
Abstract
Abstract
Halobacterium salinarum is an extremely halophilic archaeon that inhabits high-salinity aqueous environments in which the temperature can range widely, both daily and seasonally. An OMICS analysis of the 37°C and 49°C proteomes and transcriptomes for revealing the biomodules affected by temperature is reported here. Analysis of those genes/proteins displaying dramatic changes provided a clue to the coordinated changes in the expression of genes within five arCOG biological clusters. When proteins that exhibited minor changes in their spectral counts and insignificant p values were also examined, the apparent influence of the elevated temperatures on conserved chaperones, metabolism, translation, and other biomodules became more obvious. For instance, increases in all eight conserved chaperones and three arginine deiminase pathway enzymes and reductions in most tricarboxylic acid (TCA) cycle enzymes and ribosomal proteins suggest that complex system responses occurred as the temperature changed. When the requirement for the four proteins that showed the greatest induction at 49°C was analyzed, only CctA (chaperonin subunit α), but not Hsp5, DpsA, or VNG1187G, was essential for thermotolerance. Environmental stimuli and other perturbations may induce many minor gene expression changes. Simultaneous analysis of the genes exhibiting dramatic or minor changes in expression may facilitate the detection of systems level responses.
Introduction
T
HSPs, which can be induced not only by thermal shock but also by other stimuli, such as radiation, hypoxia, salinity, and parasitism, are ubiquitously present in eukaryotic, bacterial, and archaeal cells (Sørensen et al., 2003). The lethal damage caused by heat includes the unfolding or misfolding of proteins, which can lead to protein denaturation or aggregation (Nguyen et al., 1989; Richter et al., 2010). Therefore, the primary functions of HSPs are to monitor and respond to the state of protein folding and homeostasis through the chaperone and protease systems in cells (Mathew and Morimoto, 1998). Molecular chaperones are the predominant group of HSPs and are broadly conserved among the three domains of life. Previous analyses of several complete genome sequences revealed that the HSP100 and HSP90 families are absent from the Archaea. Therefore, in the Archaea, the HSP70/DnaK, HSP40/DnaJ, small HSPS (sHSPs), and HSP60/chaperonin families are considered to assist in folding proteins into their native functional states ( Lund et al., 2003; Macario et al., 1999). If damaged proteins cannot be repaired, then well-known proteolytic factors, such as Lon and Clp, or proteasomes play very important roles in preventing the accumulation of these proteins (Goldberg, 2003). The requirement for proteasomes in the Archaea under heat shock conditions has been demonstrated in previous studies. Thermoplasma acidophilum cells became nonviable after extended exposure to carboxybenzyl-leucyl-leucyl-leucine vinyl sulfone (Z-L3VS), a potent proteasome inhibitor, at a heat shock temperature (Ruepp et al., 1998). Haloferax volcanii 20S core and proteasome-activating nucleotidase (panA and panB) gene deletion mutants (ΔpsmA, ΔpsmAΔpanA, and ΔpsmAΔpanB), which carry a common deletion of the 20S proteasomal core α1 subunit gene (psmA), display reduced thermotolerance (Zhou et al., 2008).
Although the amino acid sequences and functions of HSPs are highly conserved, the mechanisms regulating the expression of the heat shock protein genes (hsp) are strikingly different across the three domains. For instance, in the prokaryotic model organism Escherichia coli, the transactivation of the hsp genes is triggered by elevated levels of the σ32 factor, which replaces the major transcription initiation factor σ70 and recruits RNA polymerase to hsp promoters to induce gene expression (Arsene et al., 2000; Wang and deHaseth, 2003). Eukaryote hsp genes are mainly modulated by interactions between the heat shock factor (HSF) and a heat shock element (HSE) in the hsp promoter regions. In higher eukaryotes, inactive HSFs are monomeric, preexist in the cytoplasm, and are activated by their heat-induced homotrimerization. The trimers then accumulate and bind to HSEs inside the nucleus, stimulating hsp gene expression after their phosphorylation (Mager and De Kruijff, 1995). Neither genes homologous to those encoding bacterial sigma factors nor genes homologous to those encoding eukaryotic HSF/HSE components have yet been identified in the Archaea (Hickey et al., 2002). A recent study demonstrated that the novel archaeal protein Phr specifically binds the transcription initiation sites of hsp20, an AAA+ ATPase gene, and to phr itself. Moreover, inhibition of the transcription of these genes can be reversed by incubation at high temperatures in vitro (Vierke et al., 2003). However, the exact mechanisms that regulate the expression of these genes are yet to be clarified.
This study, in which we compared the cellular expression changes that occur at temperatures of 37°C and 49°C, complements previous transcriptomic and two-dimensional (2D) gel-based proteomic analyses of H. salinarum grown at 42°C and 49°C ( Coker et al., 2007; Shukla, 2006). The induction of five proteins and eight genes, including several conserved HSPs, at 49°C was documented in those studies. This suggests that H. salinarum has a heat shock-like response at the optimal growth temperature of 49°–50°C, revealed by a growth kinetic analysis based on Arrhenius plots (Robinson et al., 2005). Because some of the conserved hsp mRNAs increased at 42°C and 49°C but remained low at 30°C and 37°C, it was interesting to compare the differences in their expression in cells incubated at 49°C or 37°C. Since proteins are the terminal functional products of the bioinformation pathway, the expression changes triggered by a temperature shift were initially examined with a label-free quantitative proteomics approach in this study. A microarray analysis was also performed to correlate the mRNA levels with the protein changes in the apparently affected systems. Finally, five of the most affected nonmetabolic protein genes were selected for deletion to investigate their requirement for cell survival and growth at a sublethal high temperature.
