
Abstract
Abstract
Network-biology inspired modeling of interactome data and computational chemistry have the potential to revolutionize drug discovery by complementing conventional methods. We consider asthma, a complex disease characterized by intricate molecular mechanisms, for our study. We aim to integrate prediction of potent drug targets using graph-theoretical methods and subsequent identification of small molecules capable of modulating activity of the best target. In this work, we construct the protein interactome underlying this disease: Asthma Protein Interactome (API). Using a strategy based on network analysis of the interactome, we identify a set of potential drug targets for asthma. Topologically and dynamically, v-src sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog (SRC) emerges as the most central target in API. SRC is known to play an important role in promoting airway smooth muscle cell growth and facilitating migration in airway remodeling. From interactome analysis, and with the reported role in respiratory mechanisms, SRC emerges as a promising drug target for asthma. Further, we proceed to identify leads for SRC from a public database of small molecules. We predict two potential leads for SRC using ligand-based virtual screening methodology.
Introduction
Asthma, a common chronic inflammatory medical condition of the airways, is characterized by variable and recurring symptoms: lung inflammation, reversible airflow obstruction, and enhanced airway responsiveness to a variety of environmental stimuli. Numerous asthma susceptibility genes have been identified from linkage, candidate gene association, genome-wide association studies, and microarray expression studies. In most of these approaches, large-scale molecular interaction details have not been included. Computational methods of protein interactome network analysis could be useful in understanding complex cellular processes (Salwinski, 2003). Hence, in addition to the classical techniques of target gene identification, protein interactome analysis could be used as an important strategy for the identification of novel drug targets.
Airway remodeling is one of the major hallmarks of asthma. Increased airway smooth muscle (ASM) cell mass due to the proliferation of ASM cells characterizes chronic severe asthma. In the asthmatic airway, ASM cell mass is one of the characteristic structural changes that lead to airflow obstruction (Davies et al., 2003). In asthmatic conditions, ASM hyperplasia and hypertrophy are observed in the airway wall remodeling (Ebina et al., 1993).
Experimental studies (Russello, 2004) have shown that c-SRC tyrosine kinase is upregulated in many types of human tumors (Summy and Gallick, 2003; Tsygankov and Shore, 2004). Apart from its role in cancers, SRC is reported to be necessary for the respiratory mechanisms associated with asthma (Krymskaya et al., 2005a), by promoting the ASM cell growth and migration that occur in airway remodeling, making it a promising drug target. SRC has also been reported to be involved in the regulation of ASM through the inhibition of p42/p44 MAPK (Conway et al., 1999; Rakhit et al., 1999). PP1, a pharmacological inhibitors of SRC, has also been reported to be potentially involved in regulating smooth muscle cell growth and migration (Waltenberger et al., 1999). It has been reported that inhibitors of SRC can modulate thrombin-induced ASM cell proliferation (Tsang et al., 2002). These reports lend credence to the key role of SRC in asthma.
Kinases make use of adenosine triphosphate (ATP) for phosphorylation of a specific Tyr/Ser/Thr residue in proteins. Hence, ATP-competitive small molecules are of interest as potential inhibitors of kinases. Structural studies of kinase complexes have revealed the presence of a clustered residues pattern and conserved hydrogen bond interactions around the active site, providing a basis for the understanding of ATP binding and competitive inhibitors (Zheng et al., 1993a, 1993b). This hydrogen bond network has been widely exploited to develop a novel chemical series of ATP-competitive kinase inhibitors (Beattie et al., 2003; Breault et al., 2003; Engh et al., 1996; Hennequin et al., 1999; Laird and Cherrington, 2003; Palmer et al., 1997; Plé et al., 2004; Schulze-Gahmen et al., 1996; Tong et al., 1997; Traxler et al., 1996, 1997; Wilson et al., 1997).
In the SRC kinase complex (PDB ID: 2SRC), the adenine moiety of adenosine monophosphate (AMP) is anchored to Glu339 and Met341 by hydrogen bonds. This hydrogen bonding pattern could be exploited to identify potent inhibitors for SRC. Several c-SRC kinase inhibitors have been identified to date, including dihydropyrimido-quinolinones (Dow et al., 1995), pyrrazolo-pyrimidines (Hanke et al., 1996), 4-anilinoquinazolines (Myers et al., 1997), and others (Klutchko et al., 1998).
Virtual screening is an important in silico technique that complements traditional high-throughput screening for novel pharmaceutically active compounds (Oprea and Matter, 2004; Shoichet, 2004). A number of approaches are used for searching databases of small molecules to discover novel lead compounds, depending on the availability of the crystal structure (Kitchen et al., 2004; Lengauer et al., 2004). Ligand based pharmacophore modeling is implemented to search for molecules with chemical features necessary for specific drug-receptor interactions.
In our studies, we construct the protein interactome underlying molecular mechanisms of asthma: Asthma Protein Interactome (API). By graph-theoretical analysis of API, we identify SRC as one of the most central proteins in the interactome. Further, we identify leads for SRC tyrosine kinase, using ligand-based pharmacophore modeling, from the ZINC database (Irwin and Shoichet, 2005), that may have therapeutic potential for asthma. We have verified the reliability of these lead molecules by flexible docking experiments. Final leads from our screening results are promising candidate molecules, which could be used for further evaluation.
Materials and Methods
Towards our goal of identification of potent targets and leads for asthma, we adopted the following procedure: (1) compilation of candidate asthma genes; (2) construction of the API by conjugating candidate genes with the Human Protein Reference Database (HPRD) (Keshava Prasad et al., 2009); (3) identification of therapeutic drug targets central to topology and dynamics by network analysis of the protein interactome; (4) construction of a ligand-based pharmacophore model for SRC tyrosine kinase, the most central drug target identified in API; and (5) virtual screening of drug-like molecules against SRC kinase.
Compiling candidate asthma genes
Asthma is a complex disease characterized by various interlinked factors. Changes at the genetic as well as the transcription level may contribute to the disease state in intricate ways. In order to build an inclusive and more comprehensive interactome, we compiled asthma candidate genes from DNA-level disease association data and transcript level changes (differential expression data), in addition to the literature-curated data. The Online Mendelian Inheritance in Man (OMIM) database (McKusick-Nathans Institute of Genetic Medicine, 2011), a database that catalogs literature for known diseases with associated genetic components, was searched with the keyword “asthma,” and the disease-associated genes identified were curated manually. We intended to obtain literature representing common major asthma-susceptible genes, including genes identified by genetic association, and case-specific genes, from PubMed (http://www.ncbi.nlm.nih.gov/pubmed), a repository of life sciences and biomedical literature. Hence, PubMed was searched with the keyword “asthma,” along with the terms “genetic association” or “case-control study” in order to retrieve most of the asthma-related published research papers until January 31, 2012. In addition, asthma candidate genes identified through four earlier literature searches, based on published research papers, were also included (Hoffjan et al., 2003; Ober and Hoffjan, 2006; Zhang et al., 2008) in our analyses, to make the literature search more exhaustive and complete. Further, we also included asthma candidate genes known from differential expression studies during human lung development (Melén et al., 2011). Asthma genes from expression data were considered to be significantly differentially expressed if they obtained an adjusted p value of <0.05 in the linear regression model. Finally, we created a list of unique asthma genes using all three of these approaches (See Supplementary Fig. S1 at www.liebertpub.com/omi).
Constructing the Asthma Protein Interactome
Genes encode proteins, functional macromolecules that regulate biological processes. By integrating experimental interactions data and computational methods, a relevant protein interactome could be constructed and subjected to analysis. We constructed protein–protein interaction (PPI) network-representing processes involved in the asthma disease condition. We created two types of PPI networks, a core network and an extended network. First, we built a human protein interactome using the HPRD (April 2011 release), a database containing manually-curated scientific information pertaining to human PPIs. The HPRD protein interaction data was further curated for formatting errors and missing gene symbols for some of the interacting partners. Candidate asthma genes, compiled from literature databases and microarray gene expression data, were mapped onto the HPRD interactome to obtain asthma PPIs forming the core of the API.
