
Abstract
Abstract
Cancer, a complex genetic disease involving uncontrolled cell proliferation, is caused by inactivation of tumor suppressor genes and activation of oncogenes. A vast majority of these cancer causing genes are known targets of microRNAs (miRNAs) that bind to complementary sequences in 3′ untranslated regions (UTR) of messenger RNAs and repress them from translation. Single Nucleotide Polymorphisms (SNPs) occurring naturally in such miRNA binding regions can alter the miRNA:mRNA interaction and can significantly affect gene expression. We hypothesized that 3′UTR SNPs in miRNA binding sites of proto-oncogenes could abrogate their post-transcriptional regulation, resulting in overexpression of oncogenic proteins, tumor initiation, progression, and modulation of drug response in cancer patients. Therefore, we developed a systematic computational pipeline that integrates data from well-established databases, followed stringent selection criteria and identified a panel of 30 high-confidence SNPs that may impair miRNA target sites in the 3′ UTR of 54 mRNA transcripts of 24 proto-oncogenes. Further, 8 SNPs amidst them had the potential to determine therapeutic outcome in cancer patients. Functional annotation suggested that altogether these SNPs occur in proto-oncogenes enriched for kinase activities. We provide detailed in silico evidence for the functional effect of these candidate SNPs in various types of cancer.
Introduction
C
Single Nucleotide Polymorphisms (SNPs) are the most abundant form of genetic variation (∼90%), occurring once every several hundred base pairs throughout the human genome (Cargill et al., 1999). By definition, a SNP is a genomic locus where two or more alternative bases occur with an appreciable frequency of >1%. Our understanding of the contribution of SNPs to diversity among individuals, phenotypes, traits, and diseases has greatly been enhanced by several landmark studies (Altshuler et al., 2010; Shastry, 2009; Thorisson and Stein, 2003; 1000 Genomes Project Consortium, 2010). Interestingly, results of The ENCODE Project Consortium illustrate that disease-associated SNPs are enriched within noncoding yet functional DNA elements (Bernstein et al., 2012). SNPs in coding regions of the genome can alter protein conformation or function as elucidated by a number of functional (Megaraj et al., 2011; Ueki et al., 2010), as well as molecular dynamic simulation studies (Huang et al., 2010; Kumar et al., 2013; Rajendran et al., 2012), while those in noncoding regions can impair regulation of gene expression (Haraksingh et al., 2013). To this end, SNPs occurring in miRNA target sites (miR-TS-SNPs) need special attention, as there is a rapid expansion of literature associating miRNAs (Vandenboom Ii et al., 2008) and miR-TS-SNPs (Landi et al., 2008; Manikandan et al., 2012; Saunders et al., 2007; Yu et al., 2007) to cancer progression and susceptibility. Given the wealth of data and sequence information that is currently available on the human genome, it is possible to locate genetic variations in miRNA target sites. Hence, using an integrative and systematic computational approach, we identified a panel of high confidence candidate SNPs in putative miRNA binding sites of proto-oncogenes that may lead to either complete or partial loss of miRNA mediated translational inhibition, which in turn, would result in increased expression of oncogenic proteins (Fig. 1). We speculate that the candidate SNPs identified in this study may have functional effects ranging from cancer susceptibility to therapeutic outcome.
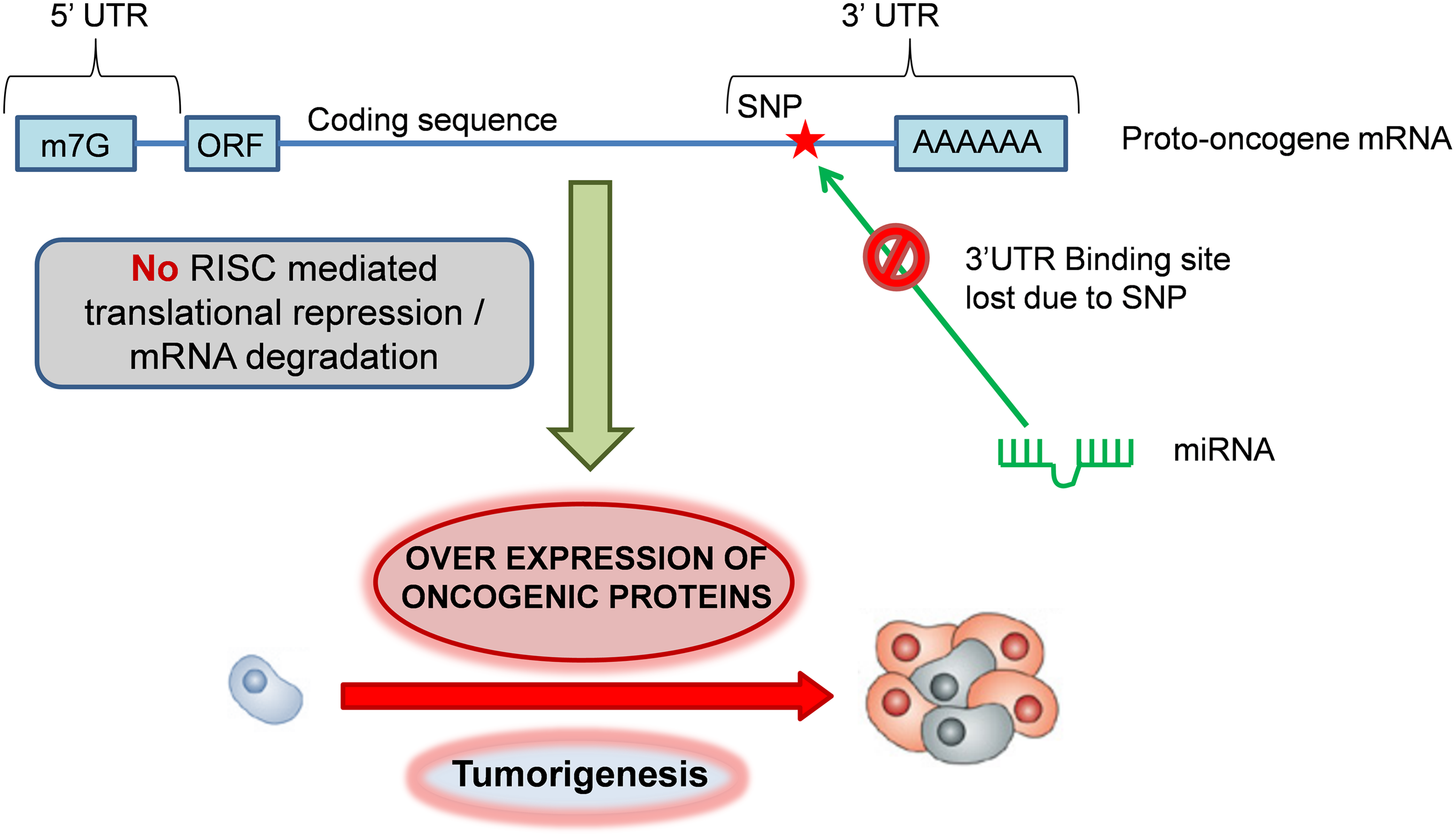
Mechanism of oncogenic activation by miR-TS-SNPs. Under normal conditions, the homeostatic expression of proto-oncogenes is governed by miRNAs, which bind to the complementary sites present in the 3′UTR of mRNAs. miR-TS-SNPs impair the binding of miRNAs to proto-oncogenes, leading to overexpression of oncogenic proteins and tumorigenesis.
