
Abstract
Abstract
The recent sequencing of the complete genome of the peach, together with the availability of new high-throughput genome, transcriptome, proteome, and metabolome analysis technologies, offers new possibilities for Prunus breeders in what has been described as the postgenomic era. In this context, new biological challenges and opportunities for the application of these technologies in the development of efficient marker-assisted selection strategies in Prunus breeding include genome resequencing using DNA-Seq, the study of RNA regulation at transcriptional and posttranscriptional levels using tilling microarray and RNA-Seq, protein and metabolite identification and annotation, and standardization of phenotype evaluation. Additional biological opportunities include the high level of synteny among Prunus genomes. Finally, the existence of biases presents another important biological challenge in attaining knowledge from these new high-throughput omics disciplines. On the other hand, from the philosophical point of view, we are facing a revolution in the use of new high-throughput analysis techniques that may mean a scientific paradigm shift in Prunus genetics and genomics theories. The evaluation of scientific progress is another important question in this postgenomic context. Finally, the incommensurability of omics theories in the new high-throughput analysis context presents an additional philosophical challenge.
Introduction
Regarding the genetic structure of Prunus species, cultivated peach (2n=16) is characterized by a narrow genetic origin with limited genetic diversity. Plum (2n=16), cherry plum (2n=16), sloe (2n=4×=32), and almond (2n=16), however, are the most polymorphic species, with the highest heterozygosity and amount of variability. Intermediate genetic variability has been observed in apricot (2n=16), prune (2n=6×=48), and sour (2n=4×=32) and sweet (2n=16) cherry. This level of genetic heterozygosity in Prunus can be attributed to the following mating system differences within species: the majority of peach cultivars are self-compatible; apricots are self-compatible in many cases; sweet and sour cherry and prune are self-incompatible in many cases; and plum, cherry plum, sloe, and almond are normally self-incompatible and thus outcrossing (Byrne, 1990; Sánchez-Pérez et al., 2006; Watkins, 1976).
Prunus production has traditionally been based on local cultivars. In addition, specific breeding programs were started during the 1920s, first in almond, and later in peach, apricot, plum, prune, and cherry. Conventional Prunus breeding consists of generating new genetic combinations through sexual hybridization and subsequently applying selection methods to the newly obtained descendants (Janick and Moore, 1996; Moore and Janick, 1983). As a result of these programs, especially during the last 3 decades, the number of new varieties has significantly increased. However, the need to introduce new cultivars to improve and sustain quality of life is indeed now even more pressing, in order to obtain better products that are safer, healthier, environmentally friendly, and attractive (Infante et al., 2011; Janick and Moore, 1996; Martínez-Gómez et al., 2003; Prohens, 2011).
Prunus breeding must address specific challenges resulting from the multiannual nature of dormancy mechanisms, which involves significant environmental influence in the expression of traits and makes the selection process more difficult. In addition, Prunus species show a long juvenile period, between 3 and 7 years, which extends the selection process and also produces differences in trait expression between juvenile and mature trees. Other specific characteristics include the narrow genetic background of commercial cultivars and the fact that vegetative propagation onto rootstock also interacts, making the selection processes more difficult (Martínez-Gómez et al., 2003; Moore and Janick, 1983). For this reason, developing new Prunus cultivars is an expensive and time-consuming process involving the generation of a large population of seedlings from which the best genotypes are selected. Whereas the capacity of breeders to generate populations from crosses is almost unlimited, the management and selection of these seedlings in different locations significantly limits the generation of new releases.
In this context, the development of efficient marker-assisted selection strategies is particularly useful in these temperate fruit species (Arús et al., 2005; Dirlewanger et al., 2004; Martínez-Gómez et al., 2003). In addition, the IPGI (international peach genome initiative) has recently released the complete peach (the model Prunus species) genome sequence [Prunus persica genome (v1.0)], which is available at http://www.rosaceae.org (Sosinski et al., 2010). This fact, together with the availability of new high-throughput omics tools, offers new possibilities for Prunus breeding and the development of molecular markers linked to the main agronomic traits (Collard and Mackill, 2008) in the postgenomic era (Leader, 2005).
In this work, omics concepts and technologies are clarified in the current postgenomic era, including both new biological and philosophical challenges as well as opportunities for the application of omics concepts and technologies in Prunus breeding.
Genomics Concepts
The discipline of genomics concerns the study of the structure and function of the genomes of a living organism (Baer et al., 1984). The plant genome can be defined as the DNA, composed of four types of subunits [adenine (A), thymine (T), guanine (G) and cytosine (C)], called nucleotides (nt) or base pairs (bp), organized into separate chromosomes inside the nucleus of a cell. We can classify this DNA into two different types: nuclear DNA (nDNA), which is properly called the genome and is located in the nucleus of the cell; and organelle DNA, including mitochondrial DNA (mDNA) and chloroplast DNA (ctDNA) coding for the RNAs and proteins in the functions of the organelles (Bendich, 2010; Bennett and Leitch, 2011). The nDNA is combined (packaging) with nuclear proteins called histones to form the chromatin that makes up the content of the nucleus of a cell. Chromatin structure is essential to compact the DNA and plays an important role in nDNA expression, including in transcription, replication, and DNA repair (Grimaud, 2011; Heslop-Harrison, 2000).
The DNA (from here we will talk about DNA instead of nDNA) includes not only the genes (coding DNA with a phenotypic function), but also the noncoding DNA sequences (Scherrer and Jost, 2007a). Two types of regions are found within the coding DNA, one called exons (expressed region transcribed into pre-messenger RNA, pre-mRNA) and the rest called introns (insertions not present in mature mRNA). The 5′ UTR is located between the transcription start site and the beginning of the coding sequence and another untraslated region (3′ UTR) is located downstream of the stop codon (Primrose and Twyman, 2004; Scherrer and Jost, 2007a, 2007b) (Fig. 1). The recent term exome has been attributed to the exon regions (around the 2% of the genome), which sligthly coincide with the transcriptome, taking into account the fact that the transcriptome is richer due to alternative splicing processes (pre-mRNA processing) (Ng et al., 2008).