Materials and Methods
Cells and culture conditions
Halobacterium salinarum strain NRC-1 (ATCC 700922) was grown in complex medium (CM+) at 37°C with shaking (200 rpm) to an optical density at 600 nm (OD600) of 0.5 (early-exponential phase), as described previously (Chu et al., 2011; DasSarma and Fleischmann, 1995). The cells were then subjected to an additional hour of incubation at 37°C (reference temperature) or 49°C (sublethal temperature) before the preparation of their proteins, or at 30°, 37°, 42°, or 49°C for different periods of time before the isolation of their total RNAs. H. salinarum Δura3 and gene deletion mutants were cultured in CM+ medium supplemented with 50 mg/L uracil at 37°C or 49°C.
Protein preparation
The cells were harvested from 400 mL of culture by centrifugation at 8700 g for 15 min at 4°C. The residual medium was spun down (200 g for 30 sec) and removed by pipetting, and the pellet was then resuspended in 5 mL of basal salt solution (DasSarma and Fleischmann, 1995). The cells were lysed by five cycles of freeze-thawing at −80°C and 37°C. After the addition of phenylmethylsulfonyl fluoride (100 mM stock solution in isopropanol) to a final concentration of 1 mM, 150 μg of DNase I, and 150 μg of RNase I (Sigma, St. Louis, MO), each lysate was incubated at room temperature for 30 min. The total proteins were desalted with a modified acetone precipitation protocol in which 300 μL of cell lysate were mixed with an equal volume of water and incubated with 600 μL of acetone:water (50:50 [v/v]) at −20°C for 2 h (Karadzic and Maupin-Furlow, 2005). The concentrations of desalted proteins were estimated using the Dc Protein Assay Kit (Bio-Rad, Hercules, CA) with bovine serum albumin (Sigma) as the standard. Aliquots (200 μg) of the proteins were digested with sequencing-grade modified trypsin (50:1 [w/w]; Promega, Madison, WI) in a final volume of 200 μL of solution containing 50 mM ammonium bicarbonate (pH 8.3) at 37°C for 16 h, and then purified with a C18 spin column (Pierce, Rockford, IL), according to the manufacturer's protocol. The samples were then lyophilized and stored at −20°C for further analysis with liquid chromatography-tandem mass spectrometry (LC-MS/MS).
RNA isolation
The cells in 100 mL of culture medium were harvested by centrifugation and lysed by freeze-thawing as described above. The total RNAs were isolated from the cell lysates with TRI Reagent (Sigma) and treated with DNase I (Sigma), according to the manufacturer's suggested procedures.
Mass spectrometry
Approximately 2 μg of peptides in 0.1% (v/v) formic acid were analyzed using an Agilent 1200 series nano-HPLC system (Agilent Technologies, Palo Alto, CA) coupled to a hybrid linear ion trap-Orbitrap (LTQ-Orbitrap Discovery) mass spectrometer (Thermo Scientific, San Jose, CA). Each sample was analyzed with five MS/MS runs in which the MS survey scans were acquired over a full m/z range of 400–2000 and by gas-phase fractionation (GPF) of four restricted windows (400–521, 516–690, 685–968, and 963–2000 m/z), as described previously (Chu et al., 2011; Yi et al., 2002). The six most intense precursor ions within 400–2000 m/z or each GPF window were sequentially isolated using the data-dependent mode (isolation width: 1 m/z) and subjected to collision-induced dissociation in the linear ion trap with a dynamic exclusion duration of 45 sec and an exclusion list size of 150.
Proteomic data analysis
RawExtract (v1.9.9.1) (McDonald et al., 2004) was used to generate the peak lists from the Thermo RAW files, and YADA (Carvalho et al., 2009) was used to curate the misassigned precursor masses. The data were then analyzed with ProLuCID (v1.0) (Xu et al., 2006) searches against a database composed of the forward and reversed sequences of 2427 unique proteins of H. salinarum NRC-1 (EMBL-EBI Integr8 database, www.ebi.ac.uk) with a precursor mass tolerance of 50 ppm, a fragment ion mass tolerance of 600 ppm, differential modifications, including oxidation of M (+15.994915 Da) and phosphorylation of STY (+79.966331 Da), and no enzyme restriction, as suggested by Xu et al. (2006). XCorr and ZScore were set as the primary and secondary scoring methods, respectively, and the results were filtered using the criterion of a false discovery rate (FDR)≤0.01 by implementing DTASelect (v2.0.41) (Tabb et al., 2002). The relative protein levels at 37°C and 49°C were estimated by spectral counting using PatternLab (v2.0), by which the data were normalized using the row sigma method and the counts were further analyzed with the TFold method (Carvalho et al., 2008). The MS proteomic data have been deposited in the PeptideAtlas (Desiere et al., 2005) with the dataset identifier PASS00217 (http://www.peptideatlas.org/repository/).
Microarray analysis
The DNA sequences of the open reading frames (ORFs) of H. salinarum strain NRC-1 (2413 unique protein-coding genes) were PCR amplified, purified, and spotted in quadruplicate onto aminosilane-coated glass slides (Corning, Corning, NY), as described previously (Baliga et al., 2002). An aliquot (10 μg) of each total RNA isolated from cells subjected to additional incubation at 37°C or 49°C for 60 min was chemically labeled with Alexa Fluor 546 or Alexa Fluor 647 dye (Molecular Probes, Eugene, OR) and subjected to dye-swapped microarray hybridization on a total of two slides. After hybridization, the slides were washed according to the manufacturer's procedure. The slides were then scanned with a DNA Microarray Scanner (Agilent). The hybridization signals were digitized using GenePix Pro 6.0 (Molecular Devices, Union City, CA) and the background intensities were extracted using the local background method. The hybridization signals were background subtracted and further processed in the R environment (Gentleman et al., 2004). The Alexa Fluor 546 and Alexa Fluor 647 intensities were normalized within the arrays with the Loess normalization method and between the slides with the scale normalization method in the LIMMA package (Smyth and Speed, 2003). The statistical significance of the differential expression between the two samples of each gene was calculated with a paired t test analysis (Cui and Churchill, 2003).