Proteins that directly interact with each other are known to play roles in similar biochemical processes or diseases (Hartwell et al., 1999). When proteins are regulated under similar experimental conditions or transcription factors, the reliability of computational links among the proteins is high. The API, an interactome underlying the asthma disease condition, was obtained by extending the core network of candidate asthma proteins to include proteins directly interacting with them. Figure 1 illustrates the API.
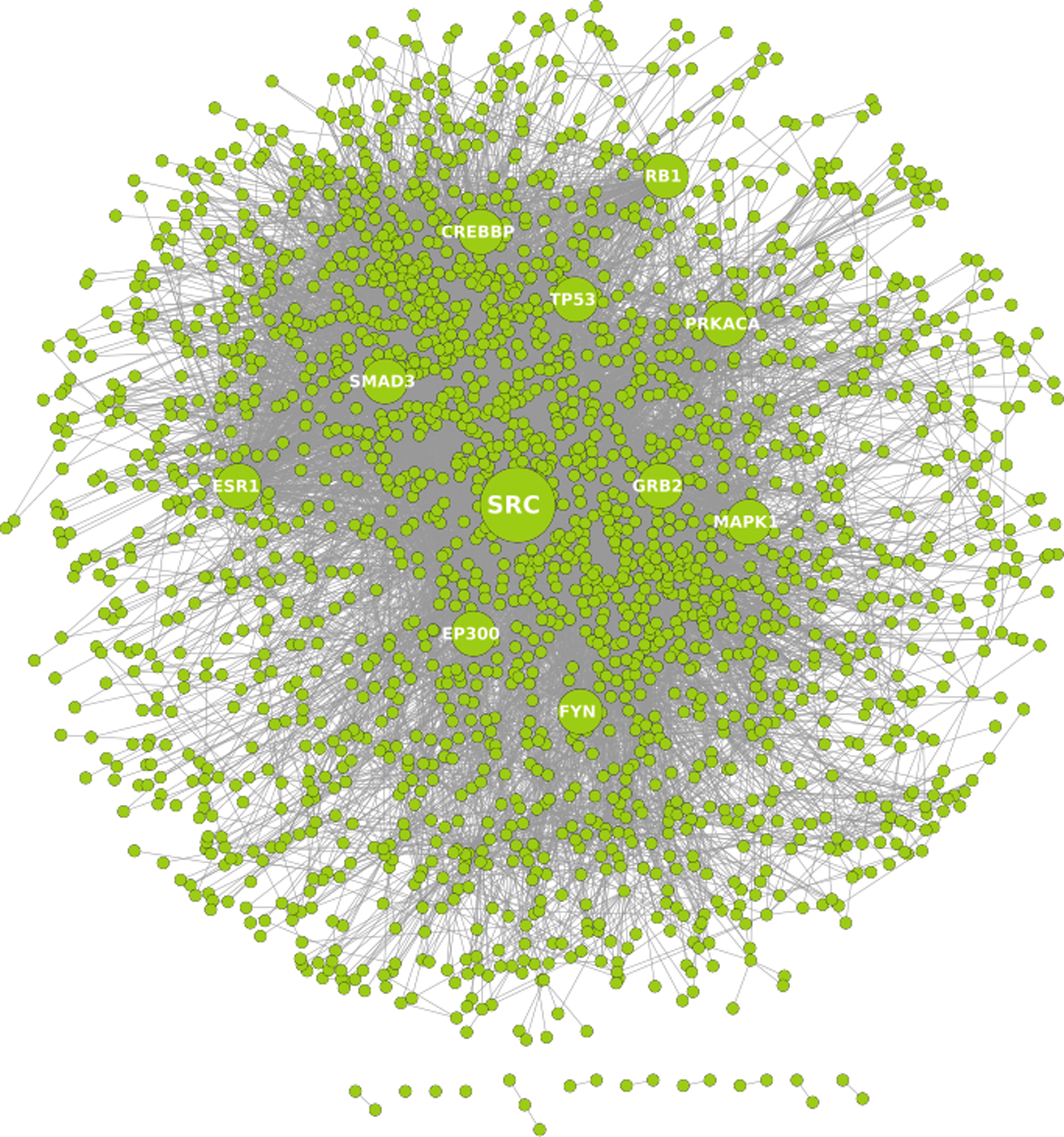
A topological view of the Asthma Protein Interactome (API). The circles represent proteins and the lines indicate an interaction between two proteins. Potential therapeutic targets identified, based on topological and dynamic features of the interactome, are highlighted by the labels. The graph was rendered using Cytoscape.
Gene ontology (GO) enrichment analysis
To describe the functional annotation of gene products, the gene ontology (GO) provides three biological domains: biological processes (BP), the molecular events to which a gene product contributes; molecular functions (MF), the biochemical activities of the gene product at the molecular level; and cellular components (CC), the subcellular locations where a gene product is active (Dimmer et al., 2008). To validate the ontological relevance of the asthma candidate proteins compiled, and to better understand the key biological processes in which they were involved, GO enrichment studies were performed. For these studies, we used GOrilla (Gene Ontology enRIchment anaLysis and visuaLizAtion tool; Eden et al., 2009), a tool to identify enriched GO terms. We performed GO enrichment using the “two unranked lists of genes” mode. The asthma candidate genes (target) were enriched against all genes present in the HPRD database (source). We identify “significantly enriched GO terms” with a p-value cutoff of 0.001, and those having at most 100 associated genes in the source data (B ≤100). The latter criterion weeds out terms those are too generic. Figure 2 illustrates details of the significantly enriched GO terms associated with asthma.