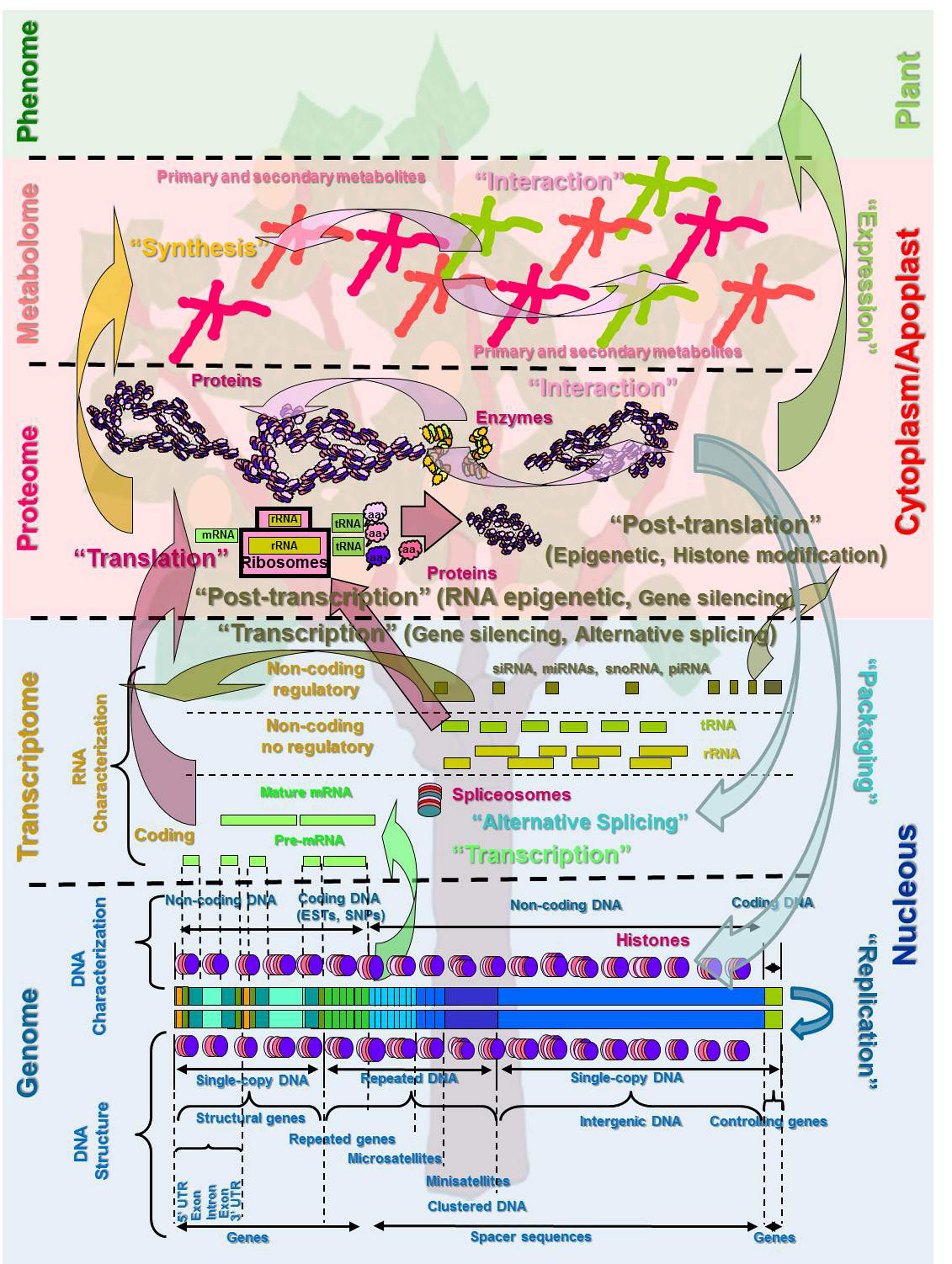
Schematic representation of the central dogma of molecular biology (Crick, 1970) in plants, including DNA (genome) organization and transcription to RNA (transcriptome) translated to protein (proteome). This figure also shows metabolite synthesis (metabolome), phenotype expression (phenome), and the DNA packaging (histones) process.
The first genomics studies in Prunus were conducted for the development of DNA markers (called molecular markers) for molecular characterization (structural genomics) and relationship with agronomic traits (functional genomics) using genetic linkage maps and QTL (quantitative trait loci) analysis. The populations typically assayed in these linkage studies were of type F1, F2, or BC1, with high diversity and low levels of linkage disequilibrium (LD), taking into account the genetic structure and heterozygosity level of Prunus species, mainly in the cases of almond, plum, cherry and prune (Arús et al., 2005; Collard and Mackill, 2008; Martínez-Gómez et al., 2003; Sorkheh et al., 2008). These DNA studies were initially performed in the 1990s using RFLPs (restriction fragment length polymorphisms), which provided an efficient method because of their codominant nature and unlimited number of markers. However, the application of these markers has been limited due to their complexity and the time-consuming nature of the process (Arús et al., 2005; Martínez-Gómez et al., 2007; Peace and Norelli, 2009) (Fig. 2).

The utilization of PCR (polymerase chain reaction)-based markers is less laborious and time consuming and has increased the possibilities of genome characterization and mapping (Martínez-Gómez et al., 2003). These markers can be considered a second generation of DNA markers. RAPDs (random amplified polymorphic DNAs) were the first PCR markers assayed, although their dominant nature and low repeatability drastically limited utilization. In addition, AFLP (amplified restriction fragment-length polymorphism) technology is a powerful DNA fingerprinting technology based on the selective amplification of a subset of genomic restriction fragments using PCR. AFLP has a number of advantages over the RAPD technique, namely, more loci analyzed per experiment and better reproducibility of banding patterns resulting from the higher specificity of primer annealing to complementary adapters. However, AFLPs are too difficult and time consuming for routine application (Sorkheh et al., 2007). For these reasons, at this moment, PCR-based SSR (simple sequence repeat) markers, also called microsatellites (Fig. 1), have become the markers of choice for cultivar identification and genetic mapping in Prunus because of their high polymorphism, abundance, codominant inheritance, and transportability (Arús et al., 2005; Gupta et al., 1996; Martínez-Gómez et al., 2003).
Recent molecular markers based on DNA sequencing (third DNA marker generation) developed in Prunus species include ESTs (expressed sequence tags) and SNPs (single nucleotide polymorphisms) (Fig. 2). Development of ESTs was initiated in peach in 2002 (Yamamoto et al., 2002), and has since been applied to other species such as apricot (Decroocq et al., 2003) and almond (Jiang and Ma, 2003). To date, a collection of more than 100,000 ESTs (more than 400,000 within the Rosaceae family) from different Prunus (mainly peach, but also almond and apricot) based on cDNA libraries has been released to public databases, and more than 25,000 putative unigenes (10,000 contigs and 15,000 singlets) have been detected. This information has been compiled in the GDR (genome database for Rosaceae) (Cabrera et al., 2009; Jung et al., 2004, 2011). On the other hand, almost 6,000 SNPs have been identified, revealing an estimated frequency of 0.07 SNPs/100 bp (Meneses et al., 2007, http://www.rosaceae.org) (Fig. 2). Due to the abundance and polymorphism of these new markers, they are becoming the markers of choice together with the SSRs for genetic mapping and QTL analysis in Prunus (Tavassolian et al., 2010).
The IPGI has recently released the complete peach genome sequence [Prunus persica genome (v1.0)] (http://www.rosaceae.org). Peach v1.0 was generated from DNA from the doubled haploid cultivar “Lovell,” which means that the genes and intervening DNA are “fixed” or identical for all alleles and both chromosomal copies of the genome. This doubled haploid nature was confirmed by the evaluation of >200 SSRs, and has facilitated a highly accurate and consistent assembly of the peach genome. The genome size (number of nucleotide bases) of Prunus was initially estimated using fluorescent techniques at around 280,000,000 bp (280 Mbp) (Baird et al., 1994), although the recent sequencing of the peach genome indicated a size of 227 Mbp for this species (Sosinski et al., 2010). The size of this genome is relatively small within the plant kingdom, where genomes range from the 125 Mbp of Arabidopsis thaliana L. Heynh (1.81 times smaller than Prunus) to the 35,817 Mbp of lily (Lilium longiflorum Duch) (157.78 times greater) (Bennett and Leitch, 2011). Gregory et al. (2007) indicated an average genome of around 6,000 Mbp per haploid in angiosperm plant genomes.
