
Abstract
Abstract
Plant growth and productivity are influenced by various abiotic stresses. Stressful conditions may lead to delays in seed germination, reduced seedling growth, and decreased crop yields. Plants respond to environmental stresses via differential expression of a subset of genes, which results in changes in omic compositions, such as transcriptome, proteome, and metabolome. Since the development of modern biotechnology, various research projects have been carried out to understand the approaches that plants have adopted to overcome environmental stresses. Advancements in omics have made functional genomics easy to understand. Since the fundamentals of classical genomics were unable to clear up confusion related to the functional aspects of the metabolic processes taking place during stress conditions, new fields have been designed and are known as omics. Proteomics, the analysis of genomic complements of proteins, has caused a flurry of activity in the past few years. It defines protein functions in cells and explains how those protein functions respond to changing environmental conditions. The ability of crop plants to cope up with the variety of environmental stresses depends on a number of changes in their proteins, which may be up- and downregulated as a result of altered gene expression. Most of these molecules display an essential function, either in the regulation of the response (e.g., components of the signal transduction pathway), or in the adaptation process (e.g., enzymes involved in stress repair and degradation of damaged cellular contents), allowing plants to recover and survive the stress. Many of these proteins are constitutively expressed under normal conditions, but when under stress, they undergo a modification of their expression levels. This review will explain how proteomics can help in elucidating important plant processes in response to various abiotic stresses.
Introduction
Proteomics has appeared as an important tool in the field of plant science, enabling us to interpret the stress responses occurring in plants. The vast range of applications of proteomics in biological fields has greatly increased its use over the last decade (Bindschedler et al., 2008; Chen et al., 2011; Evers et al., 2012; Kaufmann et al., 2011; Nanjo et al., 2011; Thelen and Peck, 2007; Yang et al., 2011; Yokthongwattana et al., 2012; Zhang et al., 2012; Zheng et al., 2012). The term “proteome” (PROTEins expressed by genOME) represents the survey of the expression of all proteins in a given time and condition. Proteomics is also described as the study of the quantitative totality of proteins of a cell, tissue, or organism, which helps in understanding the structural and developmental as well as functional aspects of the plant (Bindschedler and Cramer, 2011; Boisvert et al., 2010). Proteomic research aims at the identification of new proteins, revealing their functions and also unravelling the regulatory networks that control their expression (Acero et al., 2011).
Proteomics is now gaining momentum in three major aspects of plant science: in cellular and subcellular, structural and developmental, and physiological and genetic studies. Hossain and colleagues (2012) summarized information on organelle proteomes relevant to the plant tolerance mechanism to react to abiotic stress at the protein level. Recently, studies of proteomics have established that it has become a useful tool for studying the protein variations of the physiological events occurring in different plant organs (Kosová et al., 2011; Tran and Mochida, 2010a; Timperio et al., 2008; Yan et al. 2006). Adverse environmental conditions alter various physiological processes in plants, which are directly controlled by genes and functionalized by different proteins. Proteomics together with transcriptomics and metabolomics have enabled us to carry out large-scale analyses of genes, transcripts, proteins, and metabolites, leading to a better understanding of biological processes (Fig. 1). The long-term challenges in proteomic research include the identification and quantification of the structural and functional aspects of complete complements of proteins. Functional genomics is useful in the critical step of tackling the sets of clone generation that are representative of each protein of a proteome, and then analyzing these sets of clones on a genome-wide basis.

Proteomics is gaining momentum in all three levels of plant science. These three levels are concerned with studying the proteins at (1) subcellular and cellular levels, (2) structural and developmental levels, and (3) physiological and genetic levels.
Principal Techniques of Global Protein Analysis