Validation of differentially expressed proteins
The expression patterns of selected differentially expressed candidate proteins were indirectly confirmed by Northern blot or RT-PCR analysis. In the Northern blot analysis, approximately 20 μg of total RNA were resolved by electrophoresis on a formaldehyde agarose gel, transferred to a nylon membrane (Sambrook and Russell, 2001), and hybridized with digoxigenin (DIG)-labeled DNA probes (DIG High Prime DNA Labeling and Detection Starter Kit I, Roche Diagnostics GmbH, Mannheim, Germany), according to the manufacturer's protocol. The hybridization results were detected with an enzyme immunoassay and imaged with the Gel Doc™ EQ gel documentation system installed with the Quantity One analysis software (version 4.5.0) (Bio-Rad, Hercules, CA). The DNA probes were prepared by PCR using HotStarTaq DNA polymerase (Qiagen, Valencia, CA) and purified with the Illustra™ GFX™ PCR DNA and Gel Band Purification Kit (GE Healthcare, Piscataway, NJ). RT-PCR was performed with the ReverTra Ace Kit (Toyobo, Osaka, Japan) and HotStarTaq DNA polymerase (Qiagen, Chatsworth, CA) as described previously (Fu et al., 2010). The sequences of the PCR primers used are listed in Supplementary Table S1 (supplementary material is available online at www.liebertpub.com/omi).
Recombinant DNAs
The cctA, dpsA, hsp5, and vng1187g gene deletion plasmids were prepared with the suicide plasmid vector pNBK07, which contains the ura3, hmg, and bla genes, to construct the gene knockout mutants (Peck et al., 2000). DNAs containing the upstream and downstream regions (∼500 bp each) flanking each target gene were amplified and fused by PCR (Coker and DasSarma, 2007). To create the gene deletion plasmids, each fused flanking fragment was cloned into the StuI site of the vector and propagated in E. coli strain DH5α. The CctA expression plasmid was constructed by cloning the PCR-amplified 1,689-bp cctA ORF and its 179-bp upstream promoter sequence into the NcoI and NdeI sites of the pNBPA vector (Facciotti et al., 2007), which was used to transform E. coli (Supplementary Fig. S1) (Sambrook and Russell, 2001). The DNA sequences of the recombinant constructs were verified by DNA sequencing with the BigDye Terminator v3.1 Cycle Sequencing Kit (Life Technologies, Carlsbad, CA). The primer pairs used for PCR amplification are listed in Supplementary Table S1.
Construction of gene knockout mutants and gene complementation
Gene knockout mutants were constructed with the uracil auxotroph H. salinarum Δura3 using the homologous recombination-based pop-in-pop-out method. After the transformation of EDTA- and PEG600-treated competent host cells with the gene deletion plasmids, mevinolin-resistant integrants were selected on uracil-deprived CM+ agar plates containing 20 mg/L mevinolin. The presence of the ∼1000-bp combined flanking sequences in the genomic DNA was confirmed by PCR. Excisants were selected by inoculating the mevinolin-resistant cells onto CM+ agar plates supplemented with 300 mg/L 5-fluoroorotic acid (5-FOA). The gene deletion mutants were screened by PCR and validated by Southern blot hybridization of the purified genomic DNA. Similarly, the H. salinarum Δura3ΔcctA mutant was complemented by transforming EDTA- and PEG600-treated cells with the CctA expression plasmid (pNBPA-cctA). The primer pairs used for PCR amplification are listed in Supplementary Table S1.
Results
Protein identification and quantitation
Total proteins were prepared from triplicate cultures of H. salinarum grown to early-log phase at 37°C followed by incubation for an additional hour at the same temperature or at 49°C. Peptides from the tryptic digestion of these proteins were analyzed with a total of 90 LC-MS/MS runs. Each sample was analyzed with one full m/z window and four gas-phase fractionations (i.e., total three biological and three technical replicates). A ProLuCID analysis of the MS/MS spectra identified approximately 95,840 peptides (46,145 from 37°C and 49,695 from 49°C; FDR<0.3%) or a total of 773 proteins with ≥two unique peptide identifications (or 654 proteins with ≥two unique peptide and total peptides ≥three in either condition), which represented 32% of the predicted proteome (Supplementary Table S2). A further PatternLab spectral count analysis suggested that 60 proteins (p≤0.1; or 37 proteins with p≤0.05) were differentially expressed at these two temperatures (Table 1). Of these, 33 had a relatively higher peptide count and 27 had a relatively lower peptide count at 49°C. A gene functional category enrichment analysis according to the Archaeal Clusters of Orthologous Genes (arCOG) (Makarova et al., 2007) suggested that some of the biomodules within five of the arCOG clusters were perturbed at 49°C (Table 2). These proteins were analyzed further, together with those that displayed minor changes, to explore the systems level responses to the temperature shift.
Only the proteins with at least 10 peptides identified in the 37°C or 49°C proteome are listed. 1Conserved heat shock proteins; 2Also found differentially expressed in the microarray study by Coker et al. (2007); 3Also found differentially expressed in the 2D gel-based proteomic study by Shukla (2006).
Cell division- and translation-related protein changes
H. salinarum cultured at the two temperatures showed distinct differences in their growth curves (Supplementary Fig. S2). The cells grown at 49°C had a much lower maximum OD600 and took a relatively shorter time to achieve an OD600 of 0.5 (∼36 h) than those grown at 37°C (∼45 h). The PatternLab spectral count analysis of the proteins involved in cell division indicated that all five tubulin FtsZ superfamily proteins (FtsZ1, FtsZ2, FtsZ3, FtsZ4, and FtsZ5) showed the same increasing trend at 49°C, and FtsZ3 had the highest number of peptide identifications under both conditions (Supplementary Table S3). Of these proteins, FtsZ1 and FtsZ2 have relatively higher sequence homology (blastp and GCG “gap program” analyses) to the E. coli FtsZ ring structural protein associated with cell division (Bi and Lutkenhaus, 1991) and also to Methanococcus janaschii FtsZ1 and FtsZ2, respectively (Andreu et al., 2002; Oliva et al., 2003). In contrast, approximately 40 (81.6%) of the 49 ribosomal proteins identified showed the same trend in having a relatively smaller number of peptide identifications in the proteome of H. salinarum treated at 49°C (Table 3).