Peach v1.0 currently consists of eight pseudomolecules (scaffolds) representing the eight chromosomes of peach, numbered according to their corresponding linkage groups (Sosinski et al., 2010). In addition, this genome sequencing consisted of approximately 7.7-fold whole genome shotgun sequencing employing the accurate Sanger methodology, using an automatic sequencer and capillary electrophoresis (Sosinski et al., 2009). The assembled peach scaffolds cover nearly 99% of the peach genome, and the orientation of over 92% has been confirmed. To further validate the quality of the assembly, 74,757 Prunus ESTs were queried against the genome at 90% identity and 85% coverage, and only ∼2% were found missing. The Prunus reference genome contains a mean percentage of between about 60% A+T bases and 40% G+C (A+T / G+C: 1.5), although this varies for each scaffold (Sosinski et al., 2010). This GC content is slightly higher than that observed in other species such as Arabidopsis (about 36%), indicating greater stability and a greater percentage of protein coding genes (Barakat et al., 1998). Smarda et al. (2012) have recently described a range of GC content in plant genomes of between 33.6% and 47.5%.
Finally, gene prediction and annotation is an ongoing process, which includes the global analysis of DNA fragments, RNA fragments, proteins, and metabolites (Stadler et al., 2009). The effective annotation of such data will require the extension of current omics data developed in model organisms (Morrison et al., 2006a). Current estimation indicates that peach has 28,689 transcripts and 27,852 protein-coding genes in the reference Prunus genome (Sosinski et al., 2010). In addition, homology of query DNA sequences to known sequences and similarity searches can be inferred by BLAST (basic local alignment search tool) (Altschul et al., 1990) versus reference databases (http://blast.ncbi.nlm.nih.gov/Blast.cgi), as well as Prunus-specific databases (http://www.rosaceae.org). Finally, the Gene Ontology (GO) gene classification system enables protein function to be related to gross cellular or whole organism functions (Ashburner et al., 2000; Conesa et al., 2005), including central metabolism, nucleic acid replications, cell division, pathogenesis, etc. With the availabilty of the new high-throughput genome, transcriptome, proteome, and metabolome analysis technologies, Prunus gene prediction, annotation, and ontology will likely be revised and updated.
Transcriptomics Concepts
The discipline of transcriptomics, less studied than genomics, concerns the analysis of the transcriptome. The transcriptome can be described in living organisms including plants as the complete list of all types of RNA molecules, also composed of four types of subunits or nucleotides [adenine (A), cytosine (C), guanine (G), and uracil (U) instead of thymine], expressed in a cell, tissue, or whole organism (Velculescu et al., 1997). Unlike the genome, which is roughly fixed for a given organism, the transcriptome can vary throughout development and time with external environmental conditions. RNA, including coding (cRNA) and noncoding (ncRNA), is a less stable biological structure that selectively transmits the genetic information of the DNA at different times or by different cell types (Atkins et al., 2011; Darnell, 2011). Protein coding RNA is also called messenger RNA (mRNA) and is around 5% of the total. Noncoding RNA include nonregulatory RNA [ribosomal RNA (rRNA) (up to 85%) and transfer RNA (tRNA) (around 10%)] and regulatory RNA (less than 5%), which includes the group of small RNAs [small interfering RNA (siRNA, 21–23 nt); micro-RNA (miRNA; 20–24 nt); and small nucleolar RNA (snoRNA; 60 to 300 nt)] and other classes of RNA such as the Piwi-interacting RNA (piRNA) (Atkins et al., 2011; Bompfünewerer et al., 2004; Gingeras, 2011; Kim, 2005; Rodin et al., 2011) (Fig. 1).
The transcriptome reflects the genes that are being expressed at any given time under specific conditions. The transcription of DNA into RNA occurs in the nucleus. In this process, DNA exons can be spliced in different ways, resulting in splice variants (alternative splicing) of the same primary transcript (pre-mRNA) (Hallegger et al., 2010; Kapustin et al., 2008), to generate more variation in the mature mRNA. During nuclear maturation of the primary transcript, both ends of pre-mRNA (5′ UTR and 3′ UTR) are posttranscriptionally modified. These UTRs are involved in the posttranscriptional regulation of gene expression (Mignone and Pesole, 2011). Mature m-RNA is an intermediate template in the process of protein production from a DNA template, offering the cell the means to control the production and destination of these proteins in a complex, timely, and transient manner (Hodges, 2005). In addition, complex ribonucleoprotein machines called spliceosomes remove introns from nuclear pre-mRNA precursors by mRNA splicing. Alternative splicing, processing of pre-mRNA, not only increases proteome diversity but also regulates the level of posttranscriptional gene expression (Gingeras, 2011; Lu and Tarn, 2010; Newman, 2008).
On the other hand, the small noncoding RNAs provide well-referenced global regulation through transcriptional (including alternative splicing and gene silencing), posttranscriptional (through phenomenon such as gene silencing regulation), and posttranslational regulation with chromatin modification, including histone and other modifications described as epigenetic modifications that play a role in the transmission of gene regulation (Bompfünewerer et al., 2004; Chapman and Carrington, 2007; Grimaud, 2011; Hodges, 2005; Matzke and Matzke, 2004; Newman, 2008) (Fig. 1). Posttranscriptional RNA modifications can be dynamic and might have functions beyond fine tuning the structure and function of RNA. Understanding these RNA modification pathways and their functions may allow researchers to identify new layers of gene regulation at the RNA level in the field of RNA epigenetics (He, 2010). Many of these epigenetic mechanisms are critical to protein function. This is another explanation for the large number of proteins from a much smaller number of genes, which is similar in all living organisms. It is now known that mRNA is not always translated into protein, and the amount of protein produced for a given amount of mRNA depends on the transcription of the gene and the current physiological state of the cell. In addition, some authors suggest the possibility that RNA is the ancestral genetic material in living organisms (Darnell, 2011; Primrose and Twyman, 2004).
Forrest and Carninci (2009) described how an important part of the genome (DNA) is transcribed (RNA), although only a limited fraction is assigned to genes. Understanding the transcriptome, both as a template for protein expression and as a regulatory molecule, is essential for interpreting the functional elements of the genome and revealing the molecular constituents of cells and tissues in a biological context (Blencowe et al., 2009). Along these lines, pervasive (interleaved) transcription, a recent concept, is being applied to the transcription process (Abhinaya, 2011; Clark et al., 2011; Jacquier, 2009). These authors described pervasive transcription as the transcription of the interspersed genes that are embedded within the normal coding genes. The entire stretch of the genome is transcribed, whether it is coding for a particular protein or not. Therefore, not all coding sequences lie juxtaposed, and they may also overlap with one another. Jarvis and Robertson (2011) described this noncoding RNA phenomenon as the genomic dark matter currently emerging from the application of postgenomic tools. Inside the pervasive transcription process, noncoding small regulatory RNAs play a known regulatory role in various biological processes in plants by affecting gene expression through gene silencing (an RNA change complementary to the mature mRNA) at the posttranscriptional level (Gingeras, 2011; Hegarty and Hiscock, 2005; Ingvarsson and Street, 2011). To date, more than 2,300 genes encoding miRNAs have been annotated in Arabidopsis, rice, and other plant species belonging to 250 mi RNA families (Griffiths-Jones et al., 2008), although no information is available yet in Prunus species.