Two-dimensional polyacrylamide gel electrophoresis (2D-PAGE) is the most commonly used technique for protein separation and for obtaining quantitative data (Dooki et al., 2006). High-resolution 2D-PAGE has important applications in the resolution of a large number of proteins (Aghaei et al., 2009), and has been used to study the gene products induced in different plant species exposed to different abiotic stresses (Debnath et al., 2011), including salt treatments (Manaa et al., 2011a, 2011b; Shi et al., 2011; Sobhanian et al., 2011; Yan et al., 2006), dehydration/drought (Ge et al., 2012; Ladrera et al., 2007), high and low temperatures (Ahsan et al., 2010; Carrasco et al., 2011), heavy-metal stress (Durand et al., 2010), and herbicides (Castro et al., 2005). Differential proteomics is used to compare distinct proteomes (e.g., normal versus stressed cells and normal versus treated cells), differing both in protein quality and quantity (Ahsan et al., 2010). Thus proteome analysis connects gene expression to cell metabolism. With the growing collection of technologies available for the extraction and identification of proteins and for studying their interactions, proteomics permits clarification of the mechanisms that are expressed in the responses of cells to abiotic stresses. Different staining techniques are used to visualize the separated proteins. The color density and size of the detected spots quantifies the protein expressed. However, the accuracy of staining methods is limited due to low dynamic range. The fluorescent dyes available for proteins have overcome this limitation to a significant degree (Timms and Cramer, 2008). Mass spectroscopy (MS) for peptide mass fingerprinting (PMF) is gaining importance because of their sensitivity with partial sequencing of proteins and affordability (Wong and Cagney, 2010). High-performance liquid chromatography (HPLC)-based separation methods have also entered the field of proteomics as an alternative to 2D-PAGE. They help in identifying and functionally characterizing the recombinant proteins encoded by differentially expressed cDNA clones (Stoevesandt et al., 2009). High-throughput proteomics research would be incomplete without image-analysis software (Palmblad et al., 2007). These programs help in removing the background patterns and by quantification of spots, matching images from relative gels and comparing the intensities of relative spots (http://www.lsbi.mafes.msstate.edu/p_data.htm). Processes involved in the identification of differentially-expressed proteins under stress conditions are shown diagrammatically in Figure 2. The challenge lies in identifying proteins in the typically complex and large plant genomes. Proteome information is a direct link of genome sequence with biological activity; therefore proteome analyses rely on well-annotated sequence databases (Bindschedler and Cramer, 2011). Various bioinformatics tools are also available for plant proteome analysis (Table 1). The completion of the first plant genome in 2000 for the model plant Arabidopsis thaliana, and later for rice in 2002, also yielded information about sequencing for plant genomes like Lotus, Populus, tomato, Medicago, and maize. This amazing applicability of the tool has added information related to the functional aspects of many genes simultaneously in the gene data bank (Bindschedler and Cramer, 2011; International Rice Genome Sequence Project, 2005; Rensink and Buell, 2005; Schneider et al., 2009; Swarbreck et al., 2008; The Arabidopsis Genome Initiative, 2000; Vij and Tyagi, 2007). Increasing sequencing speed, and improvements in gene annotation and modeling, have helped to complete well-annotated plant genomes. The National Center for Biotechnology Information (NCBI) website (http://www.ncbi.nlm.nih.gov/genomes/PLANTS/PlantList.html) provides genomics data for the soybean, Medicago truncatula, the wine grape, maize, poplar, and sorghum. The UniprotKB and SwissProt protein databases are also available for plant data. However, only a small proportion of the reviewed proteins are available in these manually annotated reference databases. For example, the protein entries for Arabidopsis, rice, maize, and other “viridiplantae” organisms in Uniprot were 12,522, 10,643, 950, and approximately 8500, respectively (http://www.uniprot.org/uniprot/; accessed February 2012). However, an investigation of the Arabidopsis proteome has led to the discovery of 778 novel proteins associated with unpredicted open reading frames (ORFs), suggesting that the proteogenomic approach can greatly improve and validate the annotation of the genes (Castellana et al., 2008). Therefore, the genome-proteome-wide strategy can be used to gain insights into stress-responsive genes, thus adding to our understanding of the mechanisms involved in stress tolerance.
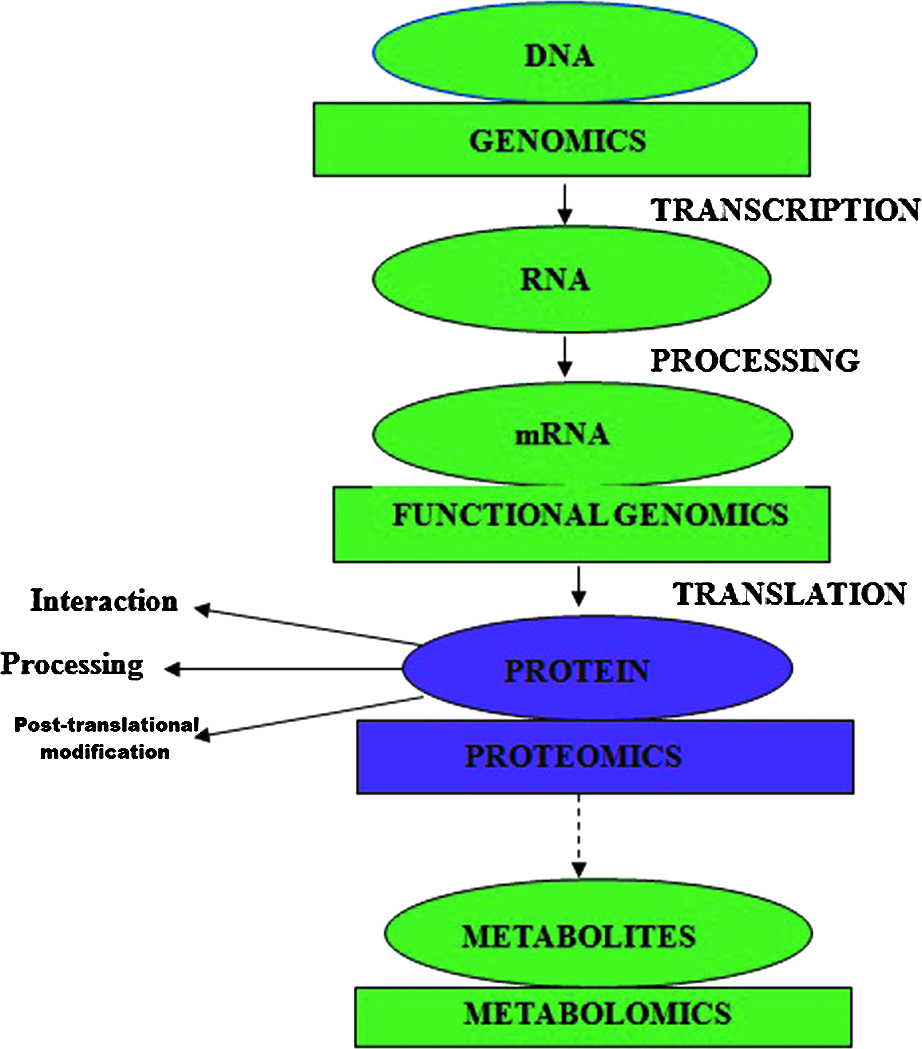
Overview of the currently available disciplines for large-scale analyses of genes, transcripts, proteins, and metabolites (PTM, post-translational modification).