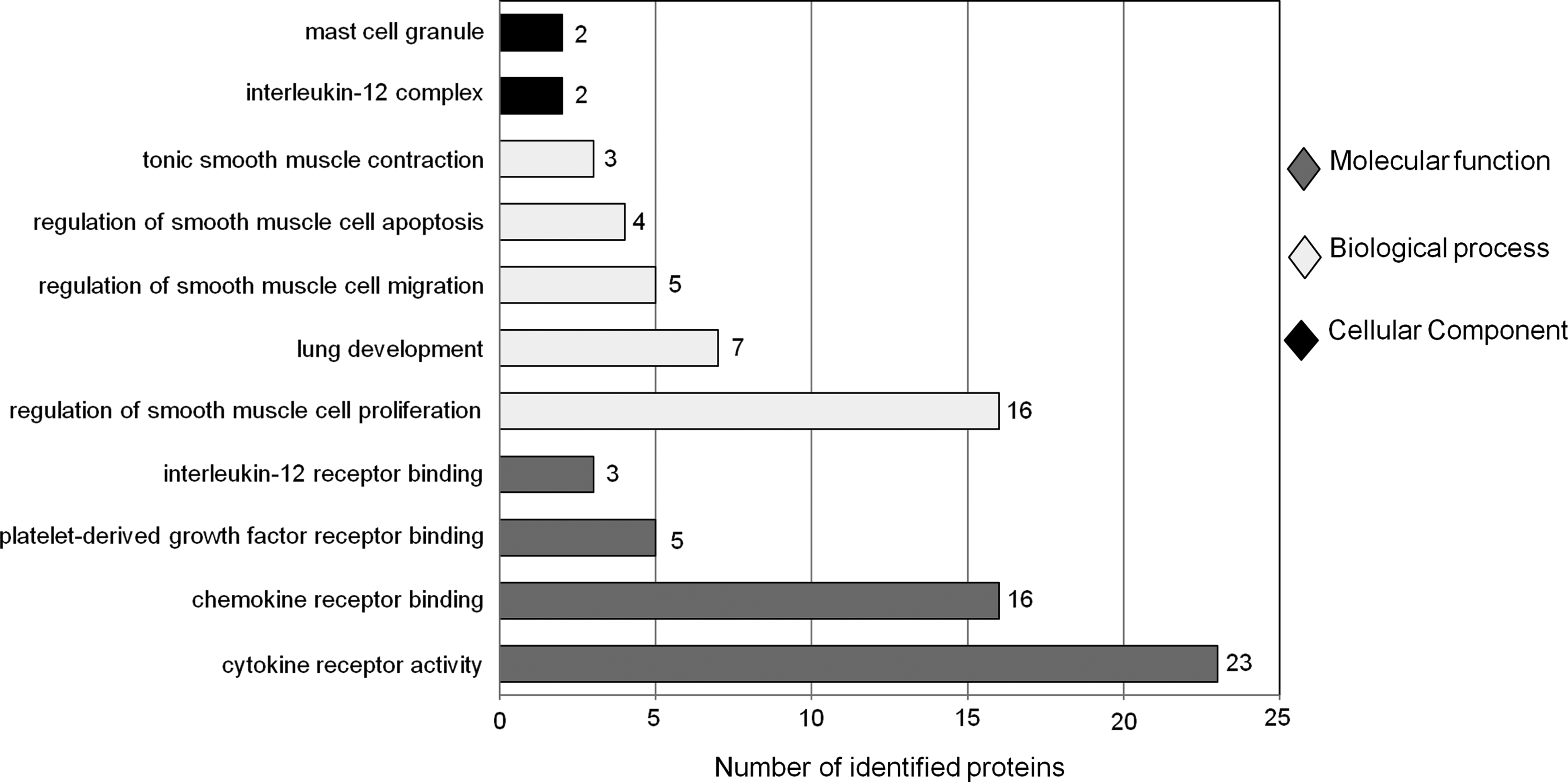
Significantly enriched gene ontology (GO) terms, for each of the GO categories (BP, MF, and CC) related to asthma, and the number of genes they are associated with, in the asthma candidate genes compiled.
Identification of therapeutic drug targets, the key nodes, for asthma
Network analysis of the API
We used graph-theoretical analysis for identifying proteins, due to their role in the structure and dynamics of the API, which are key nodes, and thus potential drug targets, for asthma. Hubs of protein interactomes are known to be more essential for survival (Jeong et al., 2001), and are also important for cellular growth (Batada et al., 2006). Proteins with high betweenness are reported to have a much higher tendency to be essential genes (Joy et al., 2005; Yu et al., 2007). We used a strategy similar to the one used by Hwang and colleagues (Hwang et al., 2008) for our studies. In their study of the asthma protein interaction network, parameters based on connectivity, betweenness, and path lengths were used to identify key nodes (Hwang et al., 2008). In our study, we used three network parameters, enumerating connectivity and flow centrality, for identifying the central proteins of the API: degree, betweenness, and stress. We performed network analysis and visualization using Cytoscape 2.8.0 software (Shannon et al., 2003).
Degree specifies the number of links (interactions) of a node (protein). Betweenness and stress are measures of a node's centrality for information transfer across the network. The degree and betweenness distribution of the API reveal the presence of hubs (See Supplementary Fig. S2 at www.liebertpub.com/omi), nodes central to topology and dynamics, respectively (Barabási and Oltvai, 2004). For the purpose of identifying key nodes in the interactome, we considered “Top10” nodes for every parameter used in this study as the hubs. Table 1 lists proteins identified as the most central nodes of the API using the three parameters.
Stress and betweenness enumerate the number of shortest paths from all pairs of vertices passing through the node of interest. In a graph G=(V, E) with N nodes, stress
where
Betweenness is a normalized measure of flow centrality. For a given graph G=(V, E), the betweenness centrality (
where
Biological relevance of the centrality measures used
For inferring the biological significance of the network parameters used, we obtained statistics of essential and non-essential genes in hubs and non-hubs. The top-ranked genes, according to each of the network metrics, were classified as “hubs,” and the rest as “non-hubs.” We divided the API into two sets, “essential” and “non-essential,” using the definition of lethality given in Mouse Genome Informatics (MGI; Blake et al., 2011). We used Mammalian Phenotype Ontologies (Smith et al., 2005) to define genes as essential when they caused embryonic, perinatal, or neonatal lethality in mouse models (Goh et al., 2007), according to the MGI. The human orthologs of murine genes were considered as essential, when the murine gene was annotated with one of the following phenotypes: neonatal lethality (MP: 0002058); embryonic lethality (MP: 0002080); perinatal lethality (MP: 0002081); postnatal lethality (MP: 0002082); lethality-postnatal (MP: 0005373); lethality-embryonic/perinatal (MP: 0005374); embryonic lethality before implantation (MP: 0006204); embryonic lethality before somite formation (MP: 0006205) or embryonic lethality before turning of embryo (MP: 0006206). Figure 3 illustrates that hubs identified by gene-ranking according to each of the network metrics (degree, betweenness, and stress) are significantly correlated with essential genes.
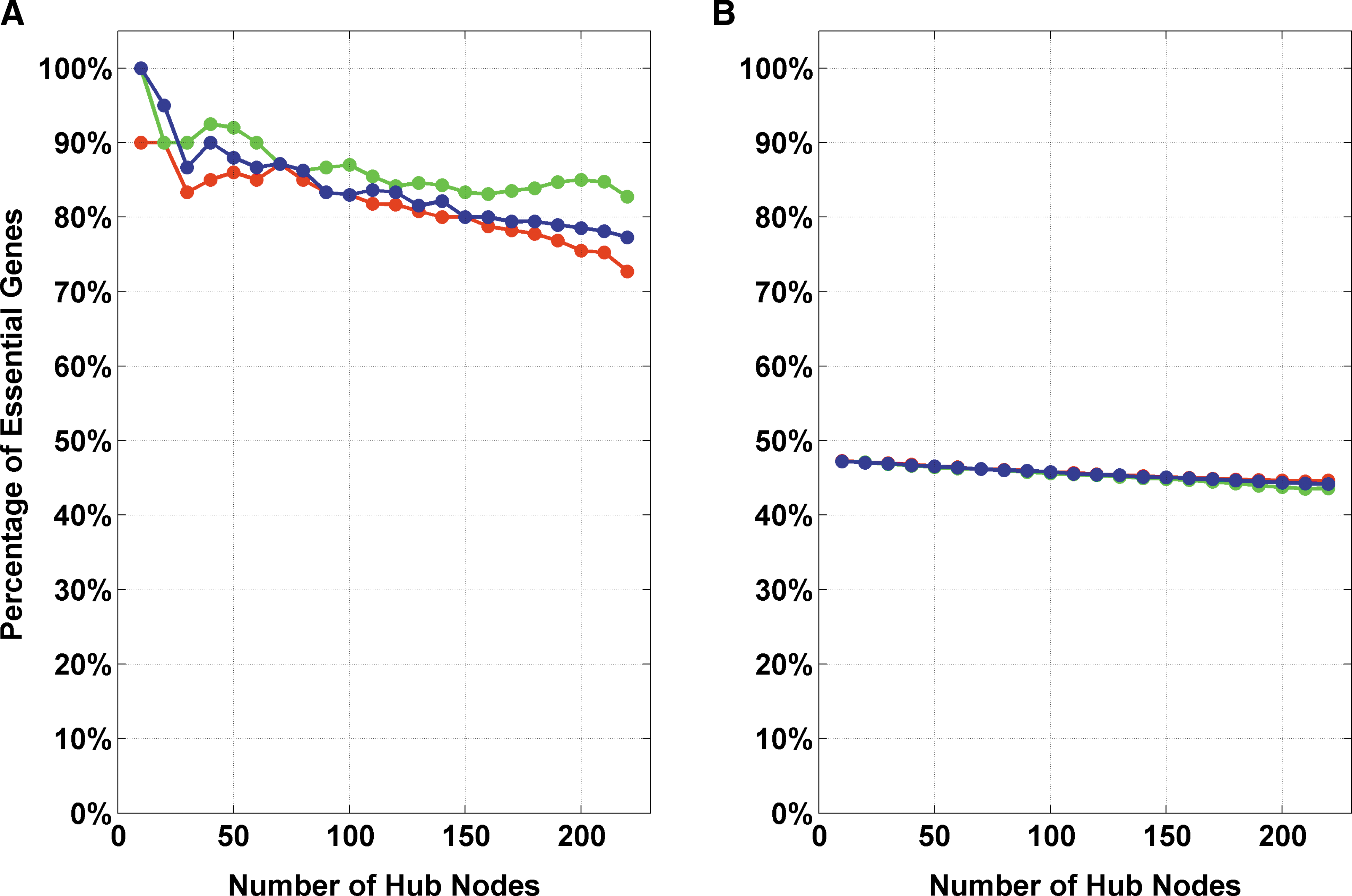
The statistics of percentages of essential genes in the (
Virtual screening of drug-like molecules against SRC tyrosine kinase
SRC tyrosine kinase was identified as top-ranked among the 11 key proteins identified (Table 1) from network analysis of the API. SRC tyrosine kinase, the most central target identified, is also known to play an important role in respiratory mechanisms (Krymskaya et al., 2005b), owing to its major role in asthma progression. We performed virtual screening of a compound library for SRC kinase to identify leads.