Materials and Methods
The proto-oncogene dataset
The list of proto-oncogenes (keyword: KW-0656) was retrieved from UniProt Knowledge Base (UniProtKB, http://www.uniprot.org/), a central hub for the collection of functional information on proteins with accurate, consistent and rich annotation (Magrane and Consortium, 2011; UniProt Consortium, 2012). Of the two sections in UniProtKB, we selected the one that brings together experimental results, computed features, and scientific conclusions (UniProtKB/Swiss-Prot) with high quality manually-annotated and nonredundant records. The other with unreviewed automatically annotated records (UniProtKB/TrEMBL) was not considered. The protein identifiers were converted to official gene symbols using the DAVID Gene ID Conversion Tool–DICT (Huang da et al., 2008), available at http://david.abcc.ncifcrf.gov/conversion.jsp.
Identification and analysis of miR-TS-SNPs with ‘oncogenic’ capability
The official gene symbols of human proto-oncogenes were uploaded in text format to MirSNP database (http://202.38.126.151/hmdd/mirsnp/search/), a collection of human SNPs in predicted miRNA target sites (Liu et al., 2012). The miR-TS-SNPs given as output were pre-organized into one of the following four categories based on their effect on miRNA binding: (i) create—the derived allele introduces a new miRNA binding site in the variant mRNA, (ii) enhance—the derived allele enhances the binding of the originally targeting miRNA to the variant mRNA (iii) break—the derived allele completely disrupts the miRNA binding site and (iv) decrease—the derived allele reduces the binding efficacy of the originally targeting miRNA to the variant mRNA. From this comprehensive list we selected SNPs of categories “break” and “decrease”, since they completely abolish or diminish the efficient binding of miRNAs to their respective mRNAs, resulting in increased or ‘leaky’ translation. These SNPs were then checked for their minor allele frequencies (MAF) in Exome Variant Server (EVS). The EVS, developed with the goal of providing sequence information of human protein coding genes across diverse, richly-phenotyped populations, is maintained by NHLBI Exome Sequencing Project and is freely accessible online at http://evs.gs.washington.edu/EVS/ (Fu et al., 2013). Only the SNPs with a MAF greater than 0.1% were considered for further analyses.
Concordant prediction of SNP-involved target sites and their putative miRNAs by TargetScanHuman 6.2 and RNAhybrid 2.1
The SNP-involved miRNA target sites together with their putative miRNAs were subjected to consistent cross-prediction with TargetScanHuman 6.2 (Grimson et al., 2007), and RNAhybrid version 2.1(Kruger and Rehmsmeier, 2006). TargetScanHuman 6.2 available at http://www.targetscan.org/ predicts both conserved and nonconserved target sites that match the seed region of each miRNA and also sites with mismatches in the seed region that are compensated by conserved 3′ pairing. RNAhybrid is an extension of classical RNA secondary structure prediction algorithm and is shown to predict functional miRNA target sites (Rehmsmeier et al., 2004). Further, RNAhybrid offers a flexible online prediction, as the user can define the position and length of the seed region with option to allow G:U wobble in seed pairing. To verify the effect of each SNP on miRNA binding and on minimum free energy (MFE) required for the formation of miRNA:mRNA duplex, the nucleotide sequence of that particular 3′UTR region containing either the ancestral or the derived allele at the SNP locus and the corresponding miRNA sequence was submitted to RNAhybrid available online at http://bibiserv.techfak.uni-bielefeld.de/rnahybrid/. The MFE of miRNA:mRNA duplex before and after introduction of the variant allele was computed and the difference (i.e., ΔMFE in kcal/mol) was calculated using the following formula:
Obtaining the expression profile of oncogenes in various cancer types
The assessment of mRNA expression profiles of oncogenes in different types of cancer provides a rational approach to study a particular miR-TS-SNP that abrogates the corresponding oncogene in a specific cancer type. Therefore, the expression profile of selective human oncogenes in various types of cancer was analyzed using NCBI's UniGene (www.ncbi.nlm.nih.gov/unigene) (Sayers et al., 2012), an organized view of the transcriptome that evaluates semi-quantitatively the expression sequence tag (EST) calculated as number of transcripts per million (TPM). The EST data for ‘Breakdown by Health State’ that shows the approximate gene expression pattern in various cancers was chosen.
Functional annotation and enrichment analysis of the proto-oncogenes harboring miR-TS-SNPs
The final list of proto-oncogenes predicted to harbor high-confidence miR-TS-SNPs was analyzed for enrichment of molecular function, KEGG pathways, and drug association using WEB-based Gene SeT AnaLysis Toolkit (WebGestalt – URL: http://bioinfo.vanderbilt.edu/webgestalt/) (Zhang et al., 2005). As obvious, the gene ID type was set to “hsapiens_gene_symbol.” The background reference was selected as “hsapiens_genome” and the statistical method was set to default “hypergeometric.” The multiple test adjustment was set to be done by Benjamini and Hochberg, the significance level was set to top 10 and the minimum number of genes for a category was two. The results of drug association analysis was visualized as a network using Cytoscape v.2.8.3 (Smoot et al., 2011) and the miR-TS-SNPs were linked with their corresponding oncogenes using the same tool.