Proteomics of Plant Physiology Under Abiotic Stress
Under a number of abiotic stress conditions, various physiological and biochemical changes occur in plants (Ahmad and Prasad, 2012a, 2012b; Ahmad and Umar, 2011; Hadiarto and Tran, 2011; Shinozaki and Yamaguchi-Shinozaki, 2007; Tran and Mochida, 2010a). Crop plants often come into contact with various abiotic stresses, including drought, high and low temperatures, salinity, heavy metals, low soil fertility, and mechanical wounding. It is reported that these stresses can reduce the crop yield to less than 60% (Hadiarto and Tran, 2011; Tester and Langridge, 2010; Tran and Mochida, 2010a). A basic step toward understanding the molecular mechanisms via which plants cope with stress conditions is gene or protein identification. The first question is the response of the plant to its changing environment, and this is dependent on the ability of the plant to detect stimuli. This leads to signal transduction within the plant cell that brings about activation of the related genetic programs (Ashraf, 2010; Hadiarto and Tran, 2011; Shinozaki and Yamaguchi-Shinozaki, 2007). Protein modification is also an important mechanism initiated in the response to stress. There are many signalling pathways that lead to these various defense responses. Signalling pathways are interconnected with one another at many levels. Only proteomics tools can help in understanding the interactions between the different signalling pathways. Several researchers have reported that proteins respond to stress in different plants differently (Komatsu et al., 2009; Kosová et al., 2011; Witzel et al., 2009). These findings are related to the different stress enzymes expressed in plant cells in response to abiotic stress. The complete sequencing of the genomes of Arabidopsis (Kaul et al., 2000), and rice (Tanaka et al., 2008), has enabled plant breeders to access large numbers of plant genes. The identification of genes or proteins functional in the plant's response to stress requires a range of genetic information and molecular tools (Rossignol et al., 2006).
2D-PAGE is used to identify the specific proteins whose accumulation is altered under abiotic stresses (Table 2). Protein expression in stress-tolerant and stress-sensitive plants can also be evaluated with the help of proteomics (Jorrín et al., 2007). Using proteomics, the effects of environmental stress on the proteins at the subcellular, cellular, and organ levels, such as mitochondria of Pisum sativum (Baginsky, 2009; Taylor et al., 2005), the nucleus of Arabidopsis (Jain et al., 2006), the anther of rice (Imin et al., 2004), and poplar leaves (Renaut et al., 2004), have also been successfully explored. Experiments by Boudsocq and associates (2007), using proteomics to study the effects of various abiotic stresses on the levels of heat shock proteins and a dnaK-type molecular chaperone, have revealed a reduced expression of these elements, whereas a 26S proteosome regulatory subunit was responsive only to osmotic stress. Regulation of gene expression is one of the strategies used by plants to fight stress. Brechenmacher and colleagues (2009) have exploited the full potential of proteomics for studying the functional genomics of soybeans. A protein reference map of soybean root-hair cells was generated, which showed a total of 1492 different proteins. These observations improved our understanding of protein functions in plant water and nutrient uptake. Also, Ahsan and Komatsu (2009) did proteome analysis at various developmental stages of soybean leaves and flowers and reported 500 and 600 proteins for the leaf and flower tissues, respectively. They found differential expression of 26 proteins in the leaves at various developmental stages, whereas in buds and flowers the proteome profile revealed 29 differentially expressed proteins. A total of 478 non-redundant proteins were identified using both 2-PAGE and multidimensional protein identification technology (MudPIT) in proteome profiling of the soybean during seed filling. The genetic nature of the overall environmental stress mechanism in plants is shown in Figure 3.

A simple model of the environmental stress responses in crop plants (ROS, reactive oxygen species).
Salinity stress
Salt is a major environmental stress that limits the crop yield, especially in arid and semiarid regions. Soil salinity is known to affect about 800 million hectares of agricultural land worldwide (Munns and Tester, 2008). This is more than 6% of the world's total land area. Due to land clearing or irrigation, salinity has affected a significant part of cultivated agricultural land. Excessive salinity of soil or water is a limitation of agriculture production in some semi-arid regions where agriculture is dependent on irrigation (Munns and Tester, 2008). Increases in various salt concentrations, especially of Na+ and Cl−, and also Ca2+, K+, (CO3)2−, (NO3 )−, and (SO4 )2−, decreases the water retentive potential of soil, which in turn decreases water uptake by roots. Therefore, elevated levels of NaCl cause osmotic stress and disturb the ion homeostasis of the cell. During osmotic stress, there is accumulation of several low-molecular-weight osmolytes, such as glycine betaine (GB) and proline- and raffinose-derived oligosaccharides, as well as of high-molecular-weight hydrophilic proteins from the late embryogenesis-abundant (LEA) superfamily (Ahmad, 2010; Ahmad and Sharma, 2010; Ahmad et al., 2010b, 2012b; Azooz et al., 2011; Cheng et al., 2009; John et al., 2009a). Also, a number of adverse processes are known to occur in plants during salinity stress, including membrane disorganization, increases in the amounts of toxic metabolites, the generation of reactive oxygen species (ROS), reduced nutrient uptake, inhibited photosynthesis, and finally the death of the cell and plant (Ahmad and Prasad, 2012a, 2012b; Ahmad and Umar, 2011; Ahmad et al., 2008, 2009, 2010a, 2010b, 2011a; John et al., 2009b; Witzel et al., 2009). Yan and associates (2006) observed 1100 spots in the proteome of rice roots that were seen upon exposure to high salt concentrations (150 mmol/L) on 2D gels, with 34 protein spots upregulated and 20 spots downregulated. Abbasi and Komatsu (2004) showed that salt stress reduces plant growth. This was also reported by Yan and associates (2006), who showed reductions in the levels of glutamate synthetase (GS), a key enzyme for nitrogen assimilation, during salt stress. It is interesting to note that many proteins respond to salt stress and also to other abiotic stresses. For instance, two proteins (peroxidase and putative nascent polypeptide-associated complex α-chain) respond to rice plants under salt stress, and also show a similar response to sugar beet leaves (Beta vulgaris) under drought stress (Hajheidari et al., 2005). Dooki and colleagues (2006) observed that 13 proteins in young rice panicles significantly changed their expression levels under salt stress. MS analysis of highly abundant proteins of the panicle led to the identification of proteins that are involved in several salt-responsive mechanisms. These proteins may help the plant to adapt to salt stress by upregulation of antioxidants and the proteins involved in translation, transcription, signal transduction, and ATP generation. Abbasi and Komatsu (2004) showed that photosystem II (PII), an oxygen-evolving enhancer protein precursor, was upregulated in the rice leaf sheath under salt stress. In wheat, ferredoxin