
Abstract
Abstract
Pseudomonas syringae pv. phaseolicola is a major plant pathogen causing halo blight disease and has world-wide importance. The emerging post-genomics field of agrigenomics, together with the availability of whole genome sequences of a number of pathogens and host organisms, offer the promise for identification of potential drug targets using sequence comparison approaches. On the other hand, lack of gene expression data for most of the phytopathogenic microbes still remains a formidable barrier. The present study aimed at the prediction of drug targets in Pseudomonas syringae pv. phaseolicola by exploiting the knowledge of Codon Usage bias for gene expression subtractively, supported by gene expression analysis and sequence comparisons. Based on screening of the Database of Essential Genes using blastx, 158 of the total 5172 genes of P. syringae pv. phaseolicola were enlisted as vitally essential genes. Similarity search for these 158 essential genes against available host–plant sequences (Phaseolous vulgaris) led to the identification of homologues of 21 genes in the host genome, thus leaving behind a subset of 137 genes. Expression analysis of these 137 genes using RSCUgene, validated by microarray gene expression data suggested 22 genes had higher expression levels in the cell, and therefore their products have been identified as putative drug targets. The gene ontology analysis of these 22 genes revealed their indispensable roles in pivotal metabolic pathways of P. syringae pv. phaseolicola. Upon comparison of the sequences of these genes with other soil bacteria, we identified two genes that were unique to P. syringae pv. phaseolicola. The products of these genes can potentially be utilized for drug development so as to control the halo blight disease and thereby accelerate translation research in the nascent field of agrigenomics.
Introduction
Extensive genetic and genomic resources are available for P. syringae pv. phaseolicola (http://www.pseudomonas-syringae.org/psp_home.html; Joardar et al., 2005), providing an ideal opportunity to elucidate mechanisms of pathogenecity, plant host susceptibility/resistance, and virulence determinants (Godfrey et al., 2010). The implications will also be useful for discovery of agricultural bactericide discovery (Hernandez-Morales et al., 2009). In recent years, various computational methods have been developed using either protein structure (Rajendran et al., 2012) or genome-based information (Bakheet and Doig, 2010; Katara et al., 2011) for prediction of drug targets in various human pathogenic bacteria. However, no such efforts for predicting drug targets in phytopathogens have been reported, primarily because of the unavailability of gene expression data from diseased conditions. The efficacy of current bactericides and fungicides can be increased in the future without affecting the yield of crops if they are designed on meticulous approaches based on gene expression data. Here we attempt to predict drug target for P. syringae pv. phaseolicola, a well-known phytopathogenic bacteria causing halo blight disease in Phaseolus vulgaris and responsible for huge economic loss worldwide. For such analysis, we developed an innovative approach using sequence similarities and codon usage biasing, subtractively. Following Katara et al. (2011), we presume that drug targets are essential for the growth and viability but highly selective as well for the pathogen with respect to the host. We have also considered their availability for interaction with the drugs (biased for their expression). To the best of our knowledge, such approaches have for the first time been applied for predicting drug targets on a phytopathogen.
Materials and Methods
Sequence resources
Gene sequences of the P. syringae pv. phaseolicola were downloaded from GenBank (ftp://ftp.ncbi.nlm.nih.gov), and available sequences of its host plant Phaseolus vulgaris and nitrogen fixing bacteria Rhizobium etli were used from online resources as database against blast using NCBI-blast facilities (http://www.ncbi.nlm.nih.gov/blast).
Identification of genes of interest
The complete sets of genes of the pathogen were subjected to blastx (Altschul et al., 1997) against the essential genes of γ-proteobacteria available at Database of Essential Genes (DEG; http://tubic.tju.edu.cn/deg/; Zhang and Lin, 2009). A random expectation value (E-value) cut-off of 0.001 and a minimum bit-score cut-off of 100 were used as the baseline to identify the housekeeping genes in the pathogen. These housekeeping genes belonging to the pathogen were subjected to blastn against their host–plant EST-sequence at the NCBI server (http://blast.ncbi.nlm.nih.gov/Blast.cgi). The homologs between pathogen and their host were excluded and the lists of nonhomologs were compiled and considered as genes of interest.