Results
Analysis of exome sequencing data reveals that 198 SNPs can impair miRNA binding sites of proto-oncogenes
The initial list of proto-oncogenes retrieved from UniProtKB (UniProt Consortium 2012) consisted of 562 entries across different species. We narrowed our dataset to the 232 entries of ‘Homo sapiens’ and queried their official gene symbols in MirSNP, a database developed by integrating information from dbSNP build 135, miRBase 18, mRNA sequences from NCBI and miRanda miRNA target prediction algorithm (Liu et al., 2012). Compilation and analysis of results showed that a total of 5452 miR-TS-SNPs impaired the binding of 1869 miRNAs to 218 proto-oncogenes. To identify functional and effective candidates from this large list, we followed a stepwise integrative and systematic computational pipeline (Fig. 2). Although numerous SNPs are documented in dbSNP database of NCBI, many of them seldom occur in the human population, and for many others the Minor Allele Frequency (MAF) is not available. Hence, to identify miR-TS-SNPs specific to the human genome with MAF >0.1%, we utilized Exome Variant Server (EVS) which serves as a repository of more than 10 million SNPs identified by sequencing 15,336 genes in 6515 individuals of European American and African American ancestry (Fu et al., 2013). Moreover, EVS also provides conservation scores (PhastCons and GERP) for each SNP-involved sequence. Upon analysis, we found that 87,906 3′UTR SNPs were documented in EVS and cross checked them with the 5452 miR-TS-SNPs. Interestingly, a minor fraction of 198 SNPs distributed across the 3′UTR of 85 proto-oncogenes overlapped between the analyzed data sets (Fig. 3), suggesting that the vast majority of miR-TS-SNPs are either very rare (MAF<0.1%) or not present in samples of the specified ancestry. As lack of sequence information in EVS can also result in false exclusion of a particular proto-oncogene, we verified the data and identified SSX2 to be one such gene. We limited our further analyses with the 198 SNPs predicted to impair the binding of 632 miRNAs to 85 proto-oncogenes.
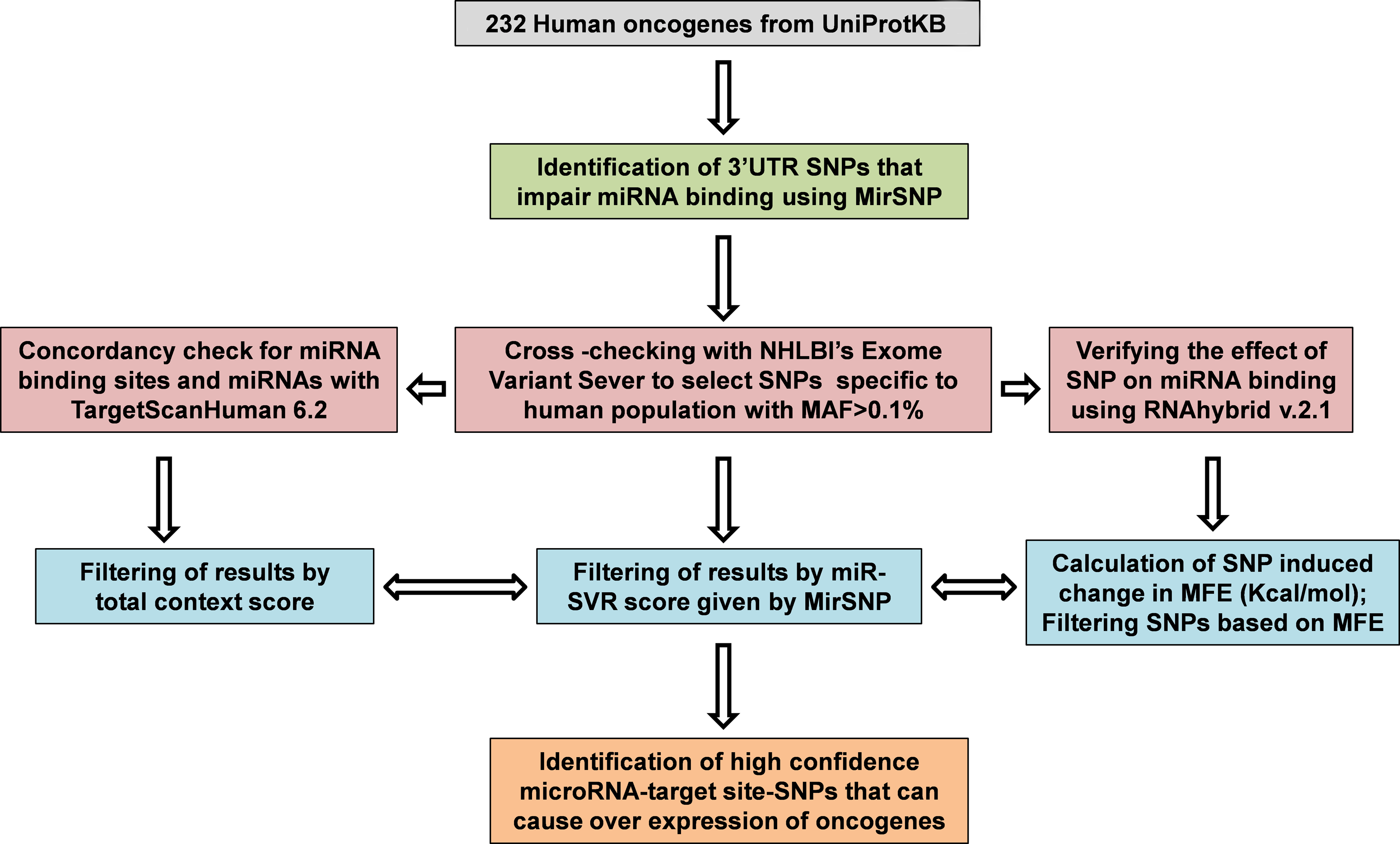
A summary of the workflow employed to identify high-confidence miR-TS-SNPs.

Venn diagram showing the significant overlap of the 3′UTR SNPs captured by the Exome Variant Server and the miR-TS-SNPs of proto-oncogenes predicted by MirSNP.