Generation of the ligand-based pharmacophore model
In a computationally-generated pharmacophore model, the selection of a suitable training set is a key factor. The pharmacophore models can only be as good as the input data. To date, there are a number of crystal structures determined for human SRC co-crystallized with different inhibitors. In this study, a set of SRC kinase inhibitors bound to SRC crystal structures with varying biological activities characterized by their IC50 values, were obtained from the Protein Data Bank (PDB; Rose et al., 2010). The following criterion was used for the selection of crystal structure-bound inhibitors in order to achieve good quality pharmacophore models: the compounds used in the training set bind to the same receptor in nearly the same fashion. The training set molecules were widely populated, covering an activity range of at least four orders of magnitude.
The SRC complexes selected in this study are as follows: 3EN7, 2HWO, 3EN6, 3EN4, 3EN5, 3F3U, 2HWP, 3SVV, 3F6X, 1YOM, 3F3T, 2QLQ, 1YOL, 3EL8, 1BYG, 2BDJ, 3G5D, 2BDF, and 3EL7 (Table 2). Crystal structures 2QLQ and 2QQ7 contain the same inhibitors in their active sites (ATP binding sites), as did 2HWP and 3LOK. Thus, in order to avoid duplication, a unique set of inhibitors were selected for the training set.
The first rule of Lipinski's “rule of five” limits the molecular weight of drug-like compounds to below 500 Da. As high-molecular-weight compounds can influence the quality of pharmacophore models, inhibitors bound to PDB structures 3F3V, 3DQW, and 2H8H were not included in the training set, as their molecular weights are 506.6, 523.25, and 542.03 Da, respectively. Finally, a set of 19 inhibitors was selected as a training set (See Supplementary Fig. S3 at www.liebertpub.com/omi), and used for merged feature pharmacophore generation with LigandScout 3.0 (Wolber and Langer, 2005) software, at default settings. The software was also used for predicting protein-ligand interactions to get an insight into the spatial organization and the nature of the bound ligands at an atomic level.
Although diverse conformation generation is a regular procedure in merged feature pharmacophore generation, this step was not performed, as we have considered only crystal-bound conformations of inhibitors in the training set. The training set compounds extracted from their crystal structures were verified for their bond orders. Ten pharmacophore models were generated (See Supplementary Table S1 at www.liebertpub.com/omi), and the best one was selected based on (1) receiver operating characteristic (ROC) curves, (2) enrichment factor (EF), and (3) the chemical features of the pharmacophore model and the proposed mechanism of SRC inhibition. The best pharmacophore model selected comprises of one hydrogen bond acceptor (HBA), one hydrogen bond donor (HBD), and two ring aromatic (RA) features (Fig. 4A). The pharmacophore model was used to perform a computational screening search in a 3D virtual compound library to identify structures with desired activity and selectivity.

(
Validation of the ligand-based pharmacophore model
The ROC curve was used to estimate the performance of ligand-based pharmacophore models. In ROC analysis, the ability of a particular pharmacophore model to correctly categorize a list of compounds as actives or inactives (“decoys”) is indicated by the area under the curve (AUC) of the corresponding ROC as well as EF values. The use of ROC curves has been observed to be advantageous over other conventional enrichment curves, since they are independent of the proportion of actives in the test set, and as they include information on sensitivity as well as specificity (Triballeau et al., 2005). ROC curves plot the sensitivity (Se) against specificity (Sp), and the AUC can be calculated for comparison. The ROC scores vary between 0 and 1.0, and the closer the ROC is to 1.0, the better the pharmacophore model is at distinguishing good samples from bad ones.
Ten pharmacophore models generated by LigandScout were investigated for their discriminatory power among actives and decoys by screening them against the Directory of Useful Decoys (DUD) dataset (Huang et al., 2006) for SRC tyrosine kinase (accessed January, 2011). The SRC tyrosine kinase ligands and decoys (159 actives and 6217 decoys) were obtained from the DUD website (http://dud.docking.org/r2/), a published dataset resource providing active molecules and decoys. The pharmacophore models were subjected to ROC analysis across a benchmark DUD dataset. During the screening against DUD set, the “maximum omitted feature” was set to zero, and the default scoring function was “pharmacophore fit.” The chemical features of the pharmacophore model and the proposed mechanism of SRC inhibition were also carefully analyzed during selection of the best pharmacophore model. Finally, the pharmacophore model with best performance in picking actives from the DUD set was selected for further studies. The ROC curve of the best pharmacophore model (pharmacophore-F) is shown in Figure 5. The model with the highest AUC and enrichment of active compounds was selected for virtual screening of the ZINC database (Irwin and Shoichet, 2005).

The performance of the pharmacophore model (pharmacophore-F) by ROC curves using the Directory of Useful Decoys (DUD) dataset for SRC tyrosine kinase. The grey curve is the ROC curve for the DUD dataset. The dotted black curve is the ROC curve for the random classification of compounds. The ROC plot was generated using LigandScout.
Drug-like compound library generation and virtual screening
Virtual screening, an in-silico tool for drug discovery, has been widely used for lead identification in drug discovery programs. Novel and potential leads can be obtained by virtual screening of chemical databases. The compounds obtained by this methodology have the advantage of being easily available for biological testing. The best pharmacophore model was used as 3D query for screening drug-relevant compounds from the ZINC database. ZINCPharmer (http://zincpharmer.csb.pitt.edu), an online interface to the Pharmer (Koes and Camacho, 2011) pharmacophore search software for the ZINC database, was used to search pharmacologically-relevant compounds from the database for novel potential inhibitors. To reduce the redundancy in the obtained compounds, the maximum number of orientations returned for each conformation and for each molecule was restricted to one. Additionally, a check for molecular weight and number of rotatable bonds was also made during the search. The molecules selected from ZINCPharmer were further checked for their drug-like properties using Lipinski's “rule of five” filters using XLOGP3 v 3.2.2 (Cheng et al., 2007), an open source tool for filtering drug-like molecules in order to remove the non-drug-like compounds. In LigandScout, conformations for the compounds obtained from ZINCPharmer were calculated using OMEGA to generate a default of 25 maximum conformations for each compound using the “best conformer generation” method. The generated conformations were translated into LigandScout database format using the “idbgen” tool.
