
Abstract
Abstract
Among the neglected tropical diseases, leishmaniasis is one of the most devastating, resulting in significant mortality and contributing to nearly 2 million disability-adjusted life years. Cutaneous leishmaniasis is a debilitating disorder caused by the kinetoplastid protozoan parasite Leishmania major, which results in disfiguration and scars. L. major genome was the first to be sequenced within the genus Leishmania. Use of proteomic data for annotating genomes is a complementary approach to conventional genome annotation approaches and is referred to as proteogenomics. We have used a proteogenomics-based approach to map the proteome of L. major and also annotate its genome. In this study, we searched L. major promastigote proteomic data against the annotated L. major protein database. Additionally, we searched the proteomic data against six-frame translated L. major genome. In all, we identified 3613 proteins in L. major promastigotes, which covered 43% of its proteome. We also identified 26 genome search-specific peptides, which led to the identification of three novel genes previously not identified in L. major. We also corrected the annotation of N-termini of 15 genes, which resulted in extension of their protein products. We have validated our proteogenomics findings by RT-PCR and sequencing. In addition, our study resulted in identification of 266 N-terminally acetylated peptides in L. major, one of the largest acetylated peptide datasets thus far in Leishmania. This dataset should be a valuable resource to researchers focusing on neglected tropical diseases.
Introduction
T
The majority of Leishmaniasis cases reported are dermatotropic infections termed as cutaneous leishmaniasis (CL). CL is caused by a complex that includes L. major, L. mexicana, and L. tropica. Upon infection with the parasite, symptoms generally start appearing within a few days and may last up to several months. Progressively increasing erythematous, and frequently pruritic, papules appear at the site of sand fly bite. Lymphatic spread with regional lymphadenopathy is common during the infection. The initial papule turns scaly and further scaly region develops into an ulcer showing inflammation. Very often, the site is self-healing, leaving a permanent scar, although the parasites persist throughout life. In few cases, this inflammatory site may acquire secondary infection leading to further complications. In a small percentage of cases, primary CL lesion may leave an individual at risk for later development of mucocutaneous leishmaniasis (Schwartz et al., 2006).
The L. major genome was the first kinetoplastid protozoan parasite genome to be sequenced, along with Trypanosoma cruzi and Trypanosoma brucei in 2005. The L. major genome size is 32.8 Mb and has 36 chromosomes ranging from 0.28 Mb to ∼2.8 Mb. According to the current annotations, L. major has 9222 genes of which 8272 are protein-coding, 911 are RNA genes, and 39 are pseudogenes (Ivens et al., 2005). In addition to L. major, another Leishmania from the old world group L. infantum (36 chromosomes) and L. braziliensis (35 chromosomes) from the new world group were sequenced in 2007 (Peacock et al. 2007). Recently, two other Leishmania species have been sequenced: L. donovani, which has 36 chromosomes with a genome size of 32.4 million base pairs encoding 8195 genes (Downing et al., 2011) and L. mexicana, which has 34 chromosomes (Rogers et al., 2011).
Annotating genomes using mass spectrometry data is a complementary approach to conventional genome annotation (Mann and Pandey, 2001; Pandey and Lewitter, 1999; Pandey and Mann, 2000). This approach of searching mass spectrometry data against EST databases (Choudhary et al., 2001) and also against six-frame translated nucleotide sequences from a genome provides the most direct evidence of protein coding genes (Yates et al., 1995). This approach has resulted in identification of novel genes and corrections to the existing gene models in Homo sapiens (Desiere et al., 2005; Fermin et al., 2006; Menon et al., 2009; Molina et al., 2005), Anopheles gambiae (Chaerkady et al., 2011; Kalume et al., 2005), Mycobacterium tuberculosis (Kelkar et al., 2011), Candida glabrata (Prasad et al., 2012), Plasmodium falciparum (Lasonder et al., 2002), Toxoplasma gondii (Xia et al., 2008), Schmidtea mediterranea (Bocchinfuso et al., 2012), Yersinia pestis (Payne et al., 2010), Pristionchus pacificus (Borchert et al., 2010), Deinococcus deserti (Baudet et al., 2010), Leishmania donovani (Nirujogi et al., 2013), and other organisms. This proteogenomics approach is also useful in identifying splice variants, extensions/truncations to the existing proteins, changes in translational start site based on identification of N-terminally acetylated peptides, and frame changes (Castellana and Bafna, 2010; Renuse et al., 2011). This approach is mostly useful in situations where complete genome sequence of the organism under study is available (Armengaud et al., 2013). However, in cases where the genome sequence is unavailable, the identification of proteins can be done by using genome sequence data from taxonomically related species; we have used comparative proteogenomics to map the proteome of L. donovani (Pawar et al., 2012).