Sequential filtering of SNP-involved miRNA target sites and their putative miRNAs
Although the miRanda algorithm employed by MirSNP was shown to have high sensitivity in detecting miRNA target sites (Alexiou et al., 2009), the accuracy of miRNA target site prediction by any single method is modest. Therefore, we strengthened the prediction of SNP-involved miRNA target sites and their putative miRNAs by combining additional databases namely TargetScan (Grimson et al., 2007) and RNAhybrid (Rehmsmeier et al., 2004). This approach ensured that different aspects of miRNA target site prediction such as pattern-based search, seed match, structural features, conservation, and hybridization energy are covered, and also enabled the identification of more conservative set of putative miRNAs by reducing spurious predictions. In addition, we refined the predictions by taking into account certain quantitative scores. The MirSNP database, as an added feature contains miRSVR score that integrates target site information and contextual features for ranking the efficiency of miRanda-predicted miRNA target sites. Betel et al. (2010) developed the miRSVR methodology by supervised training on mRNA expression data from a panel of miRNA transfection experiments and showed that miRSVR's top predictions are functional. Hence, we restricted with the top predictions that had a “good” miRSVR score of <−0.1. However, this score is not provided for all miRNA target site predictions in MirSNP, which may be traced back to SNPs (the miRSVR methodology did not consider the impact of SNPs on miRNA-mRNA bindings), the use of different UTR database and miRNA information (Liu et al., 2012). Wherever the miRSVR score was unavailable, miRNA target sites with a “total context score” of ≤−0.1 was considered to avoid exclusion of true positives to certain extent. The total context score given by TargetScan considers contextual features such as the AU content in vicinity of the miRNA target site, location of the site within the 3′UTR, and absence of secondary structures that render the target site accessible for miRNA (Grimson et al., 2007).
The miR-TS-SNPs change the minimum free energy required to form miRNA:mRNA duplex
Several reports suggest that miR-TS-SNPs can change the hybridization energy needed for the formation of miRNA:mRNA duplex (i.e., Minimum Free Energy or MFE). To address this aspect, a stretch of at least 20 nucleotides flanking either side of the SNP locus was submitted as the mRNA sequence in FASTA format to RNAhybrid, and the corresponding miRNA sequence from miRBase 19 (Kozomara and Griffiths-Jones, 2011) was also entered. The same procedure was once again repeated with the variant allele having replaced the ancestral allele in the mRNA sequence. The value of MFE before and after introduction of the SNP was noted down as illustrated in Figure 4, and the difference (ΔMFE) was calculated (see Materials and Methods). The greater the ΔMFE value, the greater is the impact of miR-TS-SNP on miRNA binding. We observed that the allelic variants increased the MFE of the corresponding RNA duplexes, leading to weaker miRNA-target site interaction. The final list consisted of 30 high-confidence miR-TS-SNPs that impaired the binding of 42 miRNAs to the 3′UTR of 54 mRNA transcripts belonging to 24 proto-oncogenes (Table 1), with the ΔMFE value of these SNPs averaging to −2.429 kcal/mol.
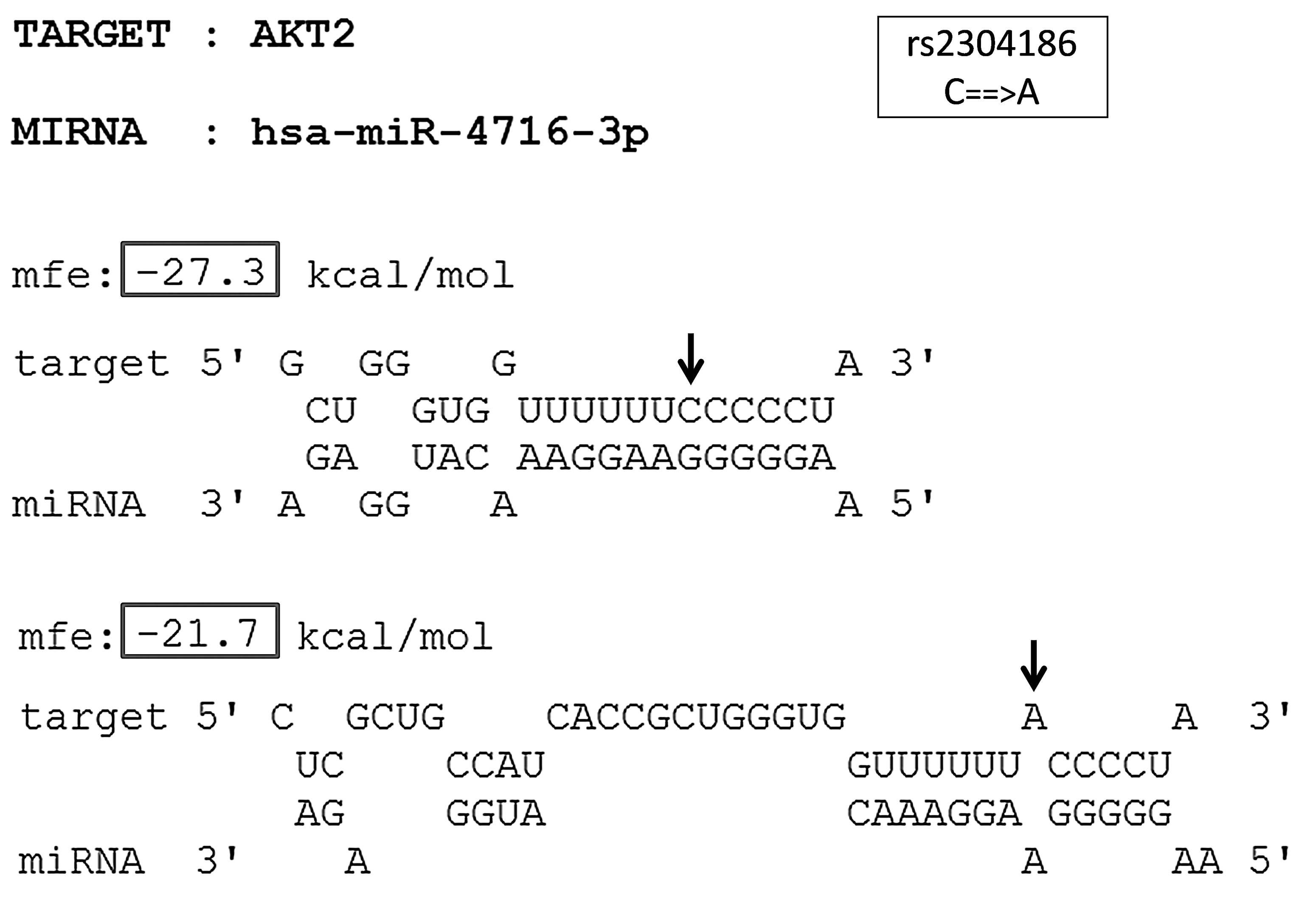
The predicted miRNA:mRNA interaction between SNP-involved target site of AKT2 and hsa-miR-4716-3p. The different alleles of the SNP rs2304186 (C→A) in the 3′UTR of AKT2 are indicated by arrow marks. The MFE of the miRNA:mRNA duplex with the ancestral allele and that with the derived allele is indicated within boxes.