NADP+ oxidoreductase was upregulated during salt stress (Caruso et al., 2008). In the roots of soybean seedlings, metabolism-related proteins are mainly downregulated under salt stress. A decrease in some glycolytic enzymes (glyceraldehyde-3-phosphate dehydrogenase; GAPDH), and several proteins involved in CO2 assimilation, were also observed as a result of salinity stress in crops (Sobhanian et al., 2010). Proteins with important functions, like membrane stabilization, proton homeostasis, and signal transduction, always show increased expression. Cheng and associates (2009) reported a new salt responding to leucine-rich-repeat type receptor-like protein kinase, OsRPK1. Wang and co-workers (2008) found some interesting results while studying the responses of the common Chinese wheat cultivar Jinan 177 and its hybrid under salt stress with a salt-tolerant Thinopyrum ponticum. Significant changes in the expression of proteins involved in signal transduction (small G proteins, ethylene signalling, and the MAP kinase cascade) were observed in both genotypes. A number of transcription and translation factors, such as the putative transcription factor BTF3 from the nascent polypeptide-associated NACA protein family, and DEAD box RNA helicase involved in the modulation of stress-inducible CBF/DREB transcriptional activators, were upregulated at high salt concentrations. Upregulation of several transporter proteins (vacuolar proton ATPase subunit E involved in Na+/H+ antiport, ABC transporters involved in the transport of secondary metabolites, and the exocyst subunits SEC1a and EXO70), and several proteins with protective functions (HSP70 and other chaperones), was observed under salt stress. Additionally, an increase in the level of transcription factor proteins by genetic engineering, such as DREBs and NACs, significantly enhanced the tolerance of both the model and crop plants to a wide range of environmental stresses, including drought, salt, and cold, even in field conditions (Ahmad and Prasad, 2012a, 2012b; Ahmad and Umar, 2011; Ahmad et al., 2010a; Hu et al., 2006; Nakashima et al., 2007; Qin et al., 2008; Tran et al., 2004, 2007b). Aghaei and colleagues (2009) reported that some proteases make their way to the cytosol from injured vacuoles under salt stress in the hypocotyls and roots of the soybean, and were found to neutralize the deleterious effects of salt on cell proteins. The expression levels of proteins also showed significant changes in grapevines under salt stress (Jelloulia et al., 2008). A total of 48 proteins showed differential expression, of which 32 showed upregulation, 9 showed downregulation, and 7 new protein spots were observed. Thus adaptation to salt stress is the result of modifications of gene expression that alter the cellular machinery. Moreover, when evaluating plant responses to salt stress, quantitative analysis of gene expression at the protein level is essential. Expression profiling at the protein level of plants under stress reflects the importance of the proteomic approach (Aghaei et al., 2009). Therefore, molecular tools are required for the identification of salt-stress-responsive genes, and proteomics is the most reproducible tool to determine the functionality of the identified genes (Afroz et al., 2011; Aghaei et al., 2009; Kosová et al., 2011; Sobhanian et al., 2011; Zhang et al., 2012). Evers and associates (2012) concluded that during cold and salt stresses in the potato the majority of photosynthesis-related genes were downregulated, whereas cell rescue and transcription factor-related genes were mostly upregulated.
Temperature stress
Temperature stress can greatly affect plant metabolism (Suzuki and Mittler, 2006). High and low (chilling and freezing) temperatures are known stressors that decrease the production of many important crops such as rice, wheat, and maize. In temperate regions low temperatures (0–12°C) are common during the growing season, and are known to decrease crop production, since chilling temperatures significantly alter plant metabolism and physiology (Foyer and Noctor, 2005). To study and understand the changes occurring inside plants due to cold stress, a proteomics approach is a wonderful tool for deciphering these changes (Afroz et al., 2011; Evers et al., 2012; Kosova et al., 2011; Zheng et al., 2012). Experiments by Yan and co-workers (2006) showed that the protein-expression pattern in rice leaf changes in response to cold stress. In this study, over 1000 protein spots were resolved when leaf proteins were separated by 2D-PAGE, and among them 31 proteins showed downregulation and 65 proteins showed upregulation. Yan and associates (2006) showed that the majority of the spots that were differentially expressed were consistently upregulated or downregulated until 24 h of recovery from temperature stress. These differentially-expressed proteins were further analyzed by PMF or matrix-assisted laser desorption ionization time-of-flight MS (MALDI-TOF/MS) and functionally classified. Another study by Kosmala and colleagues (2009) examined the functional distribution of differentially-expressed proteins in Festuca pratensis under cold stress. The functional group that was most affected by cold stress was photosynthesis, with 35.3% of cold-responsive proteins residing in this category (Kosmala et al., 2009). Lee and associates (2009) identified some novel proteins known to be involved in energy production and metabolism, vesicular trafficking, and detoxification (e.g., acetyl transferase, phosphogluconate dehydrogenase, fructokinase, NADP-specific isocitrate dehydrogenase, putative alpha-soluble N-ethylmaleimide-sensitive factor [NSF] attachment protein, and glyoxalase 1), and found that they showed higher sensitivity to cold stress in rice. Hashimoto and Komatsu (2007) observed downregulation of certain proteins at the organ level under cold stress. For example, 5-methyletetrahydropteroyl triglutamate-homocysteine S-methyl transferase was downregulated not only in the roots, but also in leaf sheaths, suggesting that common cold-stress-responsive pathways exist for the regulation of some proteins in roots and leaf sheaths. Studies carried out by Kamal and associates (2010) on wheat under cold stress revealed that some cold stress-related proteins, such as cold acclimation proteins (9000–22,000), cold shock proteins (16,000–38,000), ABA-inducible proteins (10,000–41,000), cyclophilin (13,000–18,000), low-temperature-regulated proteins (7000–14,000), kinase-like protein (6000–74,000), nitrogen-activated proteins (40,000–80,000), transcriptional adaptors (7000–29,000), and translation initiation proteins (12,000–17,000), were induced in wheat. In the winter wheat variety Cheyenne, Rinalducci and co-workers (2011) showed that out of 1000 protein spots that were reproducibly detected on each gel, 31 protein spots were downregulated and 65 were upregulated. MS analysis of these proteins showed 85 differentially-expressed proteins, including the well known and novel cold-responsive proteins, like RNA-binding proteins, lectin protein, and peptide methionine sulfoxide reductase (PMSR). PMSR is known to catalyze the reduction of protein-bound methionine sulfoxide groups to methionine, which protects the proteins from inactivation and degradation by ROS. During chilling stress many proteins, especially proteins related to photosynthetic activity, (e.g., Rubisco large subunit, of which 19 fragments were detected), showed degradation.