In Prunus species, the first transcriptomics works began at the end of the 1990s with the development of the previously mentioned ESTs, the development of cDNA-AFLP, and the analysis of several candidate genes (CGs). Later, new strategies of massive analysis (high-throughput) of transcriptomes have been applied, producing larger amounts of data in terms of expression of a large number of genes in a single experiment. One such system is massive transcriptome analysis, which uses cDNA biochips (microarrays) to analyze thousands of genes by hybridization of mRNA labeled with fluorescence on a slide. In addition, transcriptome shotgun sequencing using next-generation technologies (RNA-Seq), which has lowered the cost of DNA sequencing (in this case complementary, cDNA) and could be more suitable than the application of microarrays, is beginning to be applied to Prunus species (Martínez-Gómez et al., 2011) (Fig. 2). Furthermore, transcriptomics data results using the different approaches have been validated by quantitative real-time PCR (qRT-PCR), mainly in the case of low transcript levels. This strategy has been applied in Prunus transcriptomics studies using housekeeping genes as reference genes in the case of intermediate–high expressed genes (Martínez-Gómez et al., 2011; Tong et al., 2009; Ziliotto et al., 2008).
Regarding the transcriptional regulation and maturation of pre-mRNA (including alternative splicing) in the case of Prunus, an estimated hundreds of thousands of proteins (nearly 1 million) can be created from 27,852 protein-coding genes (described in peach) (Peace and Norelli, 2009). However, very little is known about alternative splicing processes in these species. Sosinski et al. (2010) described 838 alternative splicing processes in a total of 26,689 protein-coding genes described in the refrence Prunus genome (http://www.rosaceae.org). Finally, posttranscriptional regulation mechanisms have been described in Prunus in only two important agronomical traits: Plum pox virus resistance (Di Nicola-Negri et al., 2010; Zagrai et al., 2008) and flowering time (Leida et al., 2012).
Proteomics Concepts
The discipline of proteomics concerns the study of the proteome, which is the complete list of all proteins present in a cell, tissue, or whole living organism (including plants) and expressed by a genome (Pandey and Mann, 2000). There have been fewer proteomics studies than genomics and transcriptomics studies. Proteomics studies include the identification and cataloguing of all proteins produced in a cell (equivalent to whole genome annotation), the functional annotation of genes based on protein structure and the global analysis of protein–protein interactions (Pukkila, 2001).
Proteins are the active molecules of the life involved in structural, functional, storage, and transport processes. Proteins include enzymes that act as catalysts in starting or speeding up biochemical reactions in structural units (Cozzone, 2010; Pandey and Mann, 2000). They are assembled from 20 types of subunits (building blocks that combine to form proteins) called amino acids (aa) using information encoded in genes (DNA) (Fig. 1). These aa are naturally incorporated into a group (polypeptides) and are called proteinogenic or standard amino acids, constituting all the proteins. These 20 aa are encoded by the universal genetic code (codon) from different triplets of RNA bases (nucleotides). In addition, these proteins present a complex structure characterized as primary structure (amino acid sequence), secondary structure (regularly repeating local structures), tertiary structure (the spatial relationship of the secondary structures to one another), and quaternary structure (structure formed by several protein molecules). These complex three-dimensional shapes of proteins are crucial for proper functioning. The aa sequences (primary structure) determine the higher structural levels (secondary and tertiary) of proteins and specify their biological properties (Cozzone, 2010; Pandey and Mann, 2000; Pukkila, 2001).
The proteome has at least two levels of complexity lacking in the genome. Although the genome can be defined by the sequence of nucleotides, the proteome cannot be limited to the sum of the sequences of the proteins present. Knowledge of the proteome requires knowledge of the structure of the proteins in the proteome and the functional interaction between the proteins. This protein–protein interaction is a key of the phenotype expression (Pandey and Mann, 2000). The transcriptome can be seen as a precursor to the proteome having a strong correlation between protein domain organization and intron–exon structure (Kolman and Sdtemmer, 2001). The transcription of RNA to protein is produced in the cytoplasm through the ribosomes, although the majority of proteins are extracellular (located in the apoplast) (Fig. 1).
Although Prunus plants may have thousands of genes in total (27,852 genes have recently been estimated in the peach by Sosinski et al., 2010) and slightly more transcripts, they contain hundreds of thousands of different proteins (estimated by Peace and Norelli, 2009), including enzymes with phenotypic functions and structural units of the plant (DNA packaging, tissues, etc.). Technology for the global analysis of proteome available since the mid-1970s has largely been used in Prunus species (Fig. 2). Isoenzymes [1 D (dimension) electrophoresis] were the first proteomic markers (now also called molecular markers) widely utilized in Prunus. Isoenzymes have been used for cultivar identification in Prunus because of their environmental stability, codominant expression, and good reproducibility. Nevertheless, their utilization is limited by the small number of loci that can be analyzed with conventional enzyme staining methods as well as the low variation in some loci (Martínez-Gómez et al., 2003, 2007).
More recently, with 2D electrophoresis, proteins have been separated by molecular weight and pH as well, using PAGE (Poly Acrylamide Gel Electrophoresis) (Díaz-Vivancos et al., 2006). Additional new high-throughput proteome analysis technologies applied to Prunus include peptide mass spectrometry, to identify proteins by cleaving them into short peptides. After this peptide identification, protein identity can be deduced by matching the observed peptide masses against a protein sequence database (Díaz-Vivancos et al., 2006; Peace and Norelli, 2009).
Metabolomics Concepts
Metabolomics is the study of the metabolome, the molecules involved in metabolism (metabolites) in plants and living organisms. This is a new discipline concerning the use of genome sequence analysis to determine the capability of a cell, tissue, or whole organism to synthesize extracellular small molecules or metabolites (Fiehm, 2002) (Fig. 1). The metabolome and proteome have great potential to elucidate biological processes, and molecular biology tools have been incorporated into these fields. The biochemical pathways model of genomics regulation indicates that genes open and close different metabolic pathways involved in growth, fruit formation, climate adaptation, pest and disease resistance, and synthesis of primary (directly involved in normal growth, development, and reproduction) and secondary (not directly involved in these processes, but usually having an important biological function) metabolites (Fridman and Pichersky, 2005; Schaucher and Fernie, 2006). At this moment, gas chromatography coupled with mass spectrometry (GC-MS) is the most important tool for nontargeted metabolite analysis (Shi et al., 2011; Valdyanathan et al., 2005).
Metabolome analysis is now recognized as a crucial component of functional genomic and systems biology investigations. Innovative approaches to the study of metabolic regulation in plant systems are increasingly facilitating the emergence of systems approaches in biology (Valdyanathan et al., 2005). Advances in analytical chemistry, computation, and biotechnology have led to the recent development of methodologies for broad metabolite profiling. The study of plant metabolism is one of the first beneficiaries of these new technologies, and applications have focused on plant metabolism engineering, functional genomics, and physiology (Trethewey, 2007). Further metabolome studies include unigene annotations with simple key word searches confirming each search result with BLAST searches and GO prediction (Schmidt et al., 2011; Shi et al., 2011).
This field is still undergoing development in the case of Prunus species (Peace and Norelli, 2009) (Fig. 2), and relatively little analysis of metabolic networks in Rosaceous species has been reported. Lombardo et al. (2011) recently studied peach mesocarp metabolic networks across development using metabolomics and analysis of key regulatory enzymes. These authors have suggested that posttranscriptional mechanisms are very important for metabolomics regulation in early stages, and observed a decrease in amino acid levels coupled to an induction of transcripts encoding amino acid and organic acid–catabolic enzymes during ripening. On the other hand, many studies have been performed analyzing chemical constituents in this species for medical and pharmaceutical purposes. Poonam et al. (2011) identified 569 different chemical compounds with medical properties in 23 different Prunus species. However, studies relating these constituents to the responsible genes are scarce.