The virtual screening process was carried out employing the “iscreen” package of LigandScout. The compounds obtained from ZINCPharmer were ranked according to the pharmacophore fit score in LigandScout to indicate good matches. The score was calculated for each database “hi”’ based on how well the compounds map onto the pharmacophoric feature position constraints, and their distance deviation from the pharmacophoric feature centers. Pharmacophore fit score calculates the number of matching pharmacophore features and the root mean square deviation (RMSD) for pharmacophore alignment. To refine the compounds obtained from ZINCPharmer search, a threshold of minimum pharmacophore fit score was applied to obtain “hits.” This lower limit was chosen from the lowest pharmacophore fit score obtained for the SRC actives. These hits, with good pharmacophore fit score and having drug-like properties, were further refined using protein-ligand interaction studies with molecular docking methods.
Molecular docking
The hits satisfying pharmacophoric and drug-like filters were subjected to a molecular docking process. Autodock Vina (Trott and Olson, 2010) was used for molecular docking simulations. Protein coordinates from the crystal structure of human tyrosine kinase c-SRC (PDB ID: 2SRC), in complex with AMP, determined at a resolution of 1.50 Å, was used in molecular docking studies.
During computational prediction of ligand-binding poses, the accuracy of a molecular docking program is usually measured by the RMSD between the experimentally-observed heavy atom positions of the ligand, and those predicted by the program (Cole et al., 2005). For validation of the docking protocol, the bound AMP coordinates in the crystal complex were extracted and re-docked into the ATP binding site using AutoDock Vina. The lowest-energy structure generated during the docking runs was considered as the best conformation, and was found to closely reproduce the position and orientation observed in the crystal structure (Fig. 6). The best mode obtained from the re-docking procedure was treated as the positive control. For molecular docking, AMP was removed and hits were docked into the active site of the SRC kinase structure using the same docking protocol.
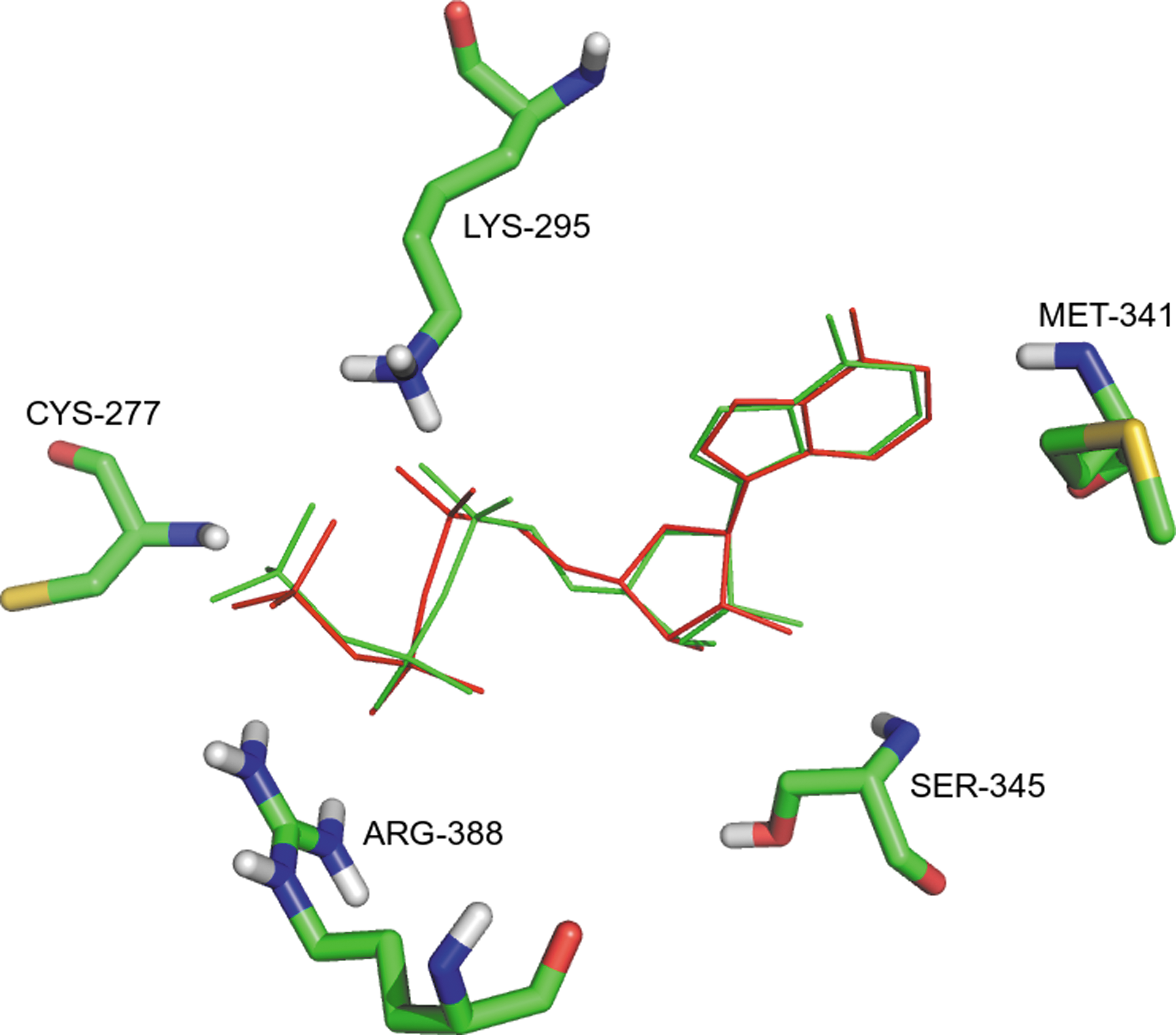
Validation of docking protocol. Overlay of the experimental (red) and predicted docked conformation (green) of ANP at the active site. The RMSD between the experimental structure and the predicted conformation was observed to be 0.437Å.
All water and solvent molecules present in the original PDB file were removed manually prior to docking, as they were not found to play any role in protein-AMP interactions. Using Autodock Tools software (Morris et al., 2009), polar hydrogen atoms and Kollman charges were added to the protein structure, followed by merging of non-polar hydrogen atoms. The protein receptor and the hits were converted from PDB to PDBQT format. All other options were kept default during docking experiments. A grid box with dimensions of 25×25×25 points along the x, y, and z axes was defined around the active site to cover the entire enzyme binding site and to accommodate free motion of the ligands. AMP was used to determine the search space size at the binding site. For each compound, several feasible binding conformations, ranked according to their binding affinities, were obtained with flexible docking simulations. The top 10 conformations were saved for each of the hits. At the end of docking run, Autodock Vina ranks all the generated docked conformations based on the binding affinity. Molecular interactions between protein and hits were observed using Ligplot (Wallace et al., 1995) software. The compounds were further selected based on binding affinity, binding mode, and molecular interactions observed at the active site. The drug-like compounds with binding affinity better than that of the positive control, and those making interactions with critical amino acids (Glu339 and Met341), were further considered for the screening procedure as leads for c-SRC kinase. We performed ROC analysis of these compounds (identified by virtual screening and molecular docking) against DUD decoys using the best pharmacophore model. We found that these drug-like compounds had very good selectivity and specificity (See Supplementary Fig. S4 at www.liebertpub.com/omi), confirming the quality of virtual screening, molecular docking, and hence that of the compounds obtained. Molecular rendering was performed using PyMOL (The PyMOL Molecular Graphics System, Version 1.5.0.1, Schrödinger, LLC.).