Prediction of Relative Synonymous Codon Usage
Complete sets of essential genes that did not share significant similarities with the ESTs of the target host were subjected to CAIcal (Puigbo et al., 2008), using Eubacterial genetic code to predict nucleotide composition, relative synonymous codon usage (RSCU), and codon usage (Agarwal and Grover, 2008; Hassan et al., 2009; Sharp and Li, 1987). These RSCU were further subjected to geometric mean to predict RSCUgene (Relative Synonymous Codon Usage for complete gene) for all genes [Eq. 2].
Where fobs (xyz) is observed frequency of a codon xyz for ‘X’ amino acid, fexp (xyz) is expected frequency (if all synonymous codon for ‘X’ amino acid were used equally) for codon xyz, L is the length, in codon, of a particular gene.
In order to measure the nonuniformity of codon usage within a group of synonymous codon for a gene, Effective number of codon (Nc) (Fuglsang, 2004; Wright, 1990)
where Codonmax is a number of maximum available codon except stop codon, and Codongene is a number of codons used in a gene.
Expression of those genes that were suggested as overexpressed genes by the help of Codon Usage biasing was also validated against cDNA microarray data observations, available at Gene Expression Omnibus (GEO) database (www.ncbi.nlm.nih.gov/geo/) under series GSE14625, GSE14983, and GSE14998 (Hernandez-Morales et al., 2009).
Annotation
The finally selected genes (probable drug targets) were subjected to Blast2GO (Conesa et al. 2005) to check their respective metabolic pathway, cellular and functional annotation.
Subtraction of genes shared by nitrogen fixing bacteria
In order to eliminate the undesired targeting of symbiotic R. etli, the finally selected genes from P. syringae pv. phaseolicola were compared for sequence similarities with protein sequences of R. etli, using NCBI-blastx (Altschul et al. 1997).
Results
Genes of interest
Similarity search for a complete set of genes in DEG led us to identify 158 essential genes for P. syringae pv. phaseolicola. However, 21 of these were found sharing considerable sequence similarity with nucleotide sequence of P. vulgaris and were thus eliminated from further analysis. The remaining 137 genes built our collection of ‘genes of interest’.
RSCU and Nc of genes of interest
All the genes could be classified into three categories: high expression (RSCUgene ≥1.1), moderate expression (RSCUgene ≥0.9) and low expression (RSCUgene ≤0.89). Evidently, only 16% (22 genes) of the genes fell into first category of high expression (Table 1). All of these genes also had gene expression ratio (Log2) ranging between+0.316 and+1.533, based on microarray data analysis (Table 1). A fairly equal number of remaining genes could be mapped to the remaining two categories of moderate and low expression.
Mean of gene expression values (test/normal) from cDNA microarray experiments (A: GSE14625, B: GSE14983, and C: GSE14998). All values are in positive “+” scale, thus indicates high expression of these genes.
For all the genes analyzed, Nc values ranged from 31 to 59, but for highly expressed genes, it ranged from 31 to 45 (Table 1). Pattern analysis for Nc and RSCUgene suggested that there is a strong negative correlation between Nc and RSCUgene (Fig. 1) with Pearson coefficient of correlation (r) being −0.89.