S: 30S small ribosomal subunit proteins; L: 50S large ribosomal subunit proteins.
Coordinated changes in energy metabolism
An apparent increase in the activity of the arginine deiminase pathway is suggested by the higher counts of arginine deiminase (ArcA; p=0.03), catabolic ornithine carbamoyltransferase (ArcB; p=0.13), and carbamate kinase (ArcC; p=0.08) peptides at 49°C. This increase might lead to the production of ATP to compensate for the reduction in energy production resulting from the putative changes in the TCA cycle and oxidative phosphorylation at 49°C (Fig. 1, Table 1, and Supplementary Table S2). Several proteins involved in the TCA cycle (Icd) or in oxidative phosphorylation (CoxA2, CoxB2, VNG0582C, and AtpI) showed significant spectral count differences between the 37°C and 49°C proteomes, suggesting that these systems were affected by elevated temperature. Although the other proteins showed only minor changes, the same trend to lower representations of pyruvate metabolic proteins and most TCA cycle proteins suggested that these pathways were slightly affected at 49°C. The changes in the expression of genes involved in the arginine deiminase pathway were examined by RT-PCR using total RNAs prepared from cells grown to an OD600 of 0.5 at 37°C and then incubated at 37°C or 49°C for 1 h (Fig. 1B). The increases in arcA, arcB, and arcC mRNA levels at 49°C are more or less consistent with the changes estimated from the proteomic data.
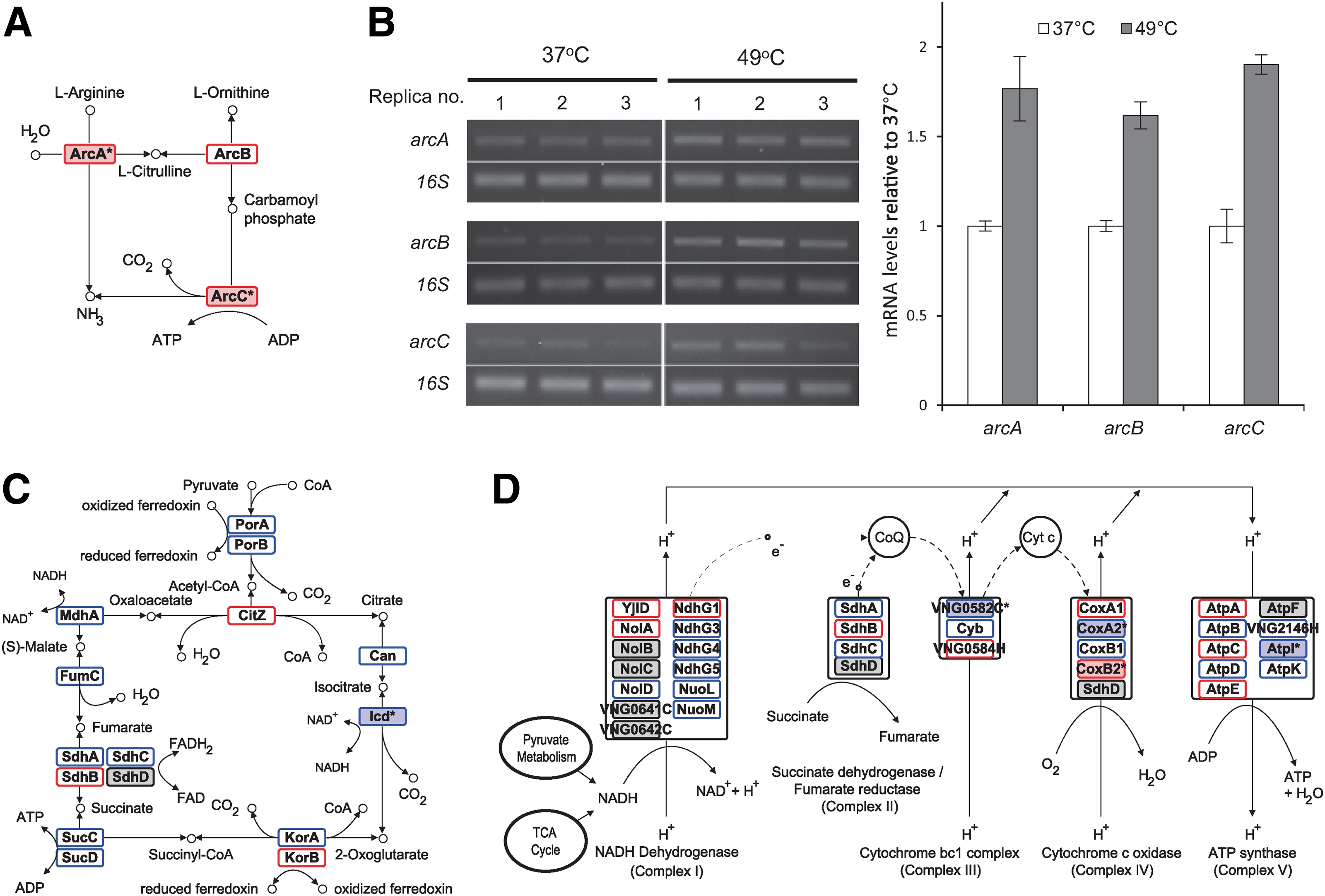
Systems view of the changes in pyruvate metabolism, TCA cycle, arginine deiminase pathway, and oxidative phosphorylation pathway. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways are used as templates to display the apparent protein expression changes in the arginine deiminase pathway
Upregulation of Conserved HSPs
Chaperone proteins, the most well-known HSPs, play an important role in restoring the proper conformation of heat-denatured proteins. The H. salinarum NRC-1 genome encodes several proteins in this category, including the chaperonin or thermosome α and β subunits (CctA and CctB, respectively), GrpE, DnaK, and DnaJ (encoded by the grpE-vng0492H-dnaK-dnaJ gene cluster), five small heat shock proteins (sHSPs), and two prefoldin subunits (PfdA and PfdB). The five putative sHSPs, which all contain an α-crystallin superfamily domain, are Hsp1 (VNG1801G), Hsp2 (VNG0097G), VNG0283C, VNG1771C, and Hsp5 (VNG6201G). A PatternLab spectral count analysis (p≤0.1) suggested that at least four conserved HSPs were upregulated at 49°C: chaperonin α and β subunits (CctA and CctB, respectively), Hsp70 (DnaK), and Hsp20 (Hsp5), which had 10–1,577 peptide identifications (Table 1). With the exception of the Lon protease, for which slightly fewer peptides were identified at 49°C, the remaining conserved HSPs had either a relatively larger number of peptides identified or were only detected at the higher temperature (Supplementary Table S4). It is not surprising that heat stress enhanced the expression of these proteins, because their primary function is to maintain protein-folding homeostasis. However, the requirement for these proteins by H. salinarum to survive in high-temperature environments has not been fully investigated.