High-temperature stress is prevalent in plants because of the emission of greenhouse gases. A large part of crop loss is due to increases in temperature worldwide, especially when combined with drought or a variety of other stressors. Heat stress is responsible for improper folding of proteins and leads to the denaturation of intracellular proteins and membrane complexes. Heat stress increases the expression of proteins with chaperon functions, such as heat-shock proteins (HSPs), and so-called small HSPs (sHSPs). Peng and associates (2004) observed that rice grain yields decreased by 10% for each 1°C increase in the minimum temperature during the growing season. Lee and colleagues (2007) identified novel high temperature stress-responsive genes and determined their expression patterns. Heat stress is also responsible for oxidative damage to biomolecules. Also, several enzymatic and non-enzymatic antioxidants are upregulated and aid in redox homeostasis. These enzymes include dehydroascorbate reductase (DHAR), glutathione-S-transferase (GST), thioredoxin h-type (Trx h), and chloroplast precursors of superoxide dismutase (SOD). Dong and colleagues (2007) also used a proteomics approach to analyzing heat-responsive proteins in rice leaves. This study showed that heat stress affects proteins, namely sHSPs. Zhang and co-workers (2010) also showed that heat stress induces profound changes in cytoskeleton composition, indicating its reorganization. In addition, an increased accumulation of some eukaryotic translation initiation factors (eIF4F and eIF5A-3) was observed, indicating profound cellular organization leading to programmed cell death (PCD) under heat stress. Ahsan and associates (2010) reported enhanced accumulation of several other proteins with chaperone functions (chaperonin 60 β subunit CPN60-β, HS90, chaperonin CPN10, and chloroplast chaperonin) in soybean seedlings under heat stress.
Heavy metal stress
Intensive industrial activity over the past 100 years has resulted in massive releases of heavy metals into the environment. Many heavy metals such as nickel (Ni), chromium (Cr), lead (Pb), cadmium (Cd), mercury (Hg), and others, are important health-threatening pollutants. Major sources of heavy metal pollution include power stations, cement factories, zinc smelting, coal burning, paint factories, and the use of phosphate fertilizers. Ions of these heavy metals are known to be mobile in the geosphere, and their accumulation in the human food chain is a cause of concern for health specialists. The main entry pathway of heavy metals into human and animal foods is through uptake by crop plants (Ahmad et al., 2011a; John et al., 2009a, 2009b). There are different ways that plants respond to heavy metal toxicity, such as immobilization, exclusion, chelation, and compartmentalization of metal ions (Ahsan et al., 2008). The formation of peptide metal-binding ligand phyto-chelatins (PCs) and metallothioneins (MTs), as well as more general stress-response mechanisms, are among the ways plants combat stress (Ahsan et al., 2009; Hradilová et al., 2010). Impairments in growth and development are among the effects of metal ions in plants. The generation of ROS through chain reactions characterized by an excess of free metals causes damage at a molecular level (Kieffer et al., 2009). It is known that increased ROS production induces the expression of HSPs and chaperones, and protects the cell from oxidative damage. If these HSPs are considered stress markers, they mark the morphological changes seen due to stress in the aerial plant parts. A study by Hradilová and colleagues (2010) showed that HSPs were able to indicate stress symptoms up to day 14, and that other toxicity symptoms were not apparent. Nearly 20% of the stressed plants had necrotic spots that were seen mostly in young leaves. Cheng and associates (2009) reported that the large number of stress proteins induced by heavy metal stress have a molecular mass of 10,000–70,000 Da in plants. Therefore proteome analysis of the proteins involved in the response to heavy metals can deliver more accurate and comprehensive information for better understanding of the plant response to heavy metal stress. Labra and co-workers (2006) reported the upregulation of proteins involved in different cellular compartments and metabolic pathways in response to the protein changes caused by potassium dichromate treatments in Zea mays. They suggested that the activation of oxidative stress mechanisms affects sugar metabolism and ATP synthesis. Additionally, patterns of protein expression of maize roots under arsenic stress was examined by Requejo and Tena (2005), who reported that about 10% of all detected maize root proteins were upregulated or downregulated by arsenic. Twenty proteins showed reproducible effects of the metal, and were selected for further analysis by MALDI-TOF/MS. Out of these 20 proteins, 11 were identified by comparing their peptide mass fingerprints to a protein and expressed sequence tag database. Their study found that maize root proteins highly responsive to arsenic exposure included a major homogenous group of seven enzymes that are involved in cellular homeostasis for redox perturbation (e.g., three superoxide dismutases, two glutathione peroxidases, one peroxiredoxin, and one p-benzoquinone reductase), in addition to four additional functionally heterogenous proteins (e.g., ATP synthase, succinyl-CoA synthetase, cytochrome P450, and guanine nucleotide-binding protein b subunit). These findings strongly suggest that the induction of oxidative stress is the main process underlying heavy metal toxicity in plants.