These compounds were further refined based on the similar chemical scaffolds and their molecular interactions at the active site. Finally, the lead compounds were selected based on their LASSO (Ligand Activity in Surface Similarity Order) score for the SRC tyrosine kinase DUD dataset using chemspider (http://www.chemspider.com/), a free chemical structure database. The LASSO score gives an appraisal of the “similarity of ligand features” to those determined in the neural network training of active ligands. The novelty of these leads was confirmed using the PubChem structure search tool. Finally, two molecules from the ZINC database were selected as the most potent leads for SRC tyrosine kinase.
All computations were performed on an HPZ600 work station and HP ProLiant DL980 server running Ubuntu 11.10 and Red Hat 4.1.2 operating systems, respectively, with Intel Xeon processors.
Results
Identification of asthma-associated therapeutic drug targets by network analysis
Compiling candidate asthma genes
To obtain an extensive list of asthma candidate genes, we used a composite strategy of a PubMed search, an OMIM search, and the use of the differential expression data available (See Supplementary Fig. S1 at www.liebertpub.com/omi). We identified 108 asthma-susceptibility genes from the PubMed literature search (up to January 31, 2012), including a list of already published asthma genes (Hoffjan et al., 2003; Ober and Hoffjan, 2006; Zhang et al., 2008), and the PubMed search returned 228 asthma genes. The OMIM literature database was also searched for relevant asthma genes, and 121 genes were obtained. A set of 71 significant differentially expressed asthma genes during human lung development (Melén et al., 2011), with an adjusted p value of <0.05, were also included in the candidate genes list. Using the above three approaches, we created a list of 291 unique core asthma candidate genes (See Supplementary Table S2 at www.liebertpub.com/omi). The Venn diagram illustrates our strategy to obtain an extensive and complete list of candidate genes (See Supplementary Fig. S1 at www.liebertpub.com/omi). We observed that the asthma gene data from differential expression studies overlapped with that of genes identified from literature searches.
Gene ontology enrichment analysis of asthma candidate genes
GO enrichment studies were performed to validate the ontological relevance of the asthma candidate proteins compiled and to identify key biological mechanisms associated with them. GO analysis revealed that biological processes and molecular functions were widely populated with “significantly enriched GO terms” related to the immune system and asthma (See Supplementary Table S3 at www.liebertpub.com/omi).
The biological process GO class was dominated by immune system-related significantly-enriched GO terms (316 out of 584). The following terms had a direct relation to asthma: regulation of smooth muscle cell proliferation (GO: 0048660), lung development (GO: 0030324), positive regulation of smooth muscle cell proliferation (GO: 0048661), regulation of smooth muscle cell migration (GO: 0014910), regulation of smooth muscle cell apoptosis (GO: 0034391), tonic smooth muscle contraction (GO: 0014820), positive regulation of smooth muscle cell apoptosis (GO: 0034393), and regulation of smooth muscle cell-matrix adhesion (GO: 2000097).
Among the significantly-enriched GO terms in the molecular function class, cytokine-, chemokine-, and interleukin-related terms dominated the ontologies. A total of 30 immune system-associated significant GO terms were obtained, of which 21 were found to be associated with asthma. Following is the list of asthma-related GO terms: cytokine receptor activity (GO: 0004896), cytokine binding (GO: 0019955), chemokine receptor binding (GO: 0042379), chemokine activity (GO: 0008009), chemokine receptor activity (GO: 0004950), platelet-derived growth factor receptor binding (GO: 0005161), C-C chemokine receptor activity (GO: 0016493), chemokine binding (GO: 0019956), platelet-derived growth factor binding (GO: 0048407), angiotensin receptor binding (GO: 0031701), CCR1 chemokine receptor binding (GO: 0031726), CCR5 chemokine receptor binding (GO: 0031730), interleukin-12 receptor binding(GO:0005143), interleukin-2 binding (GO: 0019976), interleukin-2 receptor activity (GO: 0004911), arachidonate 5-lipoxygenase activity (GO: 0004051), CCR2 chemokine receptor binding (GO: 0031727), interleukin-12 binding (GO: 0019972), interleukin-4 receptor activity (GO: 0004913), platelet-derived growth factor-activated receptor activity (GO: 0005017), and vascular endothelial growth factor binding (GO: 0038085).
Significant cellular component terms were less informative, being either too general or annotated for very few proteins. The following two terms are related for the immune system and asthma: interleukin-12 complex (GO: 0043514) and mast cell granule (GO: 0042629).
Figure 2 illustrates details of the significantly enriched GO terms associated with asthma. Details of the GO analysis are provided in the Supplementary material (See Supplementary Table S3 at www.liebertpub.com/omi).
Asthma Protein Interactome
In this study, starting from a core set of 293 asthma candidate genes, we constructed the API (Fig. 1). Of the 293 unique asthma candidate genes identified, a total of 248 were mapped on the HPRD protein interactome. The core asthma network comprises a giant cluster (147 nodes and 263 edges), 9 smaller clusters (21 nodes), and the rest fragmented into 80 isolated nodes. The API, an interactome comprised of proteins directly interacting with core asthma candidate genes, represents the underlying molecular mechanisms involved in the disease condition. The API was composed of 2212 proteins and 12,941 interactions in 9 clusters. The giant cluster contained 2192 nodes, thus constituting 99.09% of the total API. We identified potential targets, the key nodes, that are topologically and dynamically most central to the API, using graph-theoretical metrices.
Identification of asthma-associated therapeutic drug targets, the key nodes, using network analysis
Most biological networks are characterized by a small number of highly connected nodes, known as hubs, while most of the other nodes have very few connections (Barabási and Oltvai, 2004). Network-based identification of key targets has been reported in various diseases including bovine marbling (Lim et al., 2010), and Helicobacter pylori infection response (Kim and Kim, 2009).
We find that the API is characterized by scale-free degree and betweenness distributions indicating the presence of hubs (See Supplementary Fig. S2 at www.liebertpub.com/omi), as is the case for other biological molecular interaction networks. We used three network parameters (degree, betweenness, and stress), enumerating topological and dynamical features of the API, for identifying potent targets. The highly connected nodes, referred to as hubs, mediate interactions between other nodes in the network and have an important role in the topology of networks. Betweenness is an indicator of node centrality for network dynamics. Nodes with large betweenness function as bottlenecks in the network (Yu et al., 2007), even when the node's degree is low. From 2192 nodes of the largest connected cluster (giant cluster) in the API, the top 10 nodes for each network parameter were considered to be the most central nodes. From these, we obtained a list of 11 unique key nodes in the API, potential drug targets of therapeutic value for asthma. The selected nodes are about 0.50% of the total giant cluster.
Following are the 11 therapeutic drug targets identified for the API: SRC (v-src sarcoma viral oncogene homolog), CREBBP (cAMP-response-element-binding binding protein), ESR1 (estrogen receptor 1), SMAD3 (SMAD family member 3), MAPK1 (mitogen-activated protein kinase 1), PRKACA (protein kinase, cAMP-dependent, catalytic, alpha), TP53 (tumor protein p53), EP300 (E1A binding protein p300), FYN (FYN oncogene related to SRC, FGR, and YES), RB1 (retinoblastoma 1), and GRB2 (growth factor receptor-bound protein 2). Table 1 lists these potential targets and their composite ranking according to three chosen parameters. Based on the composite ranking, SRC emerges as, topologically and dynamically, the most central protein in API.
Figure 1 illustrates the API protein interactome and the central nature of SRC, making it one of the most potent targets. Additionally, we found that each of the 11 target proteins, key nodes of the API, was also reported to have a direct or indirect role in the progression of asthma (See Supplementary Table S4 at www.liebertpub.com/omi).