Protein changes supported by ancillary DNA microarray data
A DNA microarray analysis of one set of total RNAs isolated from cells (OD600=0.5) that had been treated at 37°C or 49°C for 60 min was performed to monitor the changes in mRNA expression. The mRNAs of 37 and 61 of the 654 proteins identified with LC-MS/MS (Supplementary Table S2 and Fig. 2A) were up- and downregulated, respectively, by at least twofold (p≤0.05) at 49°C (Supplementary Table S5). Of these genes, 74 showed parallel changes at both the transcript and protein levels (Fig. 2A). The 10 genes showing the most dramatic changes were arcA, arcC, cctA, cctB, coxB2, dpsA, hsp5, nirJ, vng1187g, and vng1663c (Table 4). In contrast, when the magnitude of the changes was disregarded, the common increasing or decreasing trends in the expression of these genes supported at least some of the systems changes predicted by the proteomic data (Fig. 2B). For instance, the expression of most of the ribosome, pyruvate metabolism, and TCA cycle mRNAs and proteins was likely reduced at the higher temperature, even though the transcriptomic analysis was performed with only one set of total RNA samples. The putative reduction in the mRNA levels of 35 (71%) of the 49 ribosomal subunit protein genes that had protein identification was consistent with the lower number of ribosome molecules predicted from the spectral counts. Similarly, the mRNA levels of at least eight (40%) of the 20 genes encoding pyruvate metabolism and TCA cycle enzymes were also downregulated (p≤0.05), like their protein counterparts (Fig. 2B).
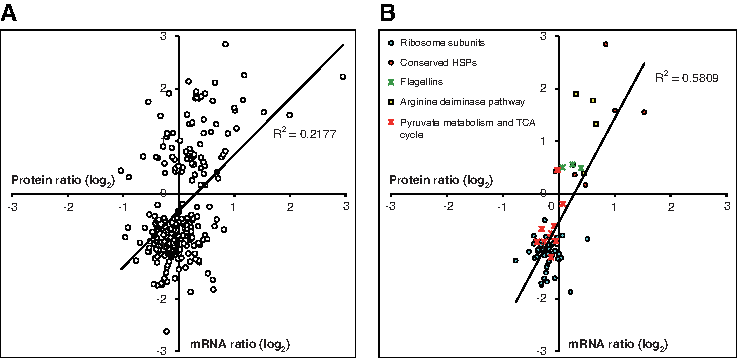
Biomodular changes are supported by the same trends of change in multiple proteins and mRNAs of genes belonging to the same module. Scatter plots and Pearson correlations of the proteomic and microarray expression ratios (log2 49°C/37°C) of (
Validation of gene expression changes
Five nonmetabolic protein genes, cctA, cctB (chaperonin α and β subunits), dpsA (starvation-induced DNA-binding protein), hsp5 (small heat shock protein), and vng1187g (putative membrane protein with copper ion-binding and oxidoreductase motifs), that showed significant changes at both the protein and mRNA levels at the higher temperature were selected from the top 10 most upregulated genes for further expression analysis (Table 4). The mRNA changes were validated by a Northern blot analysis of the total RNAs isolated from cells incubated at 30°, 37°, 42°, or 49°C for different periods of time (Fig. 3). Consistent with the proteomic and microarray data, the Northern hybridization results indicated that the mRNA levels of these genes increased at 49°C.