According to Lee and associates (2010), upregulation of enzymes cooperating with reduced glutathione (GSH) in plants was observed with increasing cadmium concentration. During the ROS-quenching process, approximately half of the upregulated proteins in the roots of rice belonged to the category involved in the oxidative stress response and GSH metabolism, including peroxidases, putative ferredoxin:NADP(H) oxidoreductase, and APX1. Accumulation of proteins like copper chaperone (CCH), ROS-scavenging enzymes, and those involved in the biosynthesis of GSH such as GS-like 1, are protective in function and are induced by elevated levels of cadmium. The activity of Rubisco, such as Rubisco LSU-binding proteins, Rubisco activase 2, a chloroplast precursor of Rubisco activase, ribulose-phosphate 3-epimerase, and carbonic anhydrase, decreased significantly in response to cadmium stress. A number of studies have also been conducted on elevated concentrations of arsenic (Ahsan et al., 2008), copper (Ahsan et al., 2007), nickel (Ingle et al., 2005), mercury (Isarankura-Na-Ayudhya et al., 2009), and zinc (Isarankura-Na-Ayudhya et al., 2009). These studies indicated the formation of necrotic spots associated with a drastic degradation of Rubisco, especially the Rubisco LSU. Cysteine synthase (CS), GST, GST-tau, and tyrosine-specific protein phosphatase proteins (TSPP) were markedly upregulated under heavy metal stress.
Thus the proteomics approach enables us to reveal molecules that play important roles in plants during exposure to heavy metals, and generates data about stress-related responses in different tissues. This information aids in the design and selection of plants that resist deleterious metal accumulations in the food chain. This may also help solve problems with essential metal deficiencies in humans.
Osmotic and water-deficit stress
Among the abiotic stresses, drought is responsible for widespread decreased crop production worldwide. Various mechanisms, including physiological and molecular mechanisms, are involved in the plant adaptation to drought. In plants, drought-induced proteins are known to be involved in physiological adaptations to water stress. Proteomics has been found to be a useful tool for the identification of the mechanisms involved in drought stress and tolerance (Hajheidari et al., 2005). A proteomics approach has been used by several groups to study drought responsiveness in many plant species, such as poplar (Durand et al., 2011), maize (Zhu et al., 2007), rice (Liu and Bennet, 2011), soybean (Yamaguchi et al., 2010), and oak trees (Sergeant et al., 2011). Zang and Komatsu (2007), and Joshep and Jini (2010), gave mannitol treatments to rice plants to evaluate the mechanisms of the responses of plants to osmotic stress using a proteomic approach. The observations of Joshep and Jini led to the identification of 327 drought-responsive proteins. Among them the levels of 12 proteins were upregulated and 3 proteins were downregulated with increases in the duration of mannitol treatment. Xu and associates (2009) observed the damage occurring in Hippophae rhamnoides (seabuckthorn) with downregulation of HsIU (a heat shock protein belonging to the Hsp100/Clp family, with ATPase and unfoldase activity), and a putative ABC transporter ATP-binding protein, due to the effect of drought stress. They also observed an increase in [Fe–S] cluster assembly that is known to occur under drought. This was implied by the accumulation of J-type co-chaperone Hsc20. From this result, they concluded that iron–sulfur [Fe–S] proteins that are commonly found in all types of organisms play important roles in processes such as redox and non-redox catalysis and signalling processes in seabuckthorn. Yamaguchi and colleagues (2010) studied the effect of drought on soybean primary roots using a proteomics approach. They observed that several enzymes and proteins related to isoflavonoid biosynthesis had increased expression levels. This may contribute to growth maintenance of roots under stressful conditions. Contrary to this, caffeoyl-CoA O-methyltransferase, an enzyme playing role in lignin synthesis, was highly upregulated. This might be associated with the enhanced accumulation of lignin, which is related to the inhibition of growth. Several proteins were increased in abundance in water-stressed roots of the soybean, and played a role in protection from oxidative damage (Yamaguchi et al., 2010). These authors were able to detect 35 proteins that exhibited significant changes in their expression profiles in drought-affected soybean primary roots. These differential expressions were observed in at least one region of water-stressed roots, compared with well-watered controls. Manaa et al. (2011a) found that during osmotic stresses, levels of ABA, phospholipid signalling, and mitogen-activated protein kinases (MAPKs) were increased in tomato. Different signalling processes were integrated to work together against osmotic stress (e.g., ABA and phospholipid molecules appeared to function upstream of the osmotic stress-activated protein kinases; Manaa et al., 2011a). Drought stresses are known to induce Ca2+-binding proteins that serve as transducers of Ca2+ signals (Kosová et al., 2011; Li et al., 2010; Sarwat et al., 2012). Ca2+-binding proteins such as calmodulin have been identified in plants (Sarwat et al., 2012). Ca2+-dependent protein kinases, calcineurin B–like proteins, and SOS3 are involved in ABA-dependent stress responses in plants. Still, the functions of many proteins induced in higher plants are unknown, and their functions can only be inferred from information available for other organisms. Proteomics can be used to analyze water-deficit-responsive proteins in various plants (Ge et al., 2012). It may also help in identifying the functions of proteins that have a significant role in the plant response to drought stress.
