for the precursors and to calculate the free energy of this model. The putative precursor is rejected if the predicted structure has secondary stems in the main stem. Any mismatch appearing in the ends of the stem structure is removed, and the free energy is calculated again.
Quality coefficient
The quality coefficient (QC) is calculated from the free energy and precursor length parameters:
The optimum QC value was estimated using values obtained from the training set of A. thaliana sequences. The QC for these miRNAs were between 0.114 and 0.581, but 95% of these known miRNAs had a value equal to or higher than 0.35. Therefore, 0.35 is used as the default value for the QC parameter, to reduce false positives.
Blast for results validation
To support the expression of the predicted miRNAs, we extracted all EST sequences of M. truncatula and A. thaliana from the dbEST database on the NCBI Web server. We obtained 269,238 sequences and 1,527,298, respectively, using the following query in the query form: “medicago truncatula” [porgn] and “arabidopsis thaliana” [porgn]. We compared all semirna-predicted precursors against ESTs and/or miRNAs in miRBase database without a low-complexity filter, only taking the best blast hit. We considered the prediction sequence to be in dbEST or miRBase when we had an alignment with at least 70 nucleotides, 90% prediction-EST overlapping, and 90% sequence identity. We also used blastx to compare M. truncatula predictions against plant division in the UniProt database, and we removed all putative miRNAs that matched known proteins (at least 23 amino acids—corresponding to 69 nucleotides, 90% prediction overlapping, and an e-value lower than 1e-05).
Results and Discussion
Semirna analyzes a user's nucleotide query sequence by searching for putative miRNAs expressed from a selected genome, which could potentially recognise the query sequence. The genome sequence has been filtered to mask low-complexity regions that could lead to biologically insignificant autocomplementarity, as this would give rise to a high number of false positive results.
First, the program searches for regions from the selected genome that have high sequence similarity to regions from the query sequence (Fig. 1). This step allows semirna to select alignments of approximately 20 nucleotides, the expected minimal length of known miRNAs.
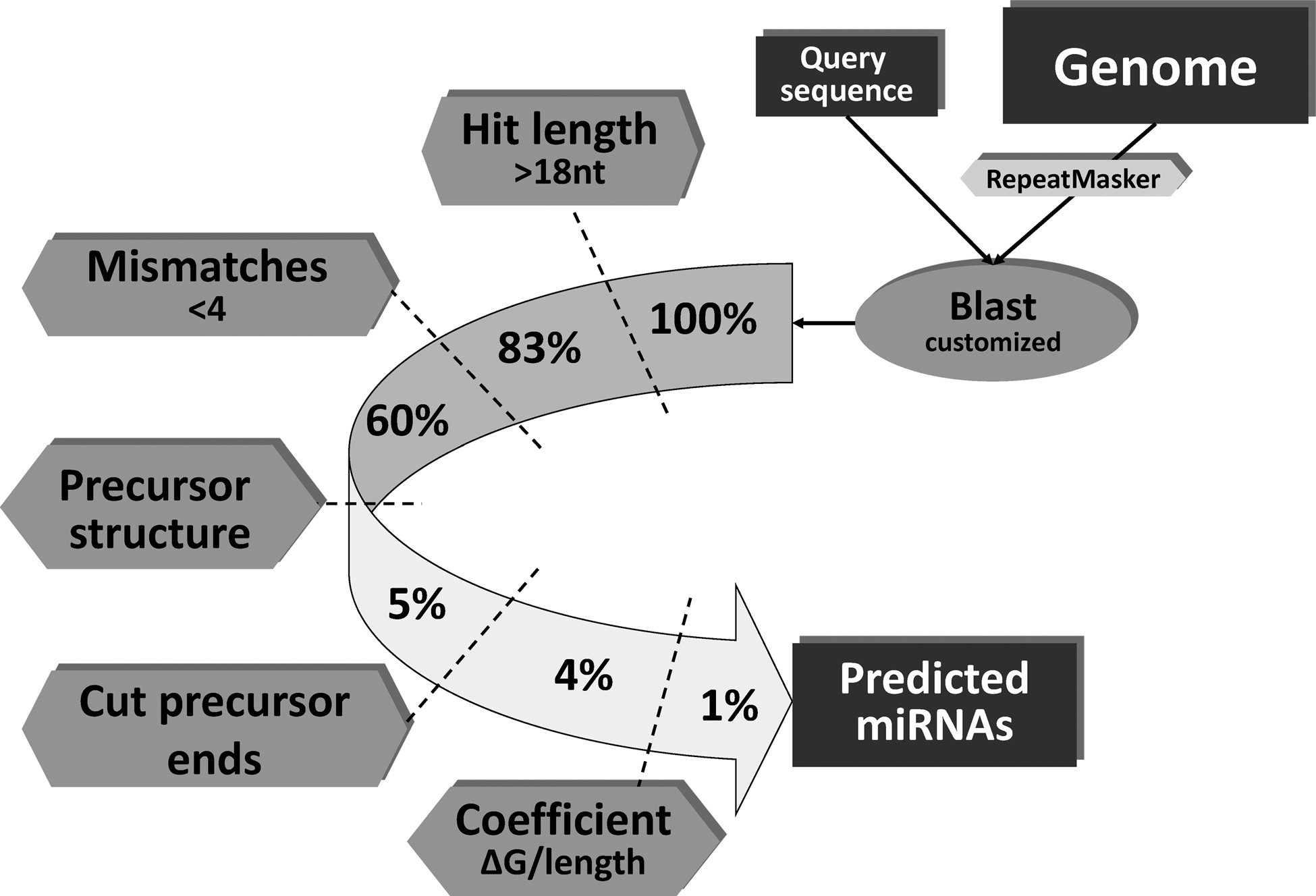