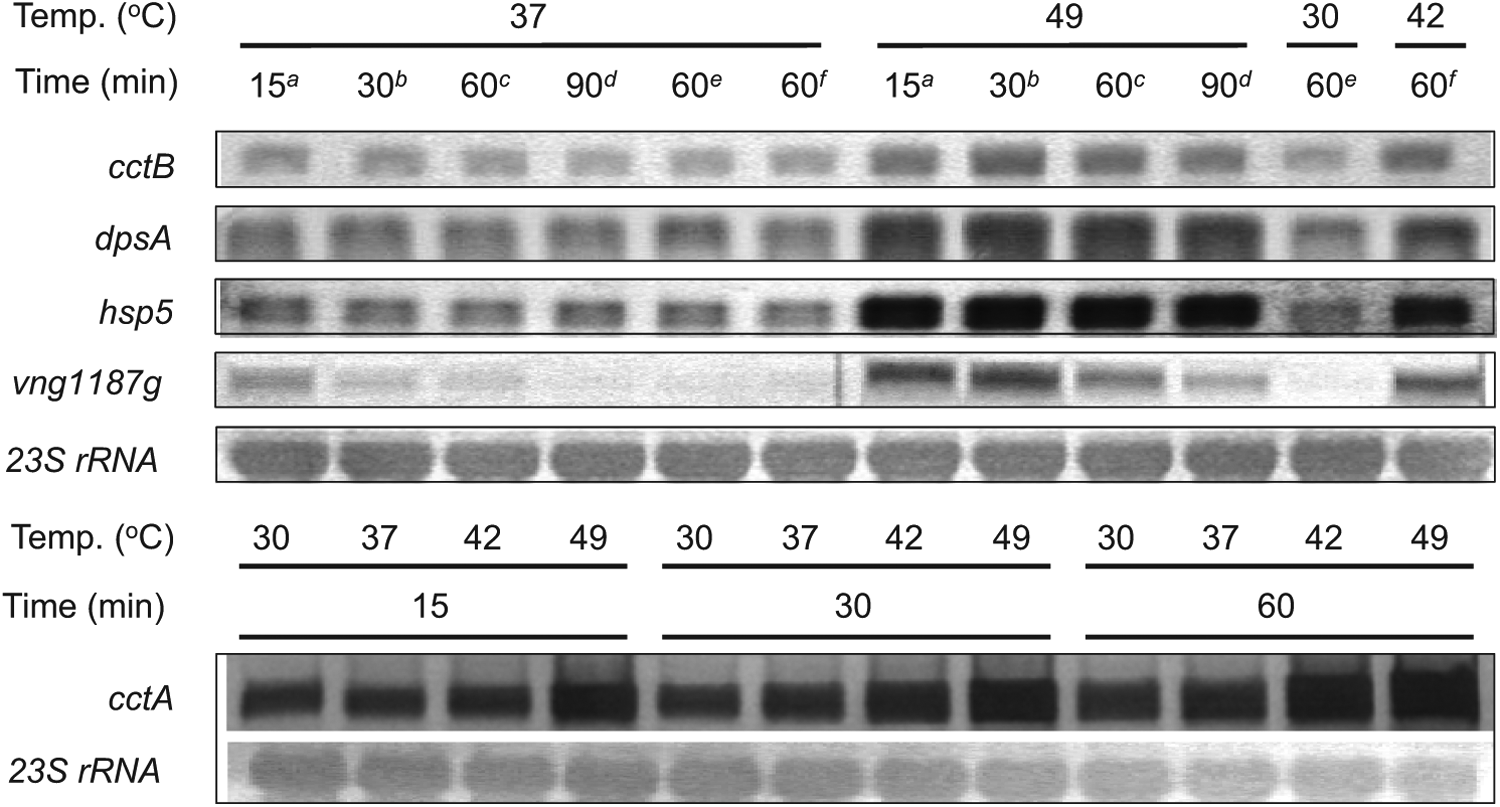
Upregulation of cctA, cctB, dpsA, hsp5, and vng1187g expression at 49°C was confirmed by Northern blot assay. Total RNAs isolated from H. salinarum NRC-1 grown at 37°C to an OD600 of 0.5 and then incubated at the indicated temperatures for the indicated time periods were analyzed by Northern hybridization. The superscripts a–f indicate the sample pairs (i.e., 37°C and 30/42/49°C) prepared at the same time from cells which had been treated at different temperatures for different period of times.
Evaluation of genes required for thermotolerance
The requirement for the expression of the cctA, cctB, dpsA, hsp5, and vng1187g genes during growth at 49°C was analyzed by gene deletion and complementation analyses. Deletion mutants lacking cctA, dpsA, hsp5, or vng1187g were successfully constructed in the uracil auxotroph Halobacterium Δura3 host strain using the homologous recombination method (Supplementary Fig. S3). Despite repeated attempts to delete the cctB gene, we could not detect any deletion mutant after screening a total of 576 colonies grown on CM+ agar plates containing 5-FOA using colony PCR. Comparison of the growth patterns of the four mutants and the host strain at 37°C and 49°C in liquid culture indicated that the deletions of the hsp5, dpsA, and vng1187g genes had no significant effect on the growth of these mutants at the high temperature (Fig. 4). However, the growth of the mutant lacking the gene encoding chaperonin subunit α (cctA) was dramatically retarded at 49°C. Complementation of the cctA deletion mutant with a CctA expression plasmid (pNBPA-cctA) regained approximately 50% of the growth of the host strain transformed with pNBPA vector at the sublethal high temperature (Fig. 5B). Only the Halobacterium Δura3ΔcctA (pNBPA-cctA) strain but not the vector (pNBPA)-transfected control showed growth in mevinolin-containing CM+ liquid medium (Fig. 5B) and agar plate (Supplementary Fig. S4, bottom right) at 49°C further supported the requirement of this gene to support thermotolerance.

Growth of the cctA deletion mutant is impaired at 49°C.