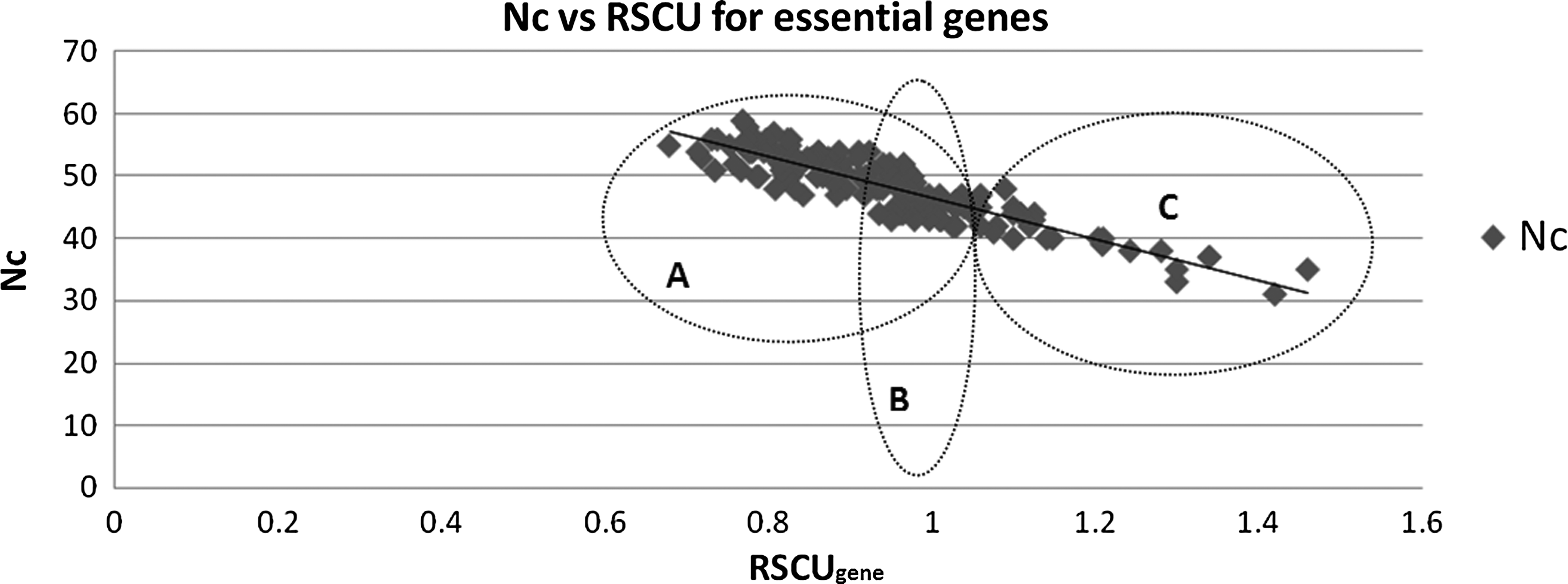
Relation between Nc and RSCUgene.
Gene ontology and subtractive comparative genomics against R. elti
The 22 genes shortlisted as targets were further subjected to gene ontology analysis using blast2GO (Table 2). More than 45% of the shortlisted genes were found coding for the proteins involved in metabolic processes of biopolymers, including cellular metabolic process, macromolecule metabolic process, and nitrogen compound metabolic process (i.e., nucleic acids and proteins). The rest of the gene products were found associated with some other vital biological processes, including biosynthesis and biogenesis processes (Fig. 2).

Summary of the biological process in which products of the predicted targets are involved.
In correspondence with the above results, the highest numbers of gene products were found carrying binding activity. Around one-fourth of the genes encoded structural constituents of the ribosome (Fig. 3). Most of the gene products were mapped as a part of the cell or organelle, and the rest of products mapped to intracellular locations (Fig. 4).