Semirna algorithm with hit percentages after each step. The miRNA search starts with a query sequence, which represents the miRNA target. This sequence is compared with a complete genome using Blast. Each Blast hit is passed through a set of filters, which take into account the hit length, the number of mismatches, the precursor structure and a quality coefficient. If a Blast hit does not pass every filter, it will be discarded. The percentage of Blast hits that pass each filter is shown in the figure.
The genome sequences found for each alignment are transformed into reverse complementarity sequences, and this initial set of candidates will then go through several filters. The filters and their parameters take into account particular characteristics of experimentally known miRNAs and their formation, establishing a rule-based system.
Filters to obtain putative miRNAs
The first filter is related to the length of the candidate miRNAs, and only sequences longer than 18 nucleotides (they can be enlarged later with G:U matches) are selected to continue through the analysis (the percentage of sequences passing each filter is shown in Fig. 1).
The second step is to remove sequences not fulfilling the following rule: in the first third of the sequence (approximately seven nucleotides), only a maximum of one mismatch can appear; in the second third a maximum of two; in the last third, again, a maximum of two. In the entire alignment, there should not be more than three mismatches in total.
The third step is to extend the miRNA candidate to get the sequence of the putative precursor. This is achieved by extending the alignment by a fixed number of nucleotides (as specified by the user) from both ends of the target mature miRNA sequence in the genome, representing the length of the flanking sequences of the precursor of the miRNA. The lengths of both flanking sequences should be different, and this will determine the final position of the miRNA within the precursor.
The lengths of the flanking sequences can be modified by the user to obtain new predictions with longer precursor sequences. Default values for the 5′ and 3′ flanking sequences are 10 and 80, respectively.
When the full putative precursor sequence is obtained, it is folded to create a secondary structure model, and its free energy is calculated. The putative precursor is rejected if the predicted structure has secondary stems in the main stem. Then, if the structure is correct but mismatches appear in the ends of the stem structure, semirna removes these ends until it returns to a matching region.