Complementation of the cctA deletion mutant suggests that CctA is required for thermotolerance. Histogram shows the growth of the complemented strain H. salinarum Δura3ΔcctA (pNBPA-cctA) and the controls H. salinarum Δura3 (pNBPA) and Δura3ΔcctA (pNBPA) (n=3) in mevinolin-containing CM+ medium at 37°C
Discussion
Responding to environmental temperature changes by reprogramming the transcriptome and proteome is important for the survival of ectothermic organisms. In this study, several biomodules containing one or more proteins with a statistically significant and/or dramatic difference in peptide identifications between the proteomes of 37°C- and 49°C-treated cultures might have been affected by the temperature change (Table 1). The systems-wide changes caused by the temperature shift became more conspicuous when proteins with subtle changes were also considered in the analysis of protein networks (Fig. 1 and Supplementary Fig. S5) or their functional categories. The most striking change was in the ribosomal machinery. Although most ribosomal components displayed a less than a 1.4-fold reduction in peptide counts, 40 of the 49 identified proteins showed the same trend, with fewer peptides identified at the higher temperature (Table 3), suggesting that there were relatively fewer ribosomes in the cells at 49°C than at 37°C. These coordinated changes were supported by the reductions in the mRNA expression of 35 of the 40 apparently downregulated ribosomal subunit proteins (Fig. 2B). Parallel changes in protein and mRNA levels were also observed in a large proportion of the components of other modules, including the TCA cycle, arginine deiminase pathway, flagella, gas vesicles, and chaperones. The reductions in expression of a number of proteins involved in pyruvate metabolism, the TCA cycle, and the oxidative phosphorylation electron transport chain might lead to a reduction in ATP production, which may be partially compensated by ATP production through the arginine deiminase pathway (Fig. 1). The increased expression of the flagellin subunit proteins (FlaA1, A2, B1, B2, and B3) (Gerl and Sumper, 1988; Ng et al., 2000) and gas vesicle structural proteins (GvpA1, C1, F1, J1, and L1) (Chu et al., 2011; Halladay et al., 1993; Shukla and DasSarma, 2004; Surek et al., 1988) might enhance the motility and buoyancy of H. salinarum, respectively, and thus allow the cells to move near the water surface to receive more sunlight for ATP production through bacteriorhodopsin light-driven proton pumping (Table 5). Interestingly, the mRNAs of the gas vesicle structural protein genes (gvpA1, C1, N1, and O1) were also upregulated at the low temperature (15°C vs. 42°C) in a previous microarray analysis (Coker et al., 2007). These results suggest that both undesirably high and low temperatures increase the synthesis of gas vesicles to increase cell buoyancy in aqueous environments.
Peptides shared by two or more proteins were excluded from peptide counting.
In terms of the proteolytic systems for protein turnover and quality control, the Archaea possess the major proteolytic machinery of both the Eukaryota and Bacteria, although the biology of proteolysis in the Archaea is poorly understood. The Archaea lack the bacterial ClpP proteases, but have a bacterial Lon-like protease and eukaryote-like proteasomes, which are responsible for cytosolic protein degradation (Fukui et al., 2002; Maupin-Furlow et al., 2005). The lower peptide count of Lon at 49°C suggested that the protein is downregulated at high temperatures, as previously reported for Pyrococcus furiosus (Shockley et al., 2003). A previous study showed that the inhibition of the peptidase activity of the T. acidophilum proteasome had only marginal effects under normal conditions, but completely blocked cell growth under heat shock conditions (Ruepp et al., 1998). Although the changes in the 20S proteasome core subunits PsmA and PsmB and the proteasome-activating nucleotidases Pan1 and Pan2 were marginal in H. salinarum, large number of their peptides (53–149) were identified in both proteomes, suggesting that they are relatively abundant proteins at both temperatures (Supplementary Table S6). In contrast, Cdc48b, which is a homologue (52% identity) of the recently characterized Cdc48 protein, which interacts with the 20S core to form a functional proteasome in T. acidophilum (Barthelme and Sauer, 2012), showed an increase of more than threefold at 49°C (p=0.0008). Because there were no parallel increases in the two 20S core subunits, the importance of the increase of Cdc48b at high temperature has yet to be clarified.
Genetic analyses of the requirement for five of the 10 proteins with the most dramatic increases at both the protein and mRNA levels indicated that CctA is important for growth at sublethal high temperatures. The ΔcctA, ΔdpsA, Δhsp5, and Δvng1187g mutants, but not the ΔcctB mutant, were successfully constructed with the homologous recombination-based pop-in-pop-out method (Peck et al., 2000). The chaperonin subunits CctA (562 amino acids) and CctB (556 amino acids) and the starvation-induced DNA-binding protein DpsA (182 amino acids) had changes of more than twofold in their peptide counts and ranked second, sixth, and fifth, respectively, among the proteins with the greatest number of peptide identifications at 49°C. These proteins were also ranked within the 13th–22th most frequently identified proteins at 37°C, suggesting that they were relatively abundant at both temperatures (Supplementary Table S2). In contrast, only 10 and 76 peptides from the sHSP Hsp5 and VNG1187 (a putative membrane protein with copper ion-binding and oxidoreductase motifs) were identified, respectively, at the higher temperature. Microarray analysis indicated that the expression of cctA, cctB, dpsA, hsp5, and vng1187g increased approximately 2.9–7.2-fold at 49°C. Short-term high temperature treatments [56°C (2 h) or 49°C (1 h) and then 56°C (2 h)] had no dramatic influence on the viability of cctA, dpsA, hsp5, and vng1187g deletion mutants (Supplementary Fig. S6). The retarded growth of the cctA, dpsA, and hsp5 deletion mutants at 37°C compared with that of the H. salinarum Δura3 parental strain suggests that these genes are at least required for the wild-type level of growth at this temperature (Fig. 4). The higher temperature (49°C) had no significant effect on the growth of the hsp5, dpsA, and vng1187g deletion mutants. In contrast, the deletion of cctA severely affected cell growth at 49°C, suggesting that it is important for cell survival at high temperatures.
The requirement of chaperonin subunit α protein CctA, but not DpsA, Hsp5, or VNG1187G, to support wild-type-level growth at the sublethal high temperature seems consistent with the “centrality-lethality rule” (i.e., proteins are more likely to be essential when they are strongly associated with other proteins in a protein interaction network) (Jeong et al., 2001). A network analysis (Search Tool for the Retrieval of Interacting Genes [STRING 9.0]; string-db.org) (Szklarczyk et al., 2011) of the five proteins selected for gene deletion analysis in the present study indicated that both CctA and CctB have a few dozen putative directly or indirectly interacting proteins, including the conserved heat shock proteins DnaJ, DnaK, GrpE, Hsp1, and Hsp5 (Supplementary Fig. S5). Compared with the chaperonin subunit proteins, DpsA, Hsp5, and VNG1187G have significantly fewer interactions with other proteins. The growth patterns of the cctA, dpsA, hsp5, and vng1187g gene deletion mutants at 49°C in this study appear to be related to their numbers of putative interacting proteins. The indispensability for growth at 49°C of CctA, which has 56 putative direct interaction partners, is further supported by the complementation of the H. salinarum Δura3ΔcctA mutant by a CctA expression construct (Fig. 5).