Interactomics Concepts
The discipline of interactomics concerns the analysis of interactions inside the proteome or the metabolome and the other omics sciences, including metabolome–metabolome interactome, protein–protein interactome, protein–DNA interactome, and protein–RNA interactome (Flavell, 2005) (Fig. 1). The interactomics discipline presents a holistic view of the biological process. Most proteins function in collaboration with other proteins, and for this reason one goal of proteomics is to identify what types of interaction exist and which proteins interact. This is especially useful in determining potential partners in cell signaling cascades. Several methods are available to probe protein–protein interactions. In the near future, the comparison of interactomes of different species might drive a powerful transformation in the study of biology (Cesareni et al., 2005; Flavell, 2005).
In this interactomics context, plant genes work through complex control networks, both transcriptional and posttranscriptional. Their RNA or protein products are similarly organized in time and space to function in networks. In addition, metabolites also work in networks. This complexity is bewildering, and it will take significant effort to understand the mechanisms and systems behind gene–trait linkages (Flavell, 2005). Toyoda et al. (2006) developed a browser (OmicBrowse, http://omicspace.riken.jp) to explore multiple datasets coordinated in the multidimensional omics space. This browser integrates omics knowledge ranging from genomes to phenomes and connects evolutional correspondence among multiple species, which in the case of the plant kingdom currently includes Arabidopsis and rice. In addition, Hu et al. (2011) have recently developed a protocol for the identification of protein–DNA interactions in vitro using microarray technology. The procedure involves double-stranding synthesized DNA oligonucleotides with a fluorescent-labeled primer, binding the labeled double-stranded DNA to the protein microarray. This approach provides simultaneous identification of protein–DNA interactions for thousands of proteins, and multiple designed DNA probes can be tested in parallel.
Nonethless, interactome studies in Prunus species have been highly limited (Martínez-Gómez et al., 2011; Peace and Norelli, 2009). Only Grimplet et al. (2004) have used proteomics to connect expressed genes (genomics and transcriptomics) with their related product (proteomics and metabolomics) in apricot.
Phenomics Concepts
The discipline of phenomics concerns the study of the phenome of a living organism (Houle et al., 2010). The phenome (classically called phenotype) can be defined as the set of observable traits in an individual resulting from the interaction between the genotype, the genetic information of an individual, and the environment (Fig. 1). The phenome defines the relationship of a portion of a genome with a specific trait of the individual (Alberch, 1991). This classical definition can be expanded according to new findings. The phenotype can be described in a holistic sense as the final expression of a genotype, taking into account the following: the transcription of DNA to RNA, including transcripts of unknown function (TUFs) with little protein coding capacity; the translation of the RNA to protein (the enzymes being the key proteins involved in the biological process); and the final interaction and expression of this proteome with the environment (Gingeras, 2011; Oti et al., 2008).
It is necessary to note the importance of accurate phenotyping (characterization of the phenotype) in terms of the power and performance of further genetic or genomic studies (Ingvarsson and Street, 2011). This accurate phenotyping can increase genetic gains obtained by breeding programs from a precision breeding approach (Collard and Mackill, 2008; Morgante and Salamini, 2003). Plant phenotypic variation is produced through a complex web of interactions between genotype and environment, and a “genotype–phenotype” map is inaccessible without the detailed phenotypic data that allow these interactions to be studied. Despite the importance of the phenotype, our ability to characterize phenomes lags behind our ability to characterize genomes (Houle et al., 2010). Phenomics should be recognized as an independent discipline in order to enable the development of accurate high-throughput phenotyping of the genotypes of interest.
We can classify the phenome (main phenotypic traits) studied in Prunus in accordance with the main objectives of breeding into three main groups: traits related to the increase of yield (flowering time, flower type, fruit size, flower compatibility, resistance to drought, and resistance to chlorosis); traits related to improvements in the quality of fresh and processed fruit (harvest maturity, peach or nectarine trait, flesh color, petiole gland shape, fruit quality, stone adherence to flesh, fruit shape, ground color, stone shape, stone adherence, color of skin, flesh color, resistance to cracking, kernel shape, softness of shell, kernel taste, twin kernel, nut shape, and juice color); and traits related to a decrease in production costs (resistance to Plum pox virus, resistance to curl leaf, Monilia and green aphid, resistance to frost, resistance to Pseudomonas, and resistance to Taphrina). Gass et al. (1996) have indicated up to 50 main phenotypic traits of interest in the different Prunus species (Fig. 2).
New Biological Challenges and Opportunities
In the new high-throughput context, there are new biological challenges and opportunities in applying the full range of omics sciences (including genomics, transcriptomics, proteomics, metabolomics, and phenomics) in the development of efficient marker-assisted selection strategies in Prunus to increase breeding gain. These opportunities are of special interest in the case of Prunus, where knowledge of the association between genes and agronomical traits is limited (Arús et al., 2005; Peace and Norelli, 2009).
It took nearly 25 years from the discovery of the structure of DNA (Watson and Crick, 1953) to the development of efficient methods for determining its sequence in the genome (Sanger et al. 1977). DNA sequencing (order of nucleotide bases in a DNA molecule) has changed our vision of plant biology and has played an important role in modern biological research. During recent years, however, the so-called next-generation sequencing technologies (high-throughput) for DNA and cDNA (DNA-Seq), have made DNA sequencing less expensive than in the past, changing DNA sequencing projects and causing a revolution in biological research. In addition, the third generation in sequencing technology, which is able to determine the base composition of single DNA cellule, has joined the race (Delseny et al., 2010; Jackson et al., 2011; Schendure and Li, 2008). Next generation DNA sequencing platforms can generate as much data in 1 day as several hundred traditional Sanger-type DNA capillary sequencers (Mardis, 2008; Schuster et al., 2008). This DNA-Seq technology allows for faster resequencing of different genotypes and species, assuming a reference-like genome exists, whereas de novo assembling does not (Jackson et al., 2011; Shendure and Li, 2008).
The development of new SNP markers for future uses in structural (molecular characterization) and functional (saturation of genetic linkage maps) genomics is an important application of genome resequencing using DNA-Seq (Jackson et al., 2011; Schuster et al., 2008) that is being applied to Prunus species (Tartarini et al., 2010; Verde et al., 2010, 2011). Recently, Ahmad et al. (2011) described the application of these new generation sequencing technologies to identify high frequency SNPs distributed throughout the peach genome. They discovered 6,654 SNPs distributed on all the genome scaffolds with ∼1 SNP/40,000 nucleotide bases. Alternatively, de novo sequencing of new genotypes from each Prunus species using DNA-Seq (de novo assembly) (Imelford and Edwards, 2009) to be used as reference in each species is another option for further new resequencing studies. The first whole genome resequencing and de novo assembly studies have been performed in prune (Dardick et al., 2011) using the reference peach genome.
On the other hand, in the past, sequencing was preceded by cloning using the Sanger methodology, and the data were therefore highly static. Today, high-throughput genome sequencing technologies are being applied to gene identification and the search for functional alleles in model plant species (Edwards and Batley, 2010; Jackson et al., 2011), although not yet in Prunus species. This newly obtained data should produce a revision in current information about gene prediction, annotation, and ontology.