The effect of the SNP on miRNA binding was given by MirSNP.↓- Decrease. X – Break. The gene symbols, transcript IDs and SNP IDs were cross verified with NCBI's dbSNP. The miRNAs that bind the SNP loci are concordantly predicted by MirSNP, TargetScanHuman 6.2, and RNAhybrid 2.1. The ΔMFE value was calculated using RNAhybrid. The conservation (PhastCons and GERP) scores are from Exome Variant Server.
Implications of the miR-TS-SNPs in various cancer types and pharmacogenomics
The high-confidence miR-TS-SNPs predicted to deregulate the expression of proto-oncogenes represent a source of reliable candidates. However, these SNPs are of least importance when the corresponding oncogenes are not expressed in cells and tissues of a specified cancer type. Hence we studied the expression of the 24 oncogenes in different types of cancer using NCBI's UniGene (Sayers et al., 2012). The currently available data demonstrate that LHX4 is overexpressed in tumors of soft tissue or muscle tissue, and hence the corresponding miR-TS-SNP rs138054044 can be analyzed primarily in tumors of the specified type (Fig. 5). In contrast, AKT2 and FUS were found to be overexpressed in all types of cancer and hence the respective miR-TS-SNPs rs2304186 and rs80301724 can be analyzed broadly in all cancers. Of note, human HOXA9 EST profile was not documented in UniGene, whereas the human FGF6 specific ESTs were absent from the total EST pool analyzed across various cancers. The proto-oncogenes were further subjected to enrichment analysis for molecular function using WebGestalt (Zhang et al., 2005) and the results evidently indicate that these SNPs occur in oncogenes functioning predominantly as kinases (Table 2).
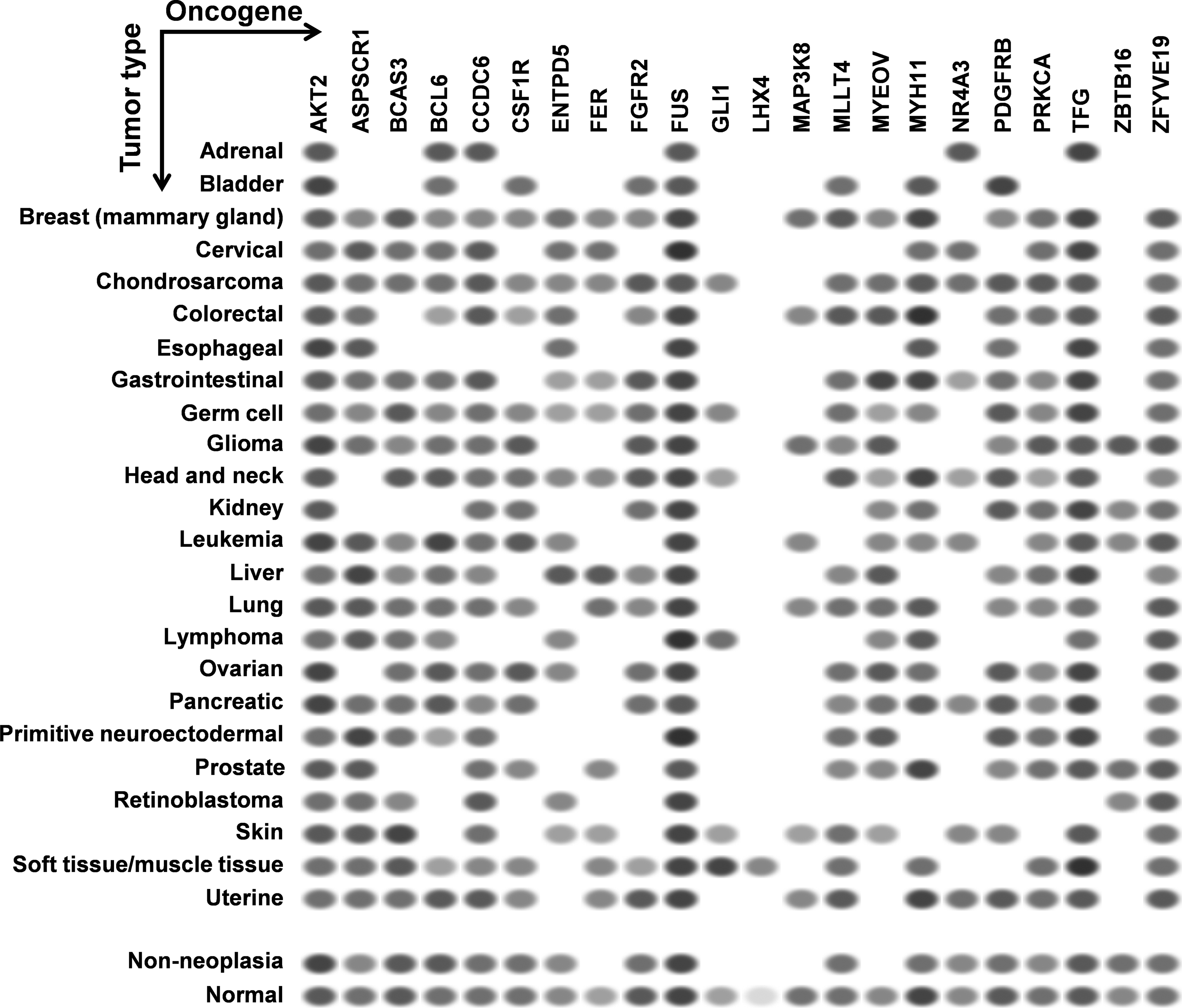
EST profiles of the oncogenes predicted to be activated by miR-TS-SNPs. Each column represents an oncogene and each row represents a particular type of cancer. The oval spots represent the expression of a particular oncogene in a corresponding type of cancer as measured by Expression Sequence Tags (ESTs). The intensity of the spot is based on the amount of transcripts per million (TPM) as captured by the ESTs. Data retrieved from NCBI's UniGene.
GO, Gene ontology; adjP, P value adjusted by multiple testing; R, Ratio of enrichment.