Finally, the precursor free energy is divided by the length of the precursor to calculate a quality coefficient (QC, see the Methods section for more details). We calculated this coefficient using the training set and fixed 0.35 as the minimum threshold at which to consider a putative miRNA true. Thus, this coefficient is used by the algorithm to discriminate between positive and negative results.
Searching for miRNAs using their target sequences
The primary use for semirna is to search for putative miRNAs targeting a user query sequence. To test this goal we used a set of 31 A. thaliana mRNAs targeted by 103 known miRNAs taken from the miRBase database. We submitted the 31 mRNAs to semirna as query sequences. Fifty-seven miRNAs were predicted (Fig. 2a), where 38 candidates can be taken as true positives and 3 matched with EST sequences. Furthermore 24 additional miRNAs were found when using flanking lengths similar to the known values for these miRNAs (Table 1).
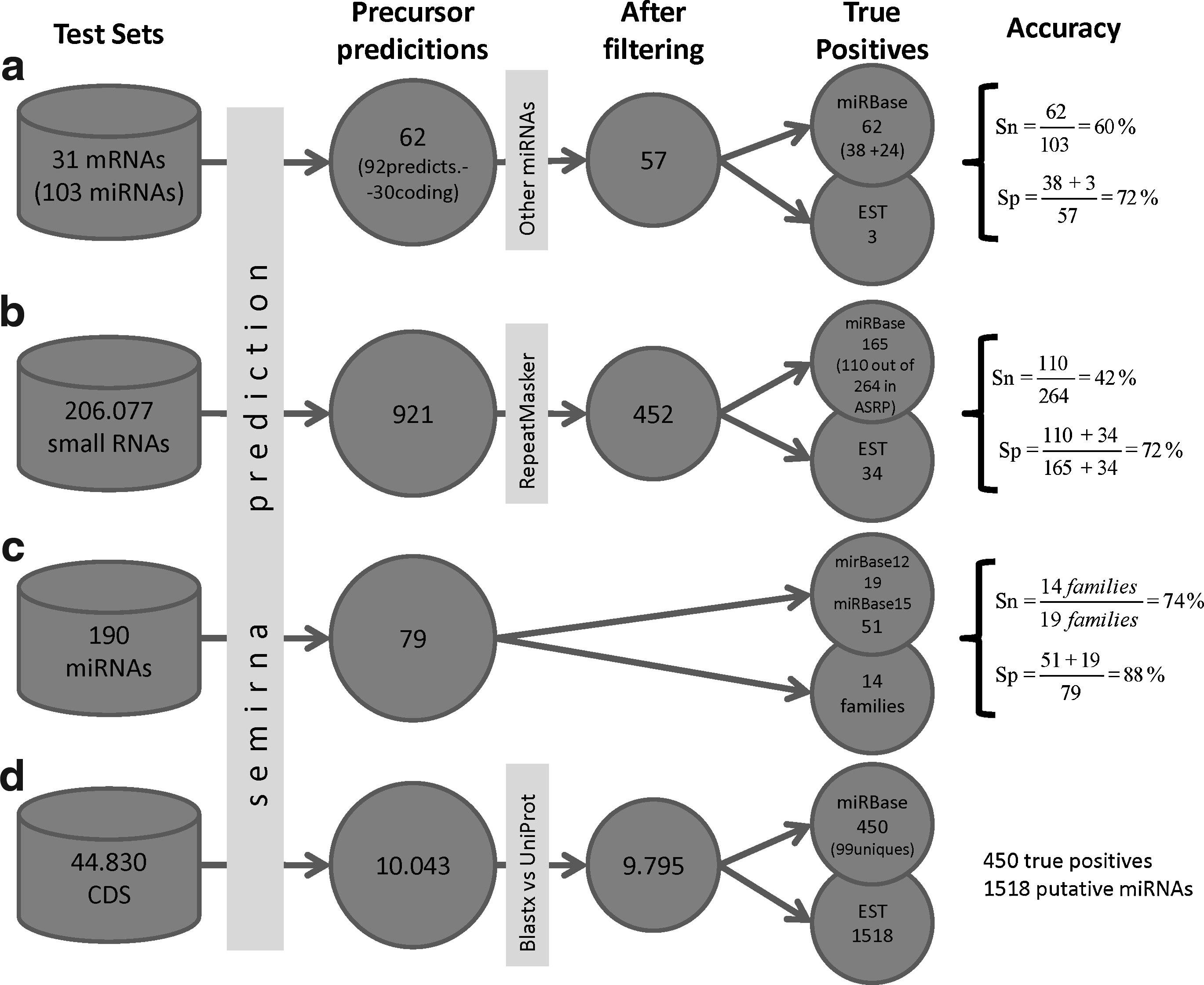
Diagram showing the steps in each acomplished tests by semirna. The first column shows the number and type of query sequences used in the test, and the remaining columns show the filters carried out on the predicted precursors (the specific filter is shown on the arrows) and the number of resultant sequences. The last column shows the number of true positives (predictions matching with sequences of miRBase), and the number of predicted precursors matching with known EST sequences. The sensitivity (Sn) and the specificity (Sp) is calculated for each test. The different query sequences used are: (
These results show an overall sensitivity of 60% and a specificity of 72% (Fig. 2a). However, the rate of false positives (specificity) can be misleading because some of the predicted miRNAs may be true positives that have not yet been experimentally validated. The test accuracy is similar to that of Lindow and Krogh (2005), with a sensitivity of 45% and a specificity of 91%. However, most of the false positives come from a small fraction of query sequences (see Table 1). If these few sequences were removed from the test, specificity would increase dramatically.