Leishmania, being a unique unicellular dimorphic pathogen, has been studied in detail by various researchers using proteomics tools and is summarized by Paape and Aebischer (2011). Proteomic profiling of Leishmanial promastigotes and amastigote stages has shown differentially regulated proteins in two different growth stages (Alcolea et al., 2011; Pawar et al., 2012; Pescher et al., 2011). Comparative proteomic analysis have been reported of antimony-resistant and -susceptible Leishmania (Biyani et al., 2011; Matrangolo et al., 2013; Walker et al., 2012), as well from oxidative and nitrosative stresses (Sardar et al., 2013). 627 phosphoproteins of L. donovani axenic promastigotes and amastigotes have been identified by proteomic analysis, which revealed leishmanial proteins have multiple phosphorylation sites and phosphorylation occurs at distinct stages of the life cycle (Tsigankov et al., 2013). Similarly large numbers of membrane proteins have been identified from promastigotes and amastigote stages that usually are difficult to detect and characterize by a proteomic approach (Brotherton et al., 2012). Few investigators have studied macrophage proteomics after infection of Leishmanial parasite and have mainly focused on cell metabolism (Menezes et al., 2013) and phagosome and exosome formation (Campbell-Valois et al., 2012). Although the L. major genome was the first genome to be sequenced, not a single study on proteomics has been reported about this pathogen.
In the present study, we carried out proteogenomics analysis of L. major promastigotes; the high resolution mass spectrometry data was searched against protein database of L. major. This resulted in identification of 23,307 unique peptides, which mapped to 3613 proteins in L. major accounting for 43% of L. major proteome. In addition to the proteomic analysis of L. major, we also carried out proteogenomics analysis by searching L. major promastigote data against six-frame translated genome of L. major. This resulted in identification of 26 genome search-specific peptides (GSSPs), which in turn resulted in identification of 3 novel genes and 15 N-terminal extensions of existing genes models in L. major based on high resolution mass spectrometry-derived data. Finally, we performed RT-PCR and sequencing to validate the existence of these novel genes and N-terminal extensions identified in L. major.
Materials and Methods
Leishmania major cell culture
We used a MHOM/IL/67/JERICHO II (ATCC 50122) strain of Leishmania major for proteomic analysis. The motile promastigote stage of L. major ATCC strain was cultured in DMEM containing 10% FBS at 25°C. The cells were grown until the L. major culture reached mid-log phase. The promastigotes were observed under a phase contrast microscope to check for the characteristic flagella that help the promastigote in whipping motility. The cells were harvested by centrifuging at 3000 rpm for 10 min and the cell pellet was washed with PBS of pH 7. This procedure was repeated six times to get rid of any contaminating serum proteins used in the cell culture media. The promastigote cells were counted using a Neubauer chamber and 1×109 cells were used for the proteomic analysis.
Sample processing, protein isolation and fractionation
Sample preparation was carried out in a similar way as described in our previous work (Pawar et al., 2012). Briefly, 1×109 promastigotes were lysed in 0.5% SDS solution post 6 sonication cycles. The lysate was centrifuged at 10,000 rpm for 10 min and the supernatant was used for further proteomic experimentation. Lysate (200 μg) was resolved using 10% SDS-PAGE, and in-gel trypsin digestion (1:20 trypsin) of different protein bands was carried out. The peptide fractions were dried using the vacuum drying process. In addition, 200 μg of lysate was subject to in-solution trypsin digestion (1:15 trypsin) as described previously (Pawar et al., 2012). This was followed by strong cation exchange (SCX) chromatography-based fractionation of tryptic peptides. These fractions were completely dried and reconstituted in 40 μL of 0.2% formic acid.
Mass spectrometry analysis
A total of 49 fractions obtained from two different fractionation methods were analyzed by LC-MS/MS. For LC-MS/MS, desalting trap column (5 