RNA processing, the connection between DNA and proteins, is the authentic Gordian knot (a metaphor for an intractable problem solved by a bold stroke) of this gene expression. Translation of transcribed mature mRNA (including splicing events) into protein (one main entrance) together with the presence of noncoding rRNA and tRNA (another main entrance), and posttranscriptional and posttranslational regulation involving small noncoding RNAs (miRNA, siRNA, piRNA, or snoRNA) (several minor but critical entrances) are shown in Figure 3. A major drawback of the cDNA microarray approach for the study of this Gordian knot is that transcriptome profiling coverage is strictly limited to the probe sets available for specific hybridization in each species as we have described before. This classical microarray usually contained EST (cDNA from mRNA) previously developed by Sanger DNA sequencing technology (Aharoni and Vost, 2001; Martínez-Gómez et al., 2011; Wullscheleger and Difacio, 2003). For this reason, and thanks to the availability of a full genome sequence for some species, new tilling microarray platforms have been developed using different spaced oligonucleotides that span the entire genome of an organism (Comai et al., 2004; Yazaki et al. 2007). At this moment, the availability of a genome reference in Prunus can open new opportunities in the development of tilling microarray slides.
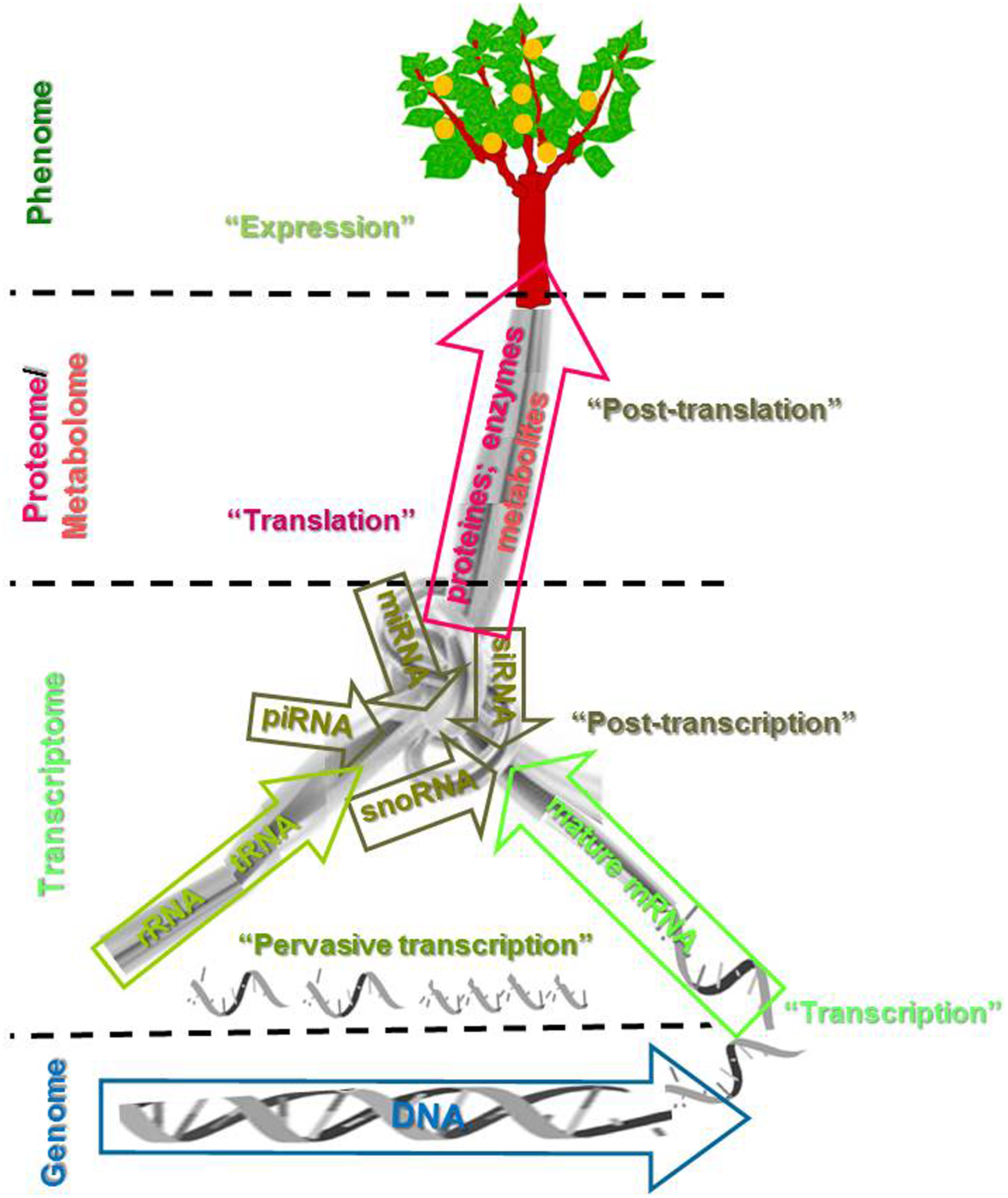
Schematic representation of the translation of coding RNA (mature mRNA including splicing events) into protein and metabolite synthesis, together with the presence of noncoding nonregulatory RNA (rRNA and tRNA), and posttranscriptional and posttranslational regulation with noncoding regulatory RNA (miRNA, siRNA, piRNA, or snoRNA).
New RNA-Seq technologies are poised to revolutionize our understanding of transcription and posttranscriptional regulation of RNA (Martínez-Gómez et al., 2011; Wang et al., 2009). RNA-Seq involves direct sequencing of cDNAs (from mRNA or miRNA) using high-throughput DNA sequencing technologies, allowing the level of transcription from a particular genomics region to be quantified from the density of corresponding reads (Flintoft, 2008). This technology represents the latest and most powerful tool for characterizing transcriptomes and continues to provide unparalleled insight into transcriptome complexity (Marguerat and Bähler, 2010; Wang et al., 2009).
The development of new gene catalogs in the different Prunus species through de novo assembly of transcriptomes is another important RNA-Seq application (Marguerat and Bähler, 2010; Zhao et al., 2011). Both new tilling microarray and RNA-Seq approaches can help solve this Gordian knot (cutting the Gordian knot).
Proteins occupy a prominent position in all biological systems, both quantitatively and qualitatively (Cozzone, 2010). As indicated before, however, proteomics is much more complicated than genomics, mostly because, whereas an organism's genome is more or less constant, the proteome differs from cell to cell and from one moment in time to another. This is because distinct genes are expressed in distinct cell types. This means that even the most basic set of proteins produced in a cell needs to be determined. The Prunus proteome is larger than the genome in the sense that there are more proteins (estimated at around 1 million) than genes (27,852 protein-coding genes described) (Peace and Norelli, 2009; Sosinski et al., 2010). Proteomics provides a much better understanding of a living organism or plant than genomics due to different factors, because the level of transcription of a gene gives only a rough estimate of its level of expression into a protein, with poor correlation between mRNA and protein abundance (Maier et al., 2009). An mRNA produced in abundance may be degraded rapidly or translated inefficiently, resulting in a small amount of protein. Although individual proteins can be readily connected to their encoding gene, connecting specific metabolites with their underlying genetic source is more difficult (Fridman and Pichersky, 2005; Schaucher and Fernie, 2006).