The increased sensitivity offered by semirna, at the cost of specificity, underlines it is intended use as a Web server: because the predictions are returned to the user as a list of results with links to the putative structure of the miRNA, and because semirna can return miRNAs with nonstandard properties (e.g., semirna can either predict miRNAs overlapping with coding genes or find highly repeated precursors in the genome), this allows the user to easily view all of the results and use their own expert opinion to decide which is the most useful results, or to extract the results and user their own custom filter, depending on their requirements. In addition, the user can adjust parameters, for example, the possible length of precursors, or could extract the results and add their own filters to improve specificity, at cost of sensitivity.
Another advantage of semirna is that unlike similar tools such as MIRFINDER (Bonnet et al., 2004), which depend on sequence conservation between A. thaliana and rice, semirna only needs the genome for which the user wants to discover miRNAs in order to obtain accurate results, meaning it can be applied to new genomes as they are sequenced.
Searching for the pre-miRNA precursors of mature miRNAs
MiRNAs are first transcribed as precursors and are then quickly processed by the DCL enzyme in plants to release the mature sequence. Thus, it is usually only possible to clone the short sequences of the mature miRNAs, which are approximately 21 nucleotides in length. When a short sequence like this is found (through an experimental assay), without flanking sequences from the precursor structure, it is difficult to discriminate between a true miRNA and the degradation product from another type of RNA.
Semirna is able to discover the putative miRNA precursor from a given small RNA. In this case, the query sequence in the Web application must be the complementary sequence to the mature miRNA, and we must select the genome from which the miRNA was cloned. Desired precursors are those that recognise the query sequence and contain a sequence 100% identical to the initial mature miRNA.
To test the utility of this strategy we took all the small RNAs in the ASRP database (Arabidopsis small RNA project) (Gustafson et al., 2005). ASRP has 206077 short sequences from cloned small RNAs, and their expression has been supported by previous studies. Several RNAs in this database correspond to known miRNAs (264 sequences), but the majority are degradation products of the most abundant classes of cellular RNAs, such as ribosomal RNAs (rRNA) and transfer RNAs (tRNA).