Biological relevance of selected network parameters
We observed that the network parameters (degree, betweenness, and stress) selected for target identification are highly correlated with essentiality. We varied the definition of hubs between the Top10 and Top230 (approximately 10% of API) genes, and computed the percentage of essential genes in the hubs and non-hubs. We found that the hubs have a very high percentage of essential genes, while the percentage of those in non-hubs was as expected from random sampling (Fig. 3). For the selected parameters, we found that the hubs are characterized by significantly high percentages (degree: between 82% and 100%; betweenness: between 72% and 90%; stress: between 77% and 100%) of essential genes. The statistics of essential genes supports our selection of network parameters for identification of drug targets.
Virtual screening of drug-like molecules against SRC tyrosine kinase
Generation of ligand-based pharmacophore model
The training set comprised of 19 active compounds in their crystal bound conformations (Table 2; See Supplementary Fig. S3 at www.liebertpub.com/omi) was used for pharmacophore generation. Most of the training set compounds were found to interact with the backbone amides of Glu339 and Met341 of the hinge region, thus indicating the importance of these two residues. The inhibitors bound to SRC crystal structures were observed to have half-maximum inhibitory concentration (IC50) ranging from 0.4 to 64,100 nM (Table 2). The variations in potency of the bound inhibitors is most likely due to variations in the hydrogen bonding interactions with the hinge region of the kinase SRC.
Based on the training set molecules, 10 pharmacophore models were generated using a merged feature pharmacophore procedure to identify the chemical features necessary to inhibit the SRC tyrosine kinase's molecular function. These pharmacophore models were characterized by the following chemical features: hydrogen bond acceptors (HBA), hydrogen bond donors (HBD), and ring aromatics (RA). Supplementary Table S1 (see www.liebertpub.com/omi) represents the pharmacophore models and their statistical parameters. The pharmacophore models generated were found to vary in their feature composition, orientation, and vector directions. An initial analysis of pharmacophore models revealed that their pharmacophoric features were sufficient, and could map all critical chemical features required for effective SRC inhibition. The biological activities of compounds depend not only on chemical features of molecules, but also their correct shapes, to fit into the active site of the protein (Adane et al., 2010). All pharmacophore models were subjected to ROC analysis to assess their abilities to accurately classify a list of compounds as actives or decoys.
Validation of the ligand-based pharmacophore model
The ROC, against a benchmark DUD actives and decoys set, was used to estimate the performance of each of the pharmacophore models, with LigandScout software. The closer the ROC score is to 1.0, the better the pharmacophore model is at distinguishing active samples from inactive ones. The ROC AUC details for the 10 pharmacophore models are provided in Supplementary Table S1 (see www.liebertpub.com/omi). ROC curve analysis of the best pharmacophore model (pharmacophore-F) yielded a ROC score of 0.84 (Fig. 5), which means that a randomly-selected active compound has a higher score than a randomly-selected decoy 8.4 times out of 10. Since the AUC value was beyond 0.5, the pharmacophore screening performed better than random discrimination of actives and decoys. The EFs of the DUD set screened by the best selected pharmacophore at 1%, 5%, 10%, and 100%, were 6.4, 11.3, 7.2, and 2.1, respectively. The best pharmacophore retrieved 131 of 159 active compounds from DUD actives, corroborating the ROC statistics. EF was also used as one of the criteria for selection. In addition to ROC and EF, the mechanism of SRC inhibition was also considered during selection of the best model. The best pharmacophore model (Fig. 4A) had the following chemical features: one HBA, one HBD, and two RAs. Figure 4B shows the overlay of the pharmacophore model over the DUD active compound (ZINC ID: ZINC01609268) with best pharmacophoric fit score.
Drug-like compound library generation and virtual screening
In our study, a virtual screening approach was used to find the potent inhibitors for SRC tyrosine kinase from ZINC, a publically-available database containing over 2.7 million commercial compounds. The selected pharmacophore model was used as a 3D query to retrieve potential inhibitors from the database. ZINCPharmer (http://zincpharmer.csb.pitt.edu) was used to search pharmacologically-relevant compounds from ZINC. ZINCPharmer retrieved 58,155 compounds using the following thresholds: (1) maximum RMSD of 0.1, (2) maximum number of orientations returned for each conformation and that for each molecule to 1, (3) molecular weight ranging between 150 and 500 Da, and (4) number of rotatable bonds between 0 and 8. However, the resulting list of molecules obtained was also found to have non-drug-like compounds as well. In order to remove non-drug-like molecules, these compounds were further filtered by applying Lipinski's “rule of five,” using XLOGP3 v 3.2.2. A total of 46,788 drug-like molecules were obtained using the “drug-like” filter criteria (H-bond donor count between 0 and 5, H-bond acceptor count between 0 and 10, partition coefficient [log P] between −2 and +5, number of rings between 0 and 10).
Up to 25 conformers were generated for each drug-like compound obtained, using the “idbgen” tool. The virtual screening process was carried out employing the “iscreen” package of LigandScout. The 46,788 compounds screened were overlaid with the best pharmacophore model. These compounds were ranked according to the pharmacophore fit score. A threshold of 35.95 pharmacophore fit value was used as the lowest limit to identify hits. This lower limit of pharmacophore fit value was assigned based on the lowest pharmacophore fit score of DUD actives over the pharmacophore model. We selected top 500 hits, with the best pharmacophore fit values, for subsequent molecular docking.
Molecular docking
To remove the false-positives and to refine the hits obtained from ZINC database screening, the top 500 compounds with best pharmacophore fit scores were docked into the active (ATP-binding) site of c-SRC kinase using the AutoDock Vina program. There are a number of crystal structures for SRC kinase-ligand complexes available in PDB. The crystal structure of the c-SRC kinase-AMP (PDB ID: 2SRC) complex was selected based on its high resolution (1.50 Å). The most critical amino acid residues of c-SRC were identified from the protein-ligand interactions observed from the c-SRC-AMP complex and published literature studies. Glu339 and Met341 were identified as the most critical amino acid residues for AMP binding.
During validation of the docking protocol, the AMP co-crystallized with c-SRC was extracted and re-docked into the same ATP binding site. When re-docked, the best binding mode of computationally docked AMP showed a binding affinity of −8.2 kcal/mol and was considered as positive control. Figure 6 shows the experimental and docked conformations inlaid into the binding site. The ability of a docking program that returns poses below a preselected RMSD value from the experimental conformation (usually 1.5 or 2 Å) is considered to be good. The heavy atom RMSD between the experimental and positive control was observed to be 0.437 Å, confirming the quality of the docking protocol used in the present study and its suitability for predicting reliable binding modes of hits. Additionally, similar protein-ligand interactions were observed for the experimental and computationally-predicted AMP conformation.
Out of 500 selected hits, a set of 269 hit compounds with binding affinity greater than that of the positive control, and having structural diversity were selected. Further, the compounds with the best binding conformations, and with molecular interactions similar to those of the positive control, were selected. Based on the prior knowledge of the existing c-SRC kinase inhibitors and the active site requirements, 55 drug-like compounds were selected based on hydrogen bond interactions, either with the Glu339 or Met341 residue. To verify the quality of these compounds obtained after virtual screening and molecular docking, they were mapped over the pharmacophore model selected for this study. Of 55 compounds, 52 were identified to have good mapping over the pharmacophore model. All four pharmacophoric features were mapped on the pharmacophore model for 51 compounds, and three of the four features were mapped for the remaining compound. These compounds were subjected to ROC analysis against DUD decoys. An AUC score of 0.95 was obtained with the selected pharmacophore model (See Supplementary Fig. S4 at www.liebertpub.com/omi). This implies that the pharmacophore screening performed better than random discriminations of actives and decoys. The EFs observed at 1%, 5%, 10%, and 100%, were 88.3, 18.6, 9.3, and 2.4, respectively. This confirms that the compounds obtained by virtual screening and molecular docking have chemical features similar to that in DUD actives.