Chaperonin belongs to the HSP60 family of proteins, which is highly conserved in nearly all organisms (Horwich et al., 2007). Previous studies have reported that most Archaea have one to three copies of cct genes in their genomes (Lund et al., 2003). Our recent survey of 20 annotated halophilic archaeal genomes (http://www.ncbi.nlm.nih.gov/genome/browse/; http://www.uniprot.org) found that each possessed 2–5 copies of thermosome subunit protein genes (Supplementary Table S7). The chaperonin complexes of organisms with multiple cct genes can form either homooligomers or heterooligomers (Phipps et al., 1991; Yoshida et al., 1997; Yoshida et al., 1998; Gutsche et al., 1999; Valpuesta et al., 2002). The requirement of cct genes to support thermotolerance has been analyzed by Kapatai et al. (2006), who found that the three cct genes (cct1, cct2, and cct3) in H. volcanii are individually dispensable, but at least one cct (cct1 or cct2) must be present for viability. The growth of the double mutant cct1::mevinolinRΔcct3 at 50°C was more strongly affected than that of the Δcct2Δcct3 strain. H. salinarum has two copies of cct, cctA and cctB, whose proteins share 79.70% and 76.64% sequence identity to H. volcanii Cct1 and Cct2, respectively. Although it is unclear why the cctB deletion mutant could not be constructed in repeated attempts, we demonstrated that the deletion of cctA alone is sufficient to impair the growth of H. salinarum at 49°C.
The expressions of conserved HSPs can also be affected by other stressors, including radiation, hypoxia, salinity, and parasitism ( Coker et al., 2007; Sørensen et al., 2003). Comparison of the proteins that showed relatively significant expression changes during the temperature shift (Table 1) with those affected by other stimuli indicated that the expression of the conserved HSP proteins CctA, CctB, DnaK, and Hsp5 might also be influenced by salinity; that Hsp5 might also be influenced by transition metal (copper and zinc) ions; and that CctA might be influenced by 0.5% isopropyl alcohol treatment (Coker et al., 2007; Ha et al., 2007; Kaur et al., 2006; Leuko et al., 2009; McCready et al., 2005; Shukla, 2006; Whitehead et al., 2006) (Supplementary Table S8). Because the primary functions of HSPs are to monitor and respond to the state of protein folding (Mathew and Morimoto, 1998), these changes may reflect the shared effects of those stressors on protein conformation. UV and γ-irradiation do not seem to affect the expression of those four conserved HSP proteins/genes. The expression of the other dramatically affected proteins identified in this study also seemed to be affected by at least one of the nonthermal treatments. However, none of them showed a common response to all of the stimuli analyzed.
The expression profiles of the proteins that showed the most dramatic changes at the elevated temperature were compared with those from previous heat shock studies (Table 1 and Supplementary Table S8). Of the five upregulated heat shock proteins (DnaJ, GrpE, sHsp1, sHsp2, and Hsp5) identified in a previous 2D gel electrophoresis-based quantitative proteomic analysis (Shukla, 2006), only Hsp5 showed a dramatic change and GrpE a minor change in our proteomic analysis (Supplementary Table S4). In the other comparisons, of the seven heat shock-induced non-transcriptional regulator genes (cctA, dnaK, hsp5, dpsA, cdc48b, cdc48d, and srl2) identified in a previous microarray analysis (Coker et al., 2007), we found that CctA, DnaK, Hsp5, DpsA, and Cdc48b proteins displayed dramatic changes in expression. Thus, most of the expression changes detected in the previous microarray analysis were also detected in our label-free quantitative proteomic analysis. The changes in DnaJ, GrpE, sHsp1, and sHsp2 expression identified by Shukla (2006) were not detected by Coker et al. (2007) or in our study, suggesting that these changes are relatively minor.
The OMICS analysis performed in this study indicated that temperature (i.e., a type of environmental stimulus) can elicit complex cellular responses. Proteomics and transcriptomics are important technologies for revealing gene expression changes, although proteins with only minor changes are often ignored in systems analyses. Here, we have shown that when these complementary technologies are used together, they can detect genes/proteins with minor changes in expression, which may facilitate the analysis of systems changes in cells exposed to environmental or other stimuli. For instance, any change in the external environment may have dramatic effects on an organism. The ability to detect both the dramatic and minor changes is important for a global view of these adaptations.
Conclusions
Advances in high-throughput expression profiling (e.g., microarrays, MS, and RNA sequencing) and bioinformatic technologies have revolutionized OMICS studies of biological responses to genetic, biochemical, physical, environmental, and other perturbations. Heat shock and temperature are relatively simple and highly reproducible stimuli that induce complex cellular reactions. In addition to providing a snapshot of the protein changes that occurred with a temperature shift, this study has also shed light on temperature-induced systems level responses. We have demonstrated here that a relatively more complete picture of the changes in each system can be captured when the components showing either dramatic or minor changes are analyzed together, to identify common trends in their expression. This data analysis strategy should be useful for detecting systems level changes in cells in response to various types of perturbations. Finally, the requirement of CctA, but not Hsp5, DpsA, or VNG1187G, for the thermotolerance of H. salinarum has been established in this study.
Footnotes
Acknowledgments
This work was supported by National Science Council Grants 95-2311-B-010-012, 96-2628-B-010-003-MY3, and 102-2320-B-010-019-MY3 and intramural funding from the Aim for the Top University Project grant awarded to National Yang Ming University by the Ministry of Education in Taiwan, Republic of China.
Author Disclosure Statement
The authors declare that there are no conflicting financial interests.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.