The sequences of small RNAs from ASRP were used as query sequences in a large-scale analysis using semirna, and only the predicted sequences of mature miRNA 100% identical to the query sequence and having a QC higher than or equal to 0.35, were taken as hits. We exclude the following sequences from the prediction to remove nonspecific precursors: redundant sequences, sequences found within coding genes, and sequences annotated by the RepeatMasker tool (http://www.repeatmasker.org), these were mainly rRNAs, transposons, pseudogenes or low-complexity sequences.
Following this strategy, semirna predicted miRNAs for 616 of the sequences in ASRP. However, sometimes the same mature miRNA was predicted with several different possible precursors, providing evidence of potential miRNA families. So, semirna predicted 452 nonredundant precursors, where 165 corresponded to known A. thaliana precursors, and these can be taken as the true positive sequences in this test. These true positive sequences corresponded to 110 ASRP's sequences out of 264 sequences annotated as miRNAs in this database, and it gives a sensitivity of 42% (Fig. 2b).
On the other hand, semirna predicted 287 putative miRNAs, which do not correspond to known sequences. But they were searched within known EST sequences from A. thaliana, where only 34 ones were found. In this way, this analysis proposes 34 high-probability putative miRNA sequences, together with the 110 true positives, with an estimated specificity of 72%.
To give a specific example, a prediction was made for ASRP11747. We found a precursor corresponding to the known ath-MIR156a (Fig. 3). However, the ASRP query sequence used is almost the complement sequence to the mature miRNA, ath-MIR156a*. This novel miRNA was also found in O. sativa when using semirna with ath-MIR156a* (validated with two known precursors from this genome in miRBase) and in M. truncatula (again with two precursors, which are not in miRBase). These results allow us to predict with confidence two new precursors of MIR156 in M. truncatula.

Predicted precursors for ASRP11747. (
Finally, two unknown precursors were predicted for the same small RNA, with identifier ASRP2121_miR416, and they are almost identical to the miR416 precursor in miRBase. However, the new precursors are within a region of A. thaliana annotated as transposon muDR, suggesting that either they do not belong to true miRNAs or they are miRNAs regulating transposon expression. Because small RNAs controlling transposon expression have previously been found, including MuDR (Slotkin et al., 2005), we should not dismiss the possibility that some of the predicted precursors participate in this intracellular regulatory pathway.
With this analysis, we show the ability of semirna to discover pre-miRNA sequences of mature miRNAs. Other tools, such as miRCat (Moxon et al., 2008), can carry out similar predictions but depend on quantitative experimental data. In contrast, semirna is able to suggest precursor sequences given only the sequence of the expected mature miRNA.
Search miRNAs by orthology
We currently know the sequences of a great number of genomes, and this number is increasing every year. When a new genome is sequenced, positional annotation of new functional elements is the first step toward acquiring new knowledge. In the case of miRNAs, interspecies sequence conservation is very strong with regard to the mature sequence, but it is weaker for the precursor. This characteristic suggests that using known mature miRNAs is a good starting point when searching for orthologues from evolutionarily close species.
This kind of analysis can be performed quickly using semirna. A mature miRNA can be used as a query sequence against a target genome different from that of the query sequence (although the same genome could also be selected to search for miRNA duplications or paralogues). Additionally, the option to search in both sense and antisense strands from the query sequence must be selected. Similar to the previous section, predicted miRNAs complementary to the query sequence should be chosen as final miRNA candidates.
The described strategy was used to discover new miRNA sequences in the M. truncatula genome, which contained only 30 known miRNAs in miRBase release 12. We use semirna to search for miRNAs in this genome and the results were later compared against M. truncatula miRNAs in miRBase release 15 where there are 375 known miRNAs. Using the 190 A. thaliana miRNAs as query sequences, semirna predicted 79 putative miRNA orthologues (removing redundant sequences and those falling within coding genes), including the majority of the known A. thaliana miRNA families (Table 2). From these new miRNAs, 19 were previously known and stored in miRBase, and we can propose 60 novel miRNAs in the M. truncatula genome as putative orthologues to corresponding sequences in A. thaliana.