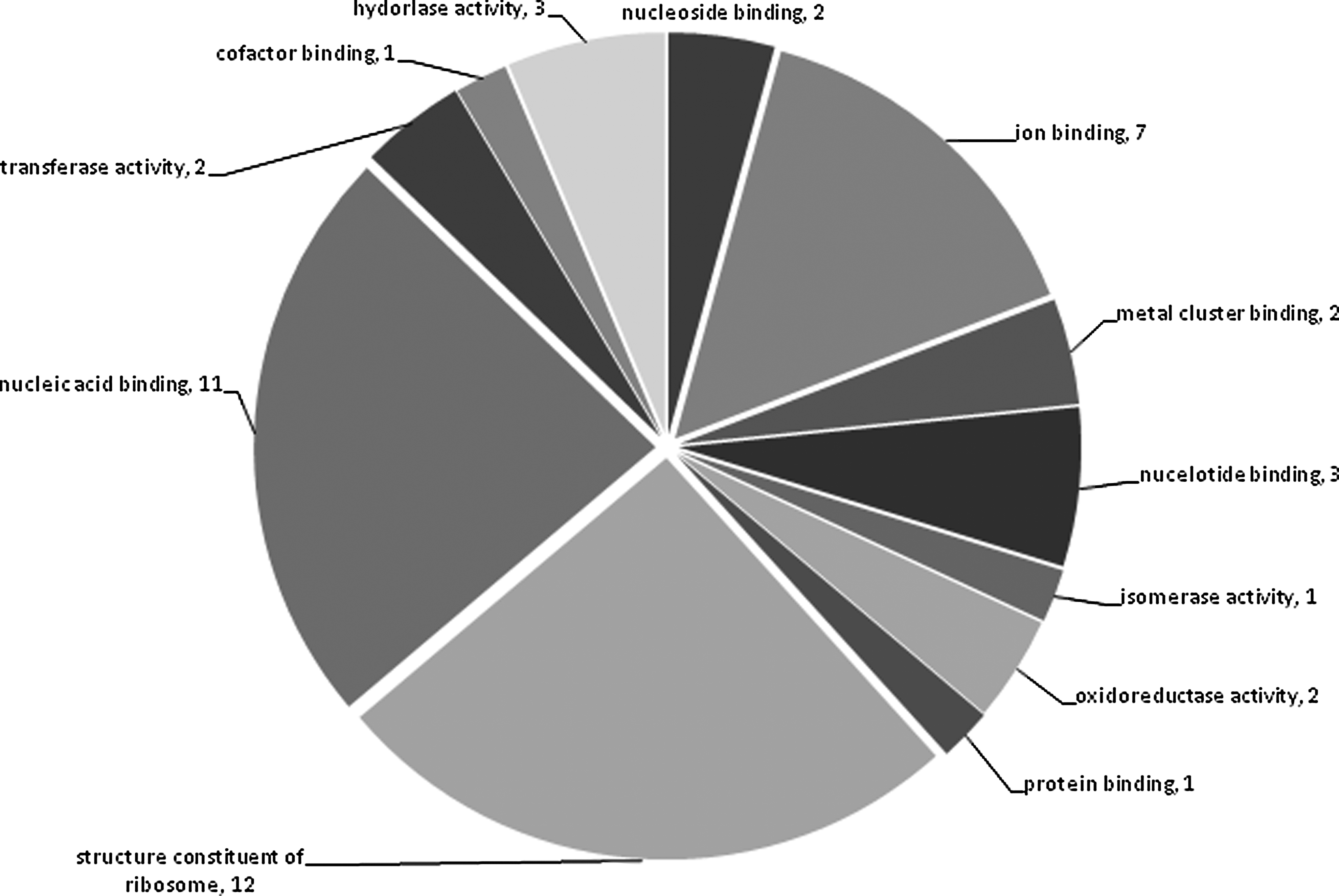
Molecular function of the products of the predicted drug target genes.