KEGG pathway enrichment analysis identified 10/24 genes as integral part of ‘Pathways in Cancer–hsa05200’ (Fig. 6) and has provided several important insights on the causal role and effect of miR-TS-SNPs in cancer. Even a single SNP that causes increased expression of the corresponding oncogene can likely activate the cancer pathway by amplifying the proliferation signals. For example, the miR-TS-SNP rs2241286 in FGF6 may increase the expression of these ligand molecules, while rs3135816 in FGFR2 and rs2066933 in CSF1R can lead to over expression of these receptor tyrosine kinases and make them sense scanty amount of ligands in the cancer microenvironment. The miR-TS-SNP rs113181701 that causes over expression of BCL6 can totally deregulate the repertoire of genes under the control of this particular transcription factor thus having an extensive effect. This suggests that studying a group of SNPs in genes of the cancer pathway can complement the single-SNP approach in understanding the aberrant molecular signaling. We also tested the proto-oncogenes for drug association, which revealed 8 miR-TS-SNPs (rs2229561 in PDGFRB, rs2066934 and rs2066933 in CSF1R, rs113181701 in BCL6, rs62621676 in PRKCA, rs78933588 and rs146333729 in ENTPD5 and rs2304186 in AKT2) to have the potential to modulate drug response in cancer patients by increasing the expression of their respective genes (Fig. 7). Further, a single miR-TS-SNP like rs113181701 can lead to resistance to multiple drugs such as Imatinib, Doxorubicin and Daunorubicin by increasing the expression of BCL6 oncoprotein. The results strongly suggest that these candidate SNPs merit further investigation in cancer cell lines from the appropriate tumor type for the drug of interest.
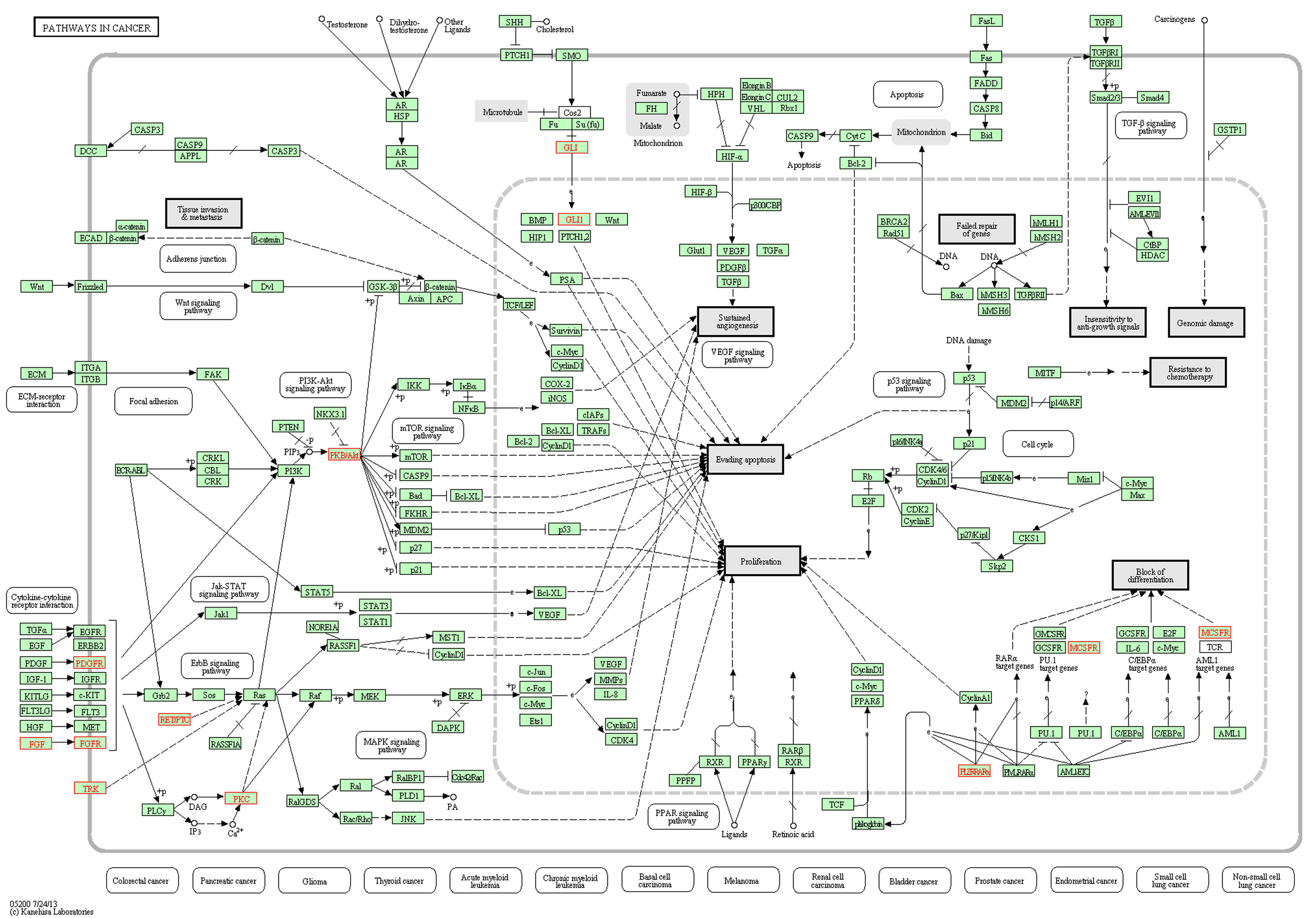
KEGG Pathways in Cancer–hs05200, mapped with the proto-oncogenes harboring miR-TS-SNP. This figure highlights the 10 proto-oncogenes that mapped to KEGG Pathways in Cancer–hs05200. Among the miR-TS-SNPs that disrupt the binding of miRNAs to these oncogenes, the occurrence of a single SNP is enough to activate the cancer pathway. Figure developed by WebGestalt KEGG enrichment analysis tool.