One of the most important new challenges for omics studies in Prunus is a large-scale identification and annotation of proteins and metabolites and their association with pomological traits, in essence, the biological meaning of the protein. New studies using high-throughput proteome (Cánovas et al., 2004; Neilson et al., 2010) and metabolome (Shi et al., 2011; Valdyanathan et al., 2005) analysis tools, including tandem mass spectrometry and gas chromatography coupled with mass spectrometry, can acquire sequence information from individual peptides by isolating them and then cataloging the fragment ions produced. These high-troughput analysis technologies should increase the protein and metabolite annotation in Prunus through the development of new amino acid databases. In addition, metabolite analysis can generate important information in applications that integrate plant sciences with nutrition and human health (Trethewey, 2007). This challenge is of special interest in the case of species whose protein and metabolite annotation is highly limited (Peace and Norelli, 2009). These results will make the search for the gene (part of the genome) responsible for an agronomic trait possible through the analysis of proteome and metabolome.
Phenotypic variation is produced through complex interactions (“genotype-phenotype”) that are inaccessible without the detailed phenotypic data that allow these interactions to be studied. In many cases, the selection and evaluation of phenotypes have been poorly developed in the experimental design of genetic and genomic studies (Houle et al., 2010). This could be the reason for the lack of accuracy in the obtained results in terms of the development of suitable markers for assisted selection in breeding programs. This question is of special interest in the case of Prunus, where truly applied marker-assisted selection strategies are highly limited (Arús et al., 2005; Dirlewanger et al., 2004; Martínez-Gómez et al., 2007; Peace and Norelli 2009).
The lack of consistency between phenomics studies limits the utility of collected data. Standardized phenotyping is a challenging approach, requiring a well-established international network of cooperation among researchers, with the coordination and agreement necessary to align the characterization of Prunus germplasm collections for further omics studies (Evans et al., 2010; Ingvarsson and Street, 2011). This is one of the main objectives of the RosBreed consortium (http://www.rosbreed.org), an international consortium created to foster research, infrastructure establishment, training, and extension for applying efficient marker-assisted selection strategies in the Rosaceae family. In this regard, Kolker (2010) recently indicated that the first decade of the 21st century has shown itself to be a time of ever-intensifying globalization and human connectivity, allowing for increased knowledge and technological progress.
Additional biological opportunities for using Prunus omics sciences include the high level of synteny among Prunus genomes (Dirlewanger et al., 2004; Jung et al., 2009). This synteny has also been described within the Rosaceae family (Arús et al., 2006; Illa et al., 2011; Shualev et al., 2008). This interesting approach was described for the first time by Vavilov in his law of homologous series in variation (Vavilov, 1922), formulated while studying the phenotypes of different plant species. Homology has been described as one of the most important concepts in plant biology, and also plays an important role in molecular and developmental biology in taxonomy, phylogeny, and evolutionary studies (Brigandt and Griffiths, 2007). In addition, syntenic linkage groups can result in similar alleles and homologous genes including ortholog genes (encoding protein with the same function) and paralog genes (encoding protein with the related but nonidentical functions) (Hammer and Schubert, 1994; Shualev et al., 2008). The high degree of synteny within the Prunus genus and the Rosaceae family should increase the efficiency of the results and conclusions obtained with the use of new high-throughput omics disciplines and their transportability across species and even genus.
Finally, the existence of biases is another new important challenge in attaining knowledge from the new high-throughput omics tools, which can affect the final obtained conclusions from a biological point of view. These biases are due to biological, methodological and statistical effects that in particular affect comparisons between data obtained with the different high-throughput genome, transcriptome, proteome, and metabolome analysis technologies. There have been several studies demonstrating the biases inherent to the DNA-Seq and RNA-Seq method as well as variation in results across protocols and platforms. Researchers have begun innovating methods to correct these biases and variances, but until now, most correction methods have involved the use of bioinformatics models for partial correction (Dohm et al., 2008; Hansen et al., 2010; Risso et al., 2011; Schwartz et al., 2011).
These biases can affect expression estimates, and it is therefore important to correct them in RNA-Seq analysis in order to have an accurate biological interpretation of the data (Roberts et al., 2011). In addition, Risso et al. (2011) described important biases related to GC content, demonstrating the existence of strong sample-specific GC content on RNA-Seq read counts, which can substantially bias differential expression analysis. On the other hand, Fernandes et al. (2010) applied the recently developed information–theoretic measures for the characterization and comparison of protein–protein interaction networks. These authors presented the results of a large-scale analysis of these networks. By quantifying the methodological biases of the experimental data, they defined an information threshold above which networks may be deemed to comprise consistent macroscopic properties, despite their small, microscopic overlaps.
New Philosophical Challenges
The discovery of the structure of DNA (Watson and Crick, 1953), with the exact definition of the gene and the central dogma of molecular biology (genetic information can be transferred among nucleic acids, and from nucleic acids to proteins, including DNA replication, transcription to RNA, and translation to protein expressed in the phenotype) (Crick, 1970; Pukkila, 2001) (Fig. 1), stands as the most important contribution to life sciences, including breeding, in the 20th century, in what has been called the genetic era. This discovery revolutionized the scientific paradigms of genetic studies in living organisms, including plants, shifting genetics from a phenomenological and statistical science to a molecular and chemistry-based science. Issues related to the mechanisms of genetic transmission, segregation, mutation, and expression of characters were reformulated in chemical and molecular terms. As a result of the DNA sequencing and analysis technologies (Sanger et al., 1977) that became available later in the genomics era, the frontiers of plant breeding have been dominated by genomics for the past 30 years. The sequencing of the complete genome of the peach represents the main landmark of the genomics era in Prunus species (Sosinski et al., 2010). At this moment, in the postgenomic era (Leader, 2005), we are looking at another scientific revolution and paradigm shift similar to the discovery of the DNA structure in the sixties and of DNA sequencing in the eighties.
The advent of these high-throughput technologies has led to previously encountered data management issues for public databases, resulting in new challenges for bioinformatics and biologists alike (Barga et al., 2011; Morrison et al., 2006b) and also contributing to the scientific paradigm shift. The rapid expansion of omics tools reflects a reinforced holistic point of view in understanding life, the expansion and differentiation of relatively simple segments of life into various encapsulated biological domains, and technical advancement in computer science, which has allowed for the integration of complex biological data through bioinformatics (Mochida and Shinozami, 2011). These extraordinary opportunities are limited by challenges in data analysis. There is an enormous gap between the capacity of an instrument to produce millions of bits of raw data per day and the ability to analyze and translate this data into applications in omics research. In this respect, bioinformatics have played the most important role in the rapid growth of omics and have effectively and completely changed conventional biology, laying a new philosophical foundation for the study of life (http://ww.omic.org).
This new context with precise and abundant information contradicts nuclear physicist Ernest Rutherford's famous sentence: “All science is either physics or stamp collecting.” In today's perspective, biological sciences can be considered real sciences. The amount of data generated in molecular biology studies at this moment is similar to the data obtained in physics studies, except for the cases in physics involving models that are much more static than in bioscience cases. The current situation in this new postgenomic, high-throughput context can be summarized by the words of poet William Blake (1757–1827): “The road to excess leads to the palace of wisdom…for we never know what enough is until we know what is more than enough.” We are currently in a situation in which we know what “more than enough” is in terms of data. A better connection between different specialists is thus necessary to exploit the massive amounts of data generated, converting this apparent excess of information into knowlege. Just as the beginning of the 20th century was the period of the great launch of physics, during which the social impact of this science, also called big science or technoscience (Feyerabend, 1962), increased, we can label this current period in the 21st century as the epoch of the great launch of bioscience (molecular biology or omics science) as the big science in relation to the development of high-throughput technologies.