The compounds were filtered based on the similar chemical scaffolds and their molecular interactions at the active site. A set of 11 representative compounds with different scaffolds were identified as representative compounds having different patterns of protein binding. The standard search was performed in chemspider (http://www.chemspider.com/), a free chemical structure database, to identify the physical and biological properties of the screened compounds.
Similarity screening was performed on the full chemspider database across whole DUD dataset, and the similarity score for each structure was obtained via the chemspider website. Final leads were selected based on their LASSO (Ligand Activity in Surface Similarity Order) score against the SRC tyrosine kinase DUD dataset. The LASSO similarity score ranges from 0 to 1.0, with 0 being not similar at all, and 1.0 being very similar to the DUD actives (or dissimilar to the decoys).
Finally, ZINC08325541 and ZINC08315590, with the best LASSO scores of 0.80 and 0.77 respectively, were selected as the most potent leads for SRC inhibition. The 2D representation of the final leads and their molecular interactions with the active site residues are depicted in Figure 7A and B. Figure 7C shows the docked conformations of leads in the active site and the pharmacophore overlay of both leads. We confirmed the novelty of these leads for SRC tyrosine kinase, using details present in the ZINC database, and the PubChem structure search tool.
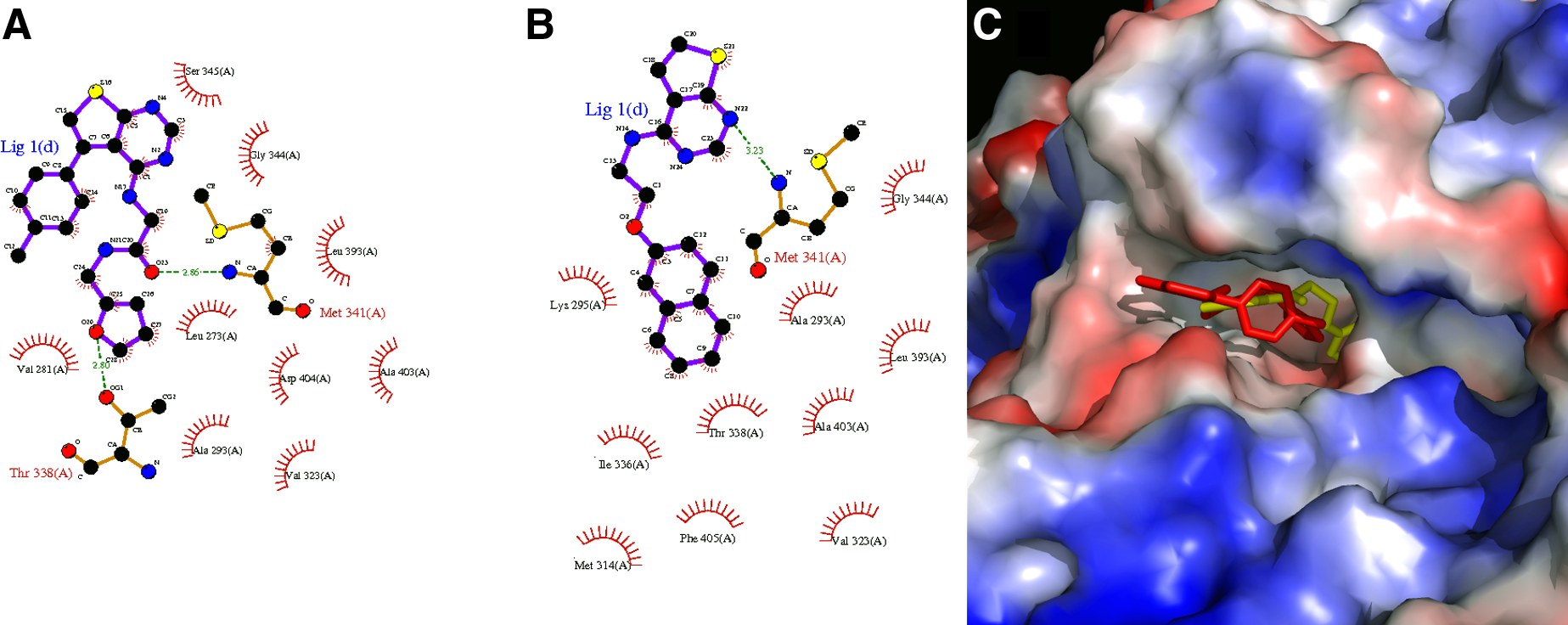
Ligplot results representing 2D interaction plots of docked leads into the active site for (
Discussion
In this work, we combine network-based molecular target identification followed by computational screening of small molecules to identify potential inhibitors of SRC tyrosine kinase, the most central target identified for asthma. We construct the Asthma Protein Interactome (API), the molecular network underlying the complex mechanisms of asthma. With graph-theoretical analysis, we identify a set of 11 key proteins, of which SRC is the most central target of API. Further, we identify two potential leads from the ZINC database using ligand-based virtual screening methodology.
In related studies, network analysis has been implemented for predicting adverse event susceptibility for new drugs in arrhythmias (Berger et al., 2010), and molecular mechanisms behind the side effects associated with several drug targets (Brouwers et al., 2011). In a significant step forward, a computational framework for the drug target prediction method, drugCIPHER, has been developed to speed up drug target identification, as well as to infer drug-target interactions (Zhao and Li, 2010).
We compiled candidate disease genes for asthma from OMIM, PubMed, and from microarray expression data. Further, we constructed the API by mapping those onto the HPRD interactome. Our analysis identifies 11 putative targets, the key nodes (SRC, CREBBP, ESR1, SMAD3, MAPK1, PRKACA, TP53, EP300, FYN, RB1, and GRB2), that are central for the topology and dynamics of API. Proteins with exceptionally large number of interactors (large degree), and those central to the information flow in the API (large betweenness and large stress), were selected as promising drug targets. These targets, by virtue of their network features, are locally and globally important nodes in the API. We identified SRC protein as the most critical node in the network architecture.
In a similar study of the asthma protein–protein interaction network, features such as degree, node betweenness, edge betweenness, and closeness centrality, were investigated to identify key genes. Seven target genes were identified that include SRC (Hwang et al., 2008). Thus SRC appears to occupy a central role in the molecular mechanisms of asthma, and hence could be treated as a potential drug target.
With our integrative strategy of the interactome-based identification of the potent targets and subsequent virtual screening studies, we identified two potential leads that could be of therapeutic relevance to asthma cure. These leads could be used for designing more potent inhibitors for SRC tyrosine kinase.
Thus we present a study reporting the use of an integrative approach of interactome-based target identification for asthma, and the subsequent implementation of ligand-based virtual screening, to identify novel leads for SRC tyrosine kinase inhibition. This strategy may be extended to identify potent targets and their inhibitors for other diseases.
Conclusions
Our network analysis method offers an integrative approach for the identification of potent targets of complex diseases, complementing traditional methods of target identification. The availability of good-quality interactome data presents a great opportunity to probe complex diseases, and complex traits in general, characterized by intricate mechanisms. Graph-theoretical parameters make it possible to screen interactome data for key molecules with meaningful features. Our integrative approach of interactome-based target identification, followed by in silico drug discovery, to obtain leads for a disease could be a useful strategy in the drug discovery process.
Footnotes
Acknowledgments
We acknowledge the financial support and infrastructure provided by the Institute of Himalayan Bioresource Technology, a constituent national laboratory of the Council of Scientific and Industrial Research, India. The authors thank Dr. Paramvir Singh Ahuja for encouragement and constant support, and Shikha Vashisht and Shivalika Pathania for technical help in manuscript preparation. The CSIR-IHBT communication number for this article is 2251.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
Supplementary Information
Supplementary data are available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.