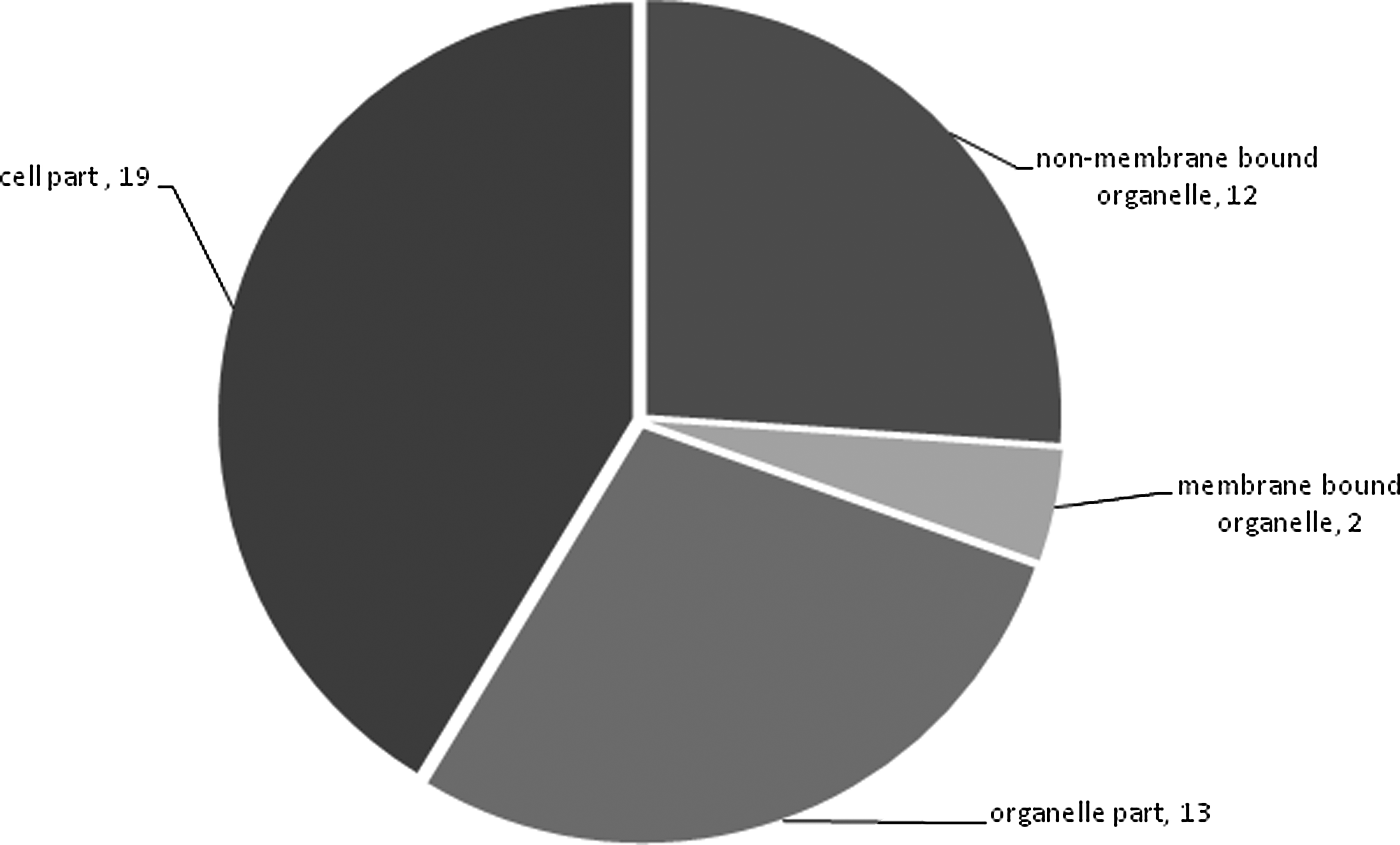
Summary of the cellular components where product of the predicted target genes are localized.
Ten pathways, as listed in Table 2, were identified in which three of the ‘target gene products' participate. For five of the gene products (encoded by PSPPH_1310, PSPPH_4343, PSPPH_0861, PSPPH_1964, and PSPPH_1976) no enzyme codes were found, and for the remaining, no corresponding data could be curated from KEGG database. Whereas 20 of the shortlisted gene homologs could be traced in R. elti, for two gene products (PSPPH_1310 and PSPPH_0723), no homolog could be found.
Discussion
The drug target identification is an important and sensitive first step in the drug discovery process that must satisfy various selection criteria to pass for the next stage (Hefti, 2008; Lipinski et al., 2001). Identification of microbe-specific ‘essential’ proteins that can be targeted while designing drugs is a possible way to control the crop diseases. We used gene sequence information for prediction of drug targets using subtractive genomics in P. syringae pv. phaseolicola, which was followed by a codon usage approach to evaluate the gene expression level. We found that the combination of these two approaches (sequence pattern and codon biasing) provides a good method to find drug targets. The combined approach used here is different from earlier methods (Bakheet and Doig, 2010; Katara et al., 2011; Sakharkar et al., 2008) and also uses a second subtraction step to avoid accidental targeting of ‘friendly species'. Extensive literature surveys suggest that such methods have never been applied earlier for phytopathogens and plant hosts.