This situation presents common philosophical challenges regarding the model of scientific change. We are facing a revolution with the use of the new high-throughput analysis techniques that may mean a change of scientific paradigm in genetics and genomics concepts and theories in plants and the less known Prunus. Another important question is how to evaluate scientific progress in this new postgenomic context. Finally, an additional philosophical challenge is the incommensurability of the new and old omics theories in the new high-throughput analysis context.
Regarding the change of the scientific paradigm, theories and data obtained using the new high-throughput omics disciplines can produce further questions in Prunus biological studies and breakthroughs with former theories. The challenge is to consider the availability of the new high-throughput technologies as either a cumulative process of improvement of the former theories, or as a revolution in which old theories are being distorted and replaced by new ones. Neopositive theories (Suppe, 1979) indicate that science is a cumulative process in which new theories (such as the new omics disciplines) do not replace previous theories, but can contribute to improving or enhancing these theories. In addition, Lakatos (1974) indicates that science is a gradual and cumulative process, and Nagel (1961) understands the cumulative process of improvement of theories as a process of reduction of theories—in other words, theories are integrated into larger ones. For Laudan (1977), scientific change is also a rational and progressive process evaluable through analysis units (research traditions). According to Popper's logic (Popper, 1959), however, science advances because old theories are distorted and replaced by new ones that contradict them. In addition, Popper (1959) and Feyerabend (1962) stated that science progress occurs in a state of permanent revolution with abrupt changes. The old theories are evaluated in hard competition with rival theories in a process analogous to Darwinian natural selection.
In an intermediate situation, Kuhn (1962) agrees with Popper in that scientific process is revolutionary and not cumulative, but says that periods of extraordinary science (revolution) are discontinuous and among them there are periods of normal science (paradigmatic periods). These paradigmatic periods change during the extraordinary science periods (such as now, with the availability of new high-throughput technologies) to new paradigmatic periods. Periods of normal science make up the bulk of scientific research, developed under the dominion of a paradigm. This paradigm is a scientific breakthrough or a theoretical model that includes a theory and some exemplary applications for an experiment that addresses some outstanding problems. Periods of stability and normal science are necessary to have a period of extraordinary science or revolution. Normal science provides extremely valuable progressive and cumulative progress (similar to the neopositivist theory), but it is during the revolution, when one paradigm shifts to another, that a real advancement in science occurs. Khun's approach differs from the Darwinian struggle of Popper's scientific theories.
Regarding the evaluation of scientific progress, the challenge is to consider the availability of the new high-throughput omics disciplines as a progressive approach to truth or as a distance from the starting point of the study. For Popper (1959), scientific progress is the search for truth as a search for a useful theory. In this progressive approach to the truth, called degree of plausibility, science seeks the truth in a search that never culminates and is a cumulative process toward a growing degree of likelihood (content of true/false) regarding the truth. Scientists are seekers but not owners of truth. To that effect, we can recall Zeno of Elea's (490 BC–430 BC) paradox of Achilles and the tortoise: in a race, the quickest runner can never overtake the slowest, because the pursuer must first reach the point whence the pursued started, so that the slowest must always hold a lead. In this respect, John Maddox (1925–2009) assured that each new discovery expands our knowledge and at the same time enlarges the boundaries of our unknowns. On the other hand, Kuhn (1962) defends scientific progress not as an approach to truth, as Popper says, but as a distance from the starting point.
In an intermediate situation, Laudan (1977) takes problem solving as a criterion for estimation of scientific progress. With the new high-throughput data, we can delve into the DNA base level in attempts to solve macrobiological questions. We are continually deepening our knowledge, distancing ourselves from the starting point of the study and continually arriving closer to the truth about the molecular control of agronomic traits, but never definitively solving the questions (given the impossibility of finding the truth). Scientific evidence does not produce certainty but rather probability, which is under constant review. The only way to continue is to put all the available information together to produce an important advance in a short period of time, thus encouraging Achilles to overcome the tortoise.
These new high-throughput approaches should also present problems of incommensurability of the new theories or data. Incommensurability must be understood as the difficulty or impossibility of comparing different data or theories and the lack of inferential connections. Scientific theories are described as commensurable if one can compare them to determine which is more accurate; if theories are incommensurable, there is no way to compare them to each other in order to determine which is more accurate. Incommensurability is a deep philosophical problem described by Kuhn (1962) and Feyerabend (1962), and it is probably one of the most important discussions in the philosophy of science of the 20th century. Khun (1962) indicated that “when paradigms change, the world changes with them.” Such incommensurability of theories in physics was described by Feyerabend (1962) in the cases of Niels Bohr's (1885–1962) new theory of the atom and Albert Einstein's (1879–1955) new theory of relativity. In addition, Laudan (1977) maintained that research traditions existing at a given time may be classified according to the progress they achieve, although they are incommensurable in terms of way they purport to explain reality.
The lack of inferential connections and incommensurability between theories and data should be the case of the data obtained with classical genetics (including genetic linkage and classical QTL analysis); classical genomics (Sanger sequencing of DNA or cDNA from RNA, and gene prediction, annotation, and ontology); classical transcriptomics (using microarray); and the data obtained with new high-throughput genome (DNA-Seq), transcriptome (tilling microarray, RNA-Seq, expressed QTLs—eQTL-), proteome (peptide mass fingerprinting), and metabolome (gas chromatography) analysis technologies. In addition, another important case in point at the moment involves the use and comparison of the large amount of data available on DNA (genomics) markers linked to agronomic traits (phenome), obtained by linkage analysis (statistical methods) and physical mapping (Sanger sequencing), in the new high-throughput sequencing of genome and transcriptome context at the base pair level.
Conclusions
The recent sequencing of the complete genome of the peach together with the availability of high-throughput genome, transcriptome, proteome, and metabolome analysis technologies offers new challenges and opportunities for Prunus breeders in what has been called the postgenomic era. These technologies will certainly generate a large amount of data and open a range of options of study. A combination approach using a full range of omics sciences and integrating their outcomes should now be an effective strategy for clarifying a wide range of biological processes to improve Prunus breeding and productivity, resulting in the development of efficient marker-assisted selection strategies to increase selection gains. From the biological point of view, new biological challenges and opportunities for the application of these omics sciences in Prunus breeding include genome resequencing, deep knowledge of RNA regulation at the transcriptional and posttranscriptional level, increased protein and metabolite annotation, and improved accuracy and standardization in genotype phenotyping. These approaches allow for the search for genes (part of the genome) responsible for agronomic traits through the analysis of the phenome, metabolome, proteome, or transcriptome. Additional biological opportunities include the high level of synteny between Prunus and Rosaceae genomes. On the other hand, from a philosophical point of view, we are facing a revolution in omics analysis techniques that may mean a change in paradigm in terms of scientific progress and a change of paradigm with regard to the concepts of Prunus genetics and genomics. Finally, an additional philosophical challenge is the incommensurability of the new and old omics theories in the new high-throughput analysis context.
Footnotes
Acknowledgments
This study was supported by the project “Gene expression analysis of the resistance to Plum pox virus, PPV (sharka) in apricot by transcriptome deep-sequencing (RNA-Seq)” of the Spanish Ministry of Science and Innovation (Project reference AGL2010-16335).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.