Waterlogging and flooding stress
One of the most damaging abiotic stresses is waterlogging caused by excessive water in the soil. Worldwide about half of crops (approximately 2 million hectares) is affected by flooding annually, causing a 25% reduction in yield (Valliyodan and Nguyen, 2008). Waterlogging also restricts the availability of oxygen to plant roots. Anaerobic metabolism occurs due to a lack of oxygen and leads to glycolysis, followed by pyruvate fermentation. In higher plants the cell wall is the first organelle to respond to flood stress signals, and transmits it to the cell's interior, subsequently affecting the cell signalling cascade, and the cell's stress tolerance or intolerance (Komatsu et al., 2003, 2010, 2012). Thus proteins in cell walls are involved in cell-wall signal transduction, structure, metabolism, and cell enlargement in response to the waterlogging stress. Numerous cell-wall proteins involved in stress tolerance have been identified in plants, such as soybean (Alam et al., 2010), chickpea (Bhushan et al., 2007), maize (Zhu et al., 2007), and rice (Cho et al., 2009). Sakata and colleagues (2009) revealed relationships among the 106 mRNAs, 51 proteins, and 89 metabolites that showed variations in the soybean under flooding stress. Another study of roots and hypocotyls in soybean seedlings (Komatsu et al. 2009) identified several inducible genes and proteins. Within 12 h of stress, genes that were associated with ethylene biosynthesis, alcohol fermentation, cell wall loosening, and pathogen defense were significantly upregulated. Altered expression of trypsin protease inhibitor and acid phosphatase were also observed both at the transcriptional and the proteome level. Several studies carried out on the soybean (Alam et al., 2010; Hashiguchi et al., 2009; Komatsu et al., 2010, 2011; Nanjo et al., 2010) under flooding stress have confirmed that proteins involved in glycolysis and fermentation pathways, such as alcohol dehydrogenase, fructose-bisphosphate aldolase, UDP-glucose pyrophosphorylase, and GAPDH, exhibit upregulated expression. These results confirm that flooding stress is also involved in stress due to oxygen starvation. A comparative proteomic approach (Ahsan et al., 2007) in tomato leaves responding to waterlogging stress resulted in increases in ion leakage and lipid peroxidation and increased H2O2 content, whereas the chlorophyll content decreased. Using MALDI-TOF/MS, the authors identified 52 protein spots that were differentially expressed in tomato. Many of the identified proteins were involved in photosynthesis, energy, metabolism, and protein biosynthesis. A number of the proteins identified also play roles in disease resistance, and stress and defense mechanisms. It was also observed that stress leads to disintegration of the fragments of large subunits of Rubisco. Proteome profiling by Alam and colleagues (2010) showed that continuous waterlogging stress caused an increase in glycolytic flux to meet ATP requirements. Anaerobic energy metabolism was upregulated to meet the objective. During flooding stress, the terminal electron acceptor (oxygen) of the electron transport chain (ETC) is unavailable, thus the intermediate electron carriers become reduced, affecting the redox characteristics of the cell (i.e., the NADH/NAD+ ratio). In this case, upregulation of GS in cells might help maintain the redox potential of the cell (Alam et al., 2010). Large protein sequence databases are available for Arabidopsis thaliana and rice, and they are therefore considered to be model plants (Kosová et al., 2011). These studies led to the identification of different pathways that are shared by different plant species under various stress conditions, as well as the pathways unique to a given stressor.
Hydrogen peroxide, ultraviolet rays, and ozone stress
ROS include singlet oxygen (O2), superoxide anions (O2−), hydrogen peroxide (H2O2), and hydroxyl radicals (HO·), are highly reactive and toxic, and lead to oxidative damage to biomolecules (Ahmad and Prasad, 2012a, 2012b; Ahmad and Umar, 2011). Nonetheless, ROS also act as important regulators of many biological processes, such as cell growth and development, hormone signaling, and stress responses (Tripathi et al., 2011). ROS imbalance in the cell is closely linked to several types of oxidative destruction; therefore cellular redox conditions should be well regulated. H2O2 is non-radical and carries no net charge, and has a comparatively longer half-life, which makes it a more likely long-distance signalling molecule (Zhou et al., 2011). H2O2 acts as a physiological indicator of stress intensity when plants are challenged with biotic and/or abiotic stresses. It can therefore be used to activate stress-responsive genes. Therefore, proteomic analysis of the H2O2 response may be of paramount importance for the understanding of the plant network to environmental stresses (Bakalova et al., 2008). Detoxification of ROS by extracellular ascorbic acid in the leaf apoplast has been identified as a potential tolerance mechanism toward H2O2 (Cheng et al., 2009). The apoplast consists of cell walls and the intercellular spaces. The root apoplast is important for a plant's interactions with its environment. It can sense environmental changes and stress signals and then transfer them into the cell's interior to trigger a cell-wide response. In addition to signal perception and transduction, apoplast proteins can also be used for cell wall modification and reconstruction, as well as for defense responses, although they comprise only 5–10% of the wall's dry weight. Using a bioinformatics approach, we have discovered that there are more than 1000 different apoplastic proteins in Arabidopsis (Jamet et al., 2008). Several reports have appeared about leaf apoplast proteomes under various abiotic stresses, such as salt in tobacco (Dani et al., 2005), dehydration in chickpea and rice (Bhushan et al., 2007), and boron deficiency in Lupinus albus (Alves et al., 2006). These studies have broadened our understanding of the complicated regulation of leaf apoplast proteins. A few studies have addressed the dynamic changes in the proteome of the plant root apoplast seen in response to oxidative stress, especially to H2O2, which serves as a physiological indicator of abiotic stress intensity. High irradiance and elevated ozone (O3) concentrations are seen with UV-B exposure (315–280 nm) under field conditions. It is well known that atmospheric UV-B and O3 are important components affecting the global climate, and the slightest change in their levels has adverse impacts on physiological and biochemical growth characteristics, as well as on the productivity of many plants (Ahsan et al., 2010). Therefore it is important to study their interactive effects on plant systems. Experiments done by Kumari and associates (2009) in Abelmoschus esculentus showed that important target sites for the action of UV-B and O3 are cell membranes, proteins, and DNA. The results of this study also indicated that UV-B and O3, individually as well as in combination, seem to have significant effects on the antioxidant system in plants. O3 uptake is an important phenomenon inside plant cells, and tends to be directly