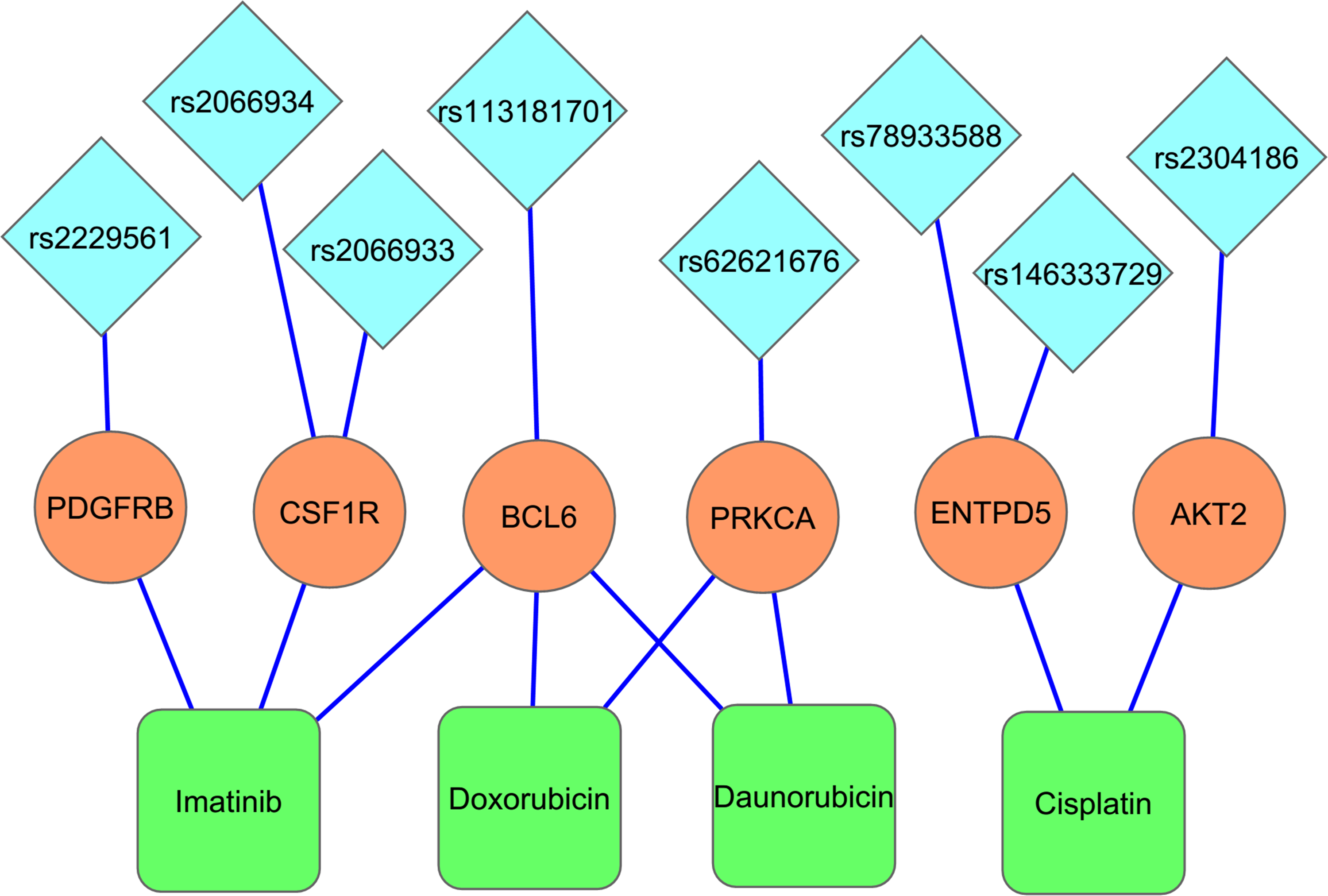
MicroRNA-target site-SNPs have the potential to modify drug response in cancer patients. The blue diamonds, the saffron circles, and the green boxes represent the miR-TS-SNPs, the respective proto-oncogene harboring the SNP and the drug associated with that particular oncogene. The lines connecting these entities represent the interactions between them. The occurrence of a miR-TS-SNP may cause overexpression of the respective oncogene, thereby leading to drug resistance/sensitivity.
Discussion
UTRs are the regulatory elements that play important roles in gene expression (Mignone et al., 2002). Greater than 60% of protein coding mRNA transcripts have target sequences in their 3′UTR to which miRNAs bind, leading to translational repression (Friedman et al., 2009). Genetic aberrations and exogenous episomal integrations that alter these miRNA target sites are of medical importance, as they may lead to severe disorders. It is becoming apparent that polymorphisms in miRNA binding sites determine disease susceptibility by causing aberrant expression of genes and can further predict treatment outcome in cancer patients (Chin et al., 2008; Ratner et al., 2011; Ryan et al., 2010; Teo et al., 2012). Although many databases have catalogued enormous number of miR-TS-SNPs, identifying truly functional candidates among them remains challenging due to insufficient knowledge on miRNA biology, as well as the limited number of experimentally validated target sites. Therefore, accurate and reliable bioinformatics prediction of SNPs that modulate miRNA binding is essential. Noteworthy to mention, previous studies have paved the way to identify not only high priority candidate miR-TS-SNPs (Landi et al., 2008; Richardson et al., 2011; Saunders et al., 2006) but also other functional SNPs located in UTR regions (Aouacheria et al., 2007) from publicly available resources.
We hypothesized that 3′UTR SNPs in miRNA target sites of proto-oncogenes could abrogate cis-regulation of gene expression, resulting in oncogenic transformation, cancer predisposition, and modulation of drug response. The methodology utilized in this study is based on the traditional miRNA–mRNA binding site complementarity and concordant cross prediction by three different algorithms. The SNP involved target sites of miRNAs were filtered based on their miRSVR and total context scores. The effect of the SNP on miRNA:mRNA duplex was verified by RNAhybrid. The results indicate that nearly 10% of the oncogenes (24/232) are subjected to expression changes which may arise due to miR-TS-SNPs. From Table 1, it can be inferred that majority of these SNPs occur in nonconserved miRNA binding sites reinforcing that conservation should be used in combination with other informative features to score target sites and not as hard filter, which may lead to a substantial loss of bona fide targets (Betel et al., 2010). The PhastCons and GERP values are given for the reader to underscore the level of conservation of the SNP involved sites. PhastCons scores are extremely useful to define broad regions of cross-species conservation using a tree-HMM approach (Siepel et al., 2005), while Genomic Evolutionary Rate Profiling or GERP assigns a conservation score to each site in alignment, independently of neighboring sites (Cooper et al., 2005). A positive GERP score represents a substitution deficit, while a negative score represents substitution surplus. Among the list of miRNAs predicted to bind the SNP-involved target sites, hsa-miR-30b, hsa-miR-34a, hsa-miR-93, and hsa-miR-182 were previously reported to be deregulated in one or more types of cancer (Xie et al., 2013). The knowledge gained by studying these cancer-associated miRNAs together with their miR-TS-SNPs will help in understanding this unique mode of gene deregulation in cancer and also microRNA pharmacogenomics (Bertino et al., 2007). The in silico approach described here therefore sets the stage for the next phase of characterization of genetic variants in the 3′UTR of cancer associated genes.