On the lines of Sakharkar et al. (2008) and Katara et al. (2011), 137 genes that are essential for P. syringae pv. phaseolicola, and do not share any significant similarity with the host genes, were identified as ‘genes of interest’. In addition to being a gene essential and unique to the pathogen, it also needs to have high expression to be of interest to plant pathologists. A highly expressing gene would code for a sufficiently high number of protein molecules available as drug targets. A protein, against which a drug is designed, should be available in sufficient stoichiometric amounts in the cells at all time points across the diversity of life stages and stress conditions. We determined 22 genes as highly expressing genes based on RSCUgene (≥1.1) analysis and verified based on microarray data analysis (Table 1). Analysis of available cDNA microarray data for expression of genes of P. syringae pv. phaseolicola under series GSE14625 (response to bean pod extract), GSE14983 (response to apoplastic fluid of bean leaf), and GSE14998 (response to bean leaf extract) (Hernandez-Morales et al., 2009), verified the theoretical statistical results for these 22 genes obtained through RSCU analysis (Table 1), and thus all of these can be used as effective targets for control of P. syringae pv. phaseolicola.
An effective number of codons (Nc) in any given gene signifies the use of a specific number of synonymous codons for their respective amino acids. When all the sense codons are used randomly, Nc has a value of 61. Similarly, it takes an extreme value of 20 when only one synonym is used for each amino acid. Thus, the lower the Nc value, the greater are the chances of codon bias and vice versa (Hassan et al., 2009). In the present study, a strong negative correlation was obtained between RSCUgene and Nc, suggesting that both these tests in combination can be used as a powerful tool for prediction of gene expression levels, especially in cases where microarray data are not available. We have further been successful in annotation of metabolic pathways for three of the 22 enzymes/proteins identified as drug targets (Table 2). Enzymes such as shikimate kinase and nucleoside diphosphate kinase, because of their indispensable utility being involved in more than one pathway, are ideal drug targets (Saidemberg et al., 2011).
It must be remembered that a drug designed to target an enzyme, which is universal in nature, can also lead to the elimination of friendly and symbiotic microbial species such as Rhizobium sp., in addition to the pathogen. In the present context, however, it must be remembered that for the control of P. syringae pv. phaseolicola, drug must be applied to seeds (Arnold et al., 2011), while the friendly and symbiotic R. etli to Phaseolus vulgaris inhabits the rhizosphere. Nevertheless, in order to further eliminate the threats of accidental targeting of R. etli by inappropriate and alternative modes of drug application (like spraying on standing crop), we compared probable target genes through NCBI_blastx with the proteome of R. etli. For two proteins out of 22 (i.e., scaffold protein and FKBP-type peptidyl-prolyl cis-trans isomerase), no significant matches were obtained between P. syringae pv. phaseolicola and R. etli.
Our results thus provide a starting material for the future discovery of drugs against P. syringae pv. phaseolicola. Twenty-two drug targets have been suggested. Considering P. syringae pv. phaseolicola mediated halo blight is a seed borne disease, seed disinfection is the most efficient method for providing pathogen free seeds of P. vulgaris. Drugs targeting scaffold protein and FKBP-type peptidyl-prolyl cis-trans isomerase can be exploited for designing drugs to be applied on standing crops. Though experimental validation of all these targets is still recommended, we believe that both the results, as well as methods discussed above, are likely to find importance among the scientists actively participating in agrigenomics translational research.
Footnotes
Acknowledgments
The authors acknowledge the DBT center for bioinformatics facility at Department of Bioscience and Biotechnology, Banasthali University, Banasthali, India, for providing essential facilities for completion of this research work. The authors also wish to thank unknown reviewers and the handling editor for constructive criticism, thereby contributing to evolution of this manuscript to its present state.
Author Disclosure Statement
No competing financial interests exist.