proportional to stomatal conductance (Tripathi et al., 2011). Therefore, once inside the plant, O3 and UV-B are known to generate ROS in cells, leading to oxidative damage. This leads to alterations in the gene expression of proteins fundamentally important to all functions of cells, to produce different proteins to cope with these stresses. Mechanisms for combating ROS depend on the interrelated activities of several antioxidant enzymes, like catalase (CAT), SOD, peroxidase (POD), ascorbate peroxidase (APX), and glutathione reductase (GR), and non-enzymatic antioxidants such as ascorbic acid, glutathione, and alpha-tocopherol (Ahmad and Umar, 2011; Ahmad et al., 2010a; Renaut et al., 2009). UV radiation may induce plants to generate signal transduction intermediates, such as nitric oxide, ROS, and ethylene, followed by downregulation of photosynthesis-related proteins, such as light-harvesting Chl a/b-binding protein, and upregulation of protective proteins, such as pathogen-related protein-1 (PRP-1) and pigment-dispersing factor 1.2 (PDF 1.2), at the mRNA level. Leaves are the main photosynthesizing organs, and they absorb nearly 90% of UV-B. A proteomic study by Du and co-workers (2011) of rice leaf total proteins under early UV stress (at 8 h) found 22 unique proteins by MALDI-TOF. This indicates how proteome analysis helps us discover the expression patterns of UV-induced genes at the protein level. Recently, Tripathi and associates (2011) observed several-fold reductions of major leaf proteins, including Rubisco, in rice plants exposed to UV-B. For the O3 effect, Sarkar and Agrawal (2010) observed high protein loss in rice plants grown in open-top chambers with elevated O3 levels. Feng and associates (2008) found a significant loss of photosynthetic proteins, including Rubisco, in rice plants exposed to O3. Besides the induction of stress-related proteins like APX and SOD, the induction of class 5 proteins (PR5) and pathogenesis-related (PR) and PR10 proteins was also observed.
Phosphoproteomics
Protein phosphorylation comprises a major area of post-translational modifications, especially in plants that mobilize a high number of genes. Many crucial physiological and biochemical functions, including responses to environmental stimuli and diseases that contribute to the complexity of the proteome, comprise part of phosphoproteomics, and play a role in the identification of phosphoproteins, quantification of phosphorylation sites, and the precise mapping of proteins and their biological utility. Recently several plants have been analyzed using systemic phosphoproteomics, and this aided in the optimization of in vitro and in vivo technologies to reveal components of the phosphoproteome within the cell/organelle (Nakagami et al., 2010, 2012). The development of a new phosphoproteomic tool, the technically improved MS-based identification of phosphorylated residues in proteins, is highly sensitive and accurate. Mass spectrometers in combination with phosphopeptide enrichment methods, mainly immobilized metal affinity purification (IMAC) and titanium dioxide (TiO2), are involved. This has led to the discovery of novel substrates that are specific for protein kinases. These methods have enabled rapid identification of kinase substrates, such as kinase assays, using plant protein microarrays. Studies have shown that phosphorylation events are involved in a wide range of biological processes taking place in plants and other organisms (Sugiyama et al., 2008). To understand the core regulatory systems, the characterization of conserved phosphoproteome features in plants is useful. Hence phosphoproteomics can promote the improvement of agronomically-important plant species. Recent research projects in phosphoproteomics technology have led to the identification of thousands of phosphorylation sites in unfractionated plant cells by simple enrichment methods. Sugiyama and colleagues (2008), using phosphoproteomics, reported more than 2000 phosphorylation sites in Arabidopsis. Research on the Medicago phosphoproteome has shed light on comparable phosphoproteomes from other plant species as well (Kersten et al., 2009). Progress in the quantitative and dynamic analysis of mapped phosphorylation sites is also occurring. A new approach, shortgun proteomics, is emerging for proteome analysis. Shortgun proteomics is useful for the identification of thousands of proteins simultaneously from a complex sample (Nakagami et al., 2012). Continuing work on phosphorylation sites in plants has prompted the creation of web resources for plant-specific phosphoproteomics data. In the era of computational biology, there is a need to significantly improve the sensitivity and specificity of the detection of phosphorylation sites in plants (Grimsrud et al., 2010).
Conclusions
Tremendous progress in the field of plant proteomics has been made in recent years. Studying the dynamic plant proteome has become comparatively simple and accurate with the advancements seen in technology and bioinformatic tools. With these developments plant physiologists are now able to solve mysteries about the physiological processes that take place inside plants. From seed germination to grain harvest, there has been tremendous growth in proteomics research around the world. Knowledge of the proteins involved in nutritional and other plant processes have widened our understanding of various metabolic processes using the proteomics approach. One remarkable achievement of proteomics is the discovery of the functions of various proteins and their expression levels in plants undergoing various types of stress. The proteomic strategy involved in the study of protein localization in cells is a necessary first step toward understanding protein functions in complex cellular networks. These fascinating events taking place during the metabolism that governs various physiological processes have been revealed by the dynamic nature of the proteome, and we believe that proteomics represents an important advance in the study of plant physiological processes.
Future Perspectives
Developments in proteomic technologies may aid researchers in understanding the plant engineering involved in important processes like nutrition, crop yields, and defense. Proteomics applications and advancements in technology such as multidimensional protein fractionation, isobaric tags for relative and absolute quantitation, label-free quantification mass spectrometry, and phosphoprotein and glycoprotein enrichment and tagging, will enable the discovery of proteins and novel regulatory mechanisms that occur during salt stress signalling and related metabolic pathways. The integration of proteomics with transcriptomics, metabolomics, and bioinformatics, has facilitated insights into the molecular networks underlying salt stress response and tolerance. These data will be used to analyze how different components interact, and generate responses to control divergent metabolic pathways. Thus, the proteomics community needs to work in concert with those working in transcriptomics, metabolomics, and bioinformatics, to reveal the mechanisms affecting plant growth and the stress response.
Footnotes
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.