The 8 miR-TS-SNPs predicted to modulate drug response in cancer patients represent reliable functional candidates for pharmacogenomic studies. There is sufficient evidence for the critical role of genetic variations altering miRNA targeting in drug treatment and therapeutic outcome of cancer patients. A report concerning the 3′UTR SNP C829T, which disrupts the binding of hsa-miR-24 to DHFR, demonstrated that the SNP caused DHFR over expression associated with methotrexate resistance. Cells with a mutant DHFR 3′ UTR exhibited a two-fold increase in mRNA half-life, expressed higher DHFR mRNA and protein, and were four times more resistant to methotrexate (Mishra et al., 2007). Zhang et al. 2011 found an association of a polymorphism in let-7 binding region of KRAS 3′-UTR (rs61764370) with treatment response to the monoclonal antibody cetuximab in metastatic colorectal cancer patients. The KRAS wild-type (KRASwt) patients harboring the heterozygous or homozygous variant allele (TG or GG) at the let-7 binding site had a 42% object response rate to cetuximab, compared to a 9% response in KRASwt patients with the homozygous TT genotype (p=0.02). Another study reported the loss of miR-519c binding site in ABCG-2 with shortened 3′UTR leading to ABCG2-overexpressing drug-resistant cell lines (To et al., 2009). Very recently, a novel 3′UTR mutation that reduced the binding affinity of miR-520a and miR-525a to PIK3CA gene was shown to be associated with increased sensitivity of colorectal cancer cell lines to the drug saracatinib (Arcaroli et al., 2012). In this context, an intriguing question remains on the contribution of multiple SNPs in the miRNA network to drug response and carcinogenesis. For instance, miR-196a-2 is predicted to bind the 3′UTR of DHFR as well as thymidylate synthase (Hu et al., 2008). A SNP in miR-196a-2 (rs11614913) is associated with cancer risk (Hu et al., 2008), while a polymorphism in the 3′UTR of thymidylate synthase was associated with response to 5-fluorouracil (Lu et al., 2006). Therefore, it would be interesting to study the different combinations of microRNA-related SNPs such as the DHFR C829T 3′UTR polymorphism, thymidylate synthase 3′UTR polymorphism, and the miR-196a-2 SNP in cancer patients to expand our understanding on the cumulative effect and interindividual variability in drug responses and disease predisposition. Application of these SNPs into treatment decisions is promising and will require further confirmation in prospective randomized trials.
Our study has carefully considered some features that determine miRNA:mRNA interaction to better understand the effect of miR-TS-SNPs and their association with disease. Despite the requirement of a perfect match between miRNA ‘seed’ and target mRNA (Lewis et al., 2005), studies have shown that G:U wobble in the seed region is acceptable (Didiano and Hobert, 2006; Lal et al., 2009). Based on this fact, we admitted G:U wobble in seed pairing while predicting the miRNA:mRNA target interactions in RNAhybrid, so that miR-TS-SNPs that simply cause G:U wobble without changing the MFE can be omitted. As the miRNA:mRNA duplex is known to contain self loops and bulge loops, SNPs in these regions may not have an effect on sequences that pair and hence they were excluded. For a subset of miR-TS-SNPs, there was ambiguity in ancestral allele when cross-checked with NCBI's dbSNP and such cases were also excluded. Wherever discrepancies existed in prediction between MirSNP-employed miRanda, TargetScan, and RNAhybrid, the results were discarded. A few functional constraints, however, were not addressed by our study. Recent projects on in vivo microRNA targets identified a significant number of non-canonical miRNA target sites by employing cross-linking and immunoprecipitation (CLIP) method (Chi et al., 2009; Hafner et al., 2010). Therefore, seed complementarity is neither necessary nor sufficient for microRNA regulation, indicating that other characteristics may also specify targeting. One such feature is the pairing between 3′ region of the miRNA (mostly nucleotides 13–16) and the UTR region complementary to this miRNA segment (Brennecke et al., 2005; Grimson et al., 2007). These “3′ supplementary sites” can enhance the efficacy of mammalian seed-matched sites and can also compensate for a single-nucleotide bulge or mismatch in the seed region, as illustrated by the let-7 miRNA sites in lin-41 and miR-196 site in HOXB8 (Vella et al., 2004; Yekta et al., 2004).
Another report highlighted the importance of the sequence surrounding miRNA target sites by mutating these regions and showing loss of miRNA mediated repression (Didiano and Hobert, 2008). Thus, SNPs falling outside the sequences that are directly involved in miRNA:mRNA hybridization could also be functional. Further, 3′UTR SNPs may significantly affect the secondary structure of mRNAs resulting in their degradation, as exemplified by the C-to-T polymorphism 14nt downstream of the miR-24 target site on DHFR gene (Mishra et al., 2007). Interestingly, data integration approach has shown that SNPs in the 3′UTR can also introduce alternative polyadenylation signals, thereby generating mRNA transcripts with shortened 3′UTR that escape miRNA binding (Thomas and Saetrom, 2012). Finally, rare variants are expected to be geographically clustered (Nelson et al., 2012) and many more SNPs await documentation in dbSNP database. Nevertheless, the miR-TS-SNPs identified in our study represent yet another mechanism of ‘loss of function’ of miRNA-mediated gene regulation and can result in overexpression of the proto-oncogenes in cancer cells.
Conclusions
There is compelling evidence that miRNA target site SNPs can modify the expression of genes and can influence cancer risk. However, no previous study has provided a cumulative list of high-confidence miR-TS-SNPs specific to oncogenes. The current study shows that our systematic and integrated computational approach is a step forward for the identification of miR-TS-SNPs that disrupt the translational control of not only oncogenes, but other genes as well. Depending on the combinatorial effect of different miRNAs that regulate a given mRNA, a SNP that disrupts a true target site may have significant functional implications on post-transcriptional regulation. The SNPs identified in this study has not been assigned any functional mechanism until now and has not formed the basis of any previous cancer case-control association studies. We provide in silico evidence for these candidate miR-TS-SNPs to activate oncogenes and functional analyses are warranted to demonstrate their contribution to carcinogenesis and therapeutic response.
Footnotes
Acknowledgments
This research was supported in part by a grant from the Department of Biotechnology (DBT), New Delhi to AKM (Grant No. BT/PR10023/AGR/36/27/2007). MM is supported by a research fellowship from the Council of Scientific and Industrial Research (CSIR), New Delhi. The infrastructural facilities of the department were provided by Department of Science and Technology, Government of India, and University Grants Commission, a statutory body of the Government of India, through DST-FIST & UGC-SAP grants respectively.
Author Disclosure Statement
The authors declare that there are no competing financial interests.