Family, A. thaliana miRNA family; Members, number of sequences in the miRNA families; Predicted, number of miRNAs predicted by semirna; miRBase12, true positives regard to miRBase release 12, compared against the number of miRNAs annotated in this release; miRBase15, true positives regard to M. truncatula miRBase release 15. The following miRNA sequences are not found in the BAC collection from M. truncatula; therefore, they cannot be predicted and are not counted in the total values: mtr-miR160, mtr-miR169a, mtr-miR169b, mtr-miR171, mtr-miR319, mtr-miR399b, mtr-miR399d.
To check these results we used the new miRNA data in miRBase release 15. To compute the specificity we searched the number of novel prediction matching with miRNAs from the new release, and 51 out of the 60 predicted precursors are now known. So, semirna gave an overall specificity of 88% (Fig. 2c). Finally we calculated the number of miRNAs families found in this prediction: 14, and it was compared with the number of miRNAs families we would expect to find (miRNA families in both A. thaliana and M. truncatula): 19. This gives a sensitivity of 74%. Thus, we show that when the query set is enriched with known miRNAs, the accuracy of semirna is even higher than in the previous predictions.
Using the A. thaliana miRNAs ath-miR160, semirna found two putative orthologues for M. truncatula. So, a new miRNA with a unique sequence, together with the already known mtr-miR160 in M. truncatula, have a sequence identity higher than 80% over all precursor sequences (Fig. 4). Therefore, these predicted miRNAs have a high probability of belonging to the same family as miR160.

Alignment of known miRNAs from M. truncatula mtr-MIR160 and predictions from semirna using ath-MIR160 as a query sequence. The asterisks mark identical positions in the four sequences, and the points mark identical positions in only three of the sequences. The mature miRNAs are highlighted in bold.
To discover additional M. truncatula miRNAs, we used a new query set with all nucleotide coding sequences downloaded from http://www.medicago.org. Using this set, semirna predicted 10043 M. truncatula unique precursors (Fig. 2d) (although many of these are redundant sequences with one or two mismatches). We wanted to support these results with known data from this genome, so we compared the predicted sequences against a collection of 269238 M. truncatula Expressed Sequence Tags (EST) from the dbEST database (Boguski et al., 1993). From the miRNAs predicted by semirna, 450 matched known miRNAs, and 1,518 were found in the EST collection. After removing the predicted precursors that were similar to protein sequences from the plant section of the UniProt database (to remove putative precursors falling within coding sequences), we have 169 putative unique precursors whose expression has already been validated by EST sequencing. For example, semirna found a new member of the mir828 family, repeated three times, with the same mature miRNA as the known one from A. thaliana (query CU024876_34.2 in the Supplementary Data), as well as a new precursor (query CR538723_7.2 in Supplementary Data) with a high QC of 0.7.
Recently, in Zhou et al. (2008), 26 miRNAs were predicted in M. truncatula from EST and GSS sequences, and we found 7 of these 26 in our test with semirna (gi: 22084052, 7675335, 66995647, 48646738, 66992800, 39849618, 44873388) (Supplementary Data: zhou_vs_semirna.blast). The semirna predictions can be validated again by checking the genome positions of the candidate miRNAs or by looking up databases of predictions like Medicago-MIRATdb (Wen et al., 2008).
Other similar tools, such as microHARVESTER (Dezulian et al., 2006), can search candidate miRNA precursors by orthology, but these need both the mature and precursor sequences as input. However, we know that precursor sequences in plant miRNAs are not well conserved, and sometimes miRNAs exist with the same mature sequence and very different precursors distributed throughout the genome, constituting miRNA families (Zhang et al., 2006b). Methods like semirna can discover all the miRNAs in a complete plant genome that are homologues to those in another plant genome.
Semirna Web server
Semirna has been implemented as a universally accessible Web server. The interface begins with a standard HTML form, which asks the user to supply all necessary information (Fig. 5a). The main parameters needed are: the query (the only mandatory parameter) and the reference genome, the flanking sequence lengths (we can estimate the length of the putative precursors by adding both the flanking and the mature miRNA lengths), the query sequence strand or strands against which we want to find miRNAs, and the quality coefficient threshold. If the given query sequence is a coding and sense sequence, the user should mark the strand option to avoid searching for miRNAs against the antisense sequence.

Semirna Web server. (
But, if the user does not know the direction of the sequence or if it is a multicoding sequence (e.g., a virus sequence), this option should not be marked. Regardless of the use of this option, the final results will show against which strand the miRNA has been found. When a query is sent, a page with the current progress will appear. After a variable amount of time (usually a few minutes), the final results will be shown.
The results are initially given as an image map (Fig. 5b). At the top, a scale bar shows the length of the query sequence, and the predicted miRNAs are shown below. Each colored bar is a predicted miRNA, which appears together with its quality coefficient and its identifier. MiRNAs with higher coefficient values are darker colored. To obtain tables with detailed information about the predicted miRNAs, the user must click on a specific bar.
Additional result files are also provided by clicking on the following links: miRNAs in FASTA format. Show all predicted mature miRNA sequences in FASTA format.
Precursors in FASTA format
Show all predicted precursor miRNA sequences in FASTA format.
Predictions in GFF format
Show a GFF format file of the predicted miRNAs, which can be useful for genome viewers, such as GBrowse.
Multiple alignment of miRNAs
Show a multiple alignment of predicted and known miRNAs all together. The identifiers have links to both miRBase and the detailed result tables from your analysis.
When the user selects a specific predicted miRNA from the image map, semirna shows a detailed table or record with all the features from the predicted miRNAs ordered in different fields (Fig. 5c). A complete description of each field is provided in the “Glossary of Terms” in the Web page.
Additionally, the user can open an image with the predicted precursor structure and a genome viewer using GBrowse, where the predicted miRNAs are placed along the selected genome, together with the known miRNAs from that organism. Using this, the user can easily check whether the predicted miRNAs may constitute a new discovery.
Conclusions
We have presented semirna, a versatile and useful tool for predicting plant miRNAs from only a target sequence and a genome. This tool has shown high accuracy throughout many different tests and could be important in the search for all miRNAs expressed in an entire plant genome.
The algorithm has been implemented as a Web server, and it is available for the scientific community and easily comparable with current knowledge.
An important contribution of the presented tool with regard to other current systems is that semirna is conditioned neither by experimental results nor by sequence conservation data. Thus, it can help to identify new miRNAs within a new genome, and it could aid in the analysis of recently sequenced genomes.
In addition, the user can use expected targets of interest and search for putative miRNAs in semirna by checking the results in the reference genome or/and in other public databases. This would allow for the creation of candidate miRNA libraries for use by the laboratory researcher, avoiding the need to perform screenings to isolate a large amount of small RNAs. To validate semirna predictions, the user could test the predicted miRNAs by means of a transcriptomic experiment, extracting RNA under different experimental conditions, which could save time.
Semirna has a good sensitivity value (42–74%), and a very high specificity value (72–88%), validated in a range of scenarios, using a number of high-throughput data sets, and will be very useful in the future analysis of plant genomes.
Supplementary Online Material
Supplementary data with results from all tests are available at http://www.bioinfocabd.upo.es/semirna/supplementary_data/
Footnotes
Acknowledgments
We thank OTRI in University Pablo de Olavide for providing the service of language editing and and Oswaldo Trelles for his critical reading of the manuscript. We also acknowledge institutional support from the Junta de Andalucía to the CABD. This research was supported by the node GNV5-UMA in the National Institute of Bioinformatics (INB), and a grant from the Spanish Ministerio de Ciencia y Tecnología (GEN2006-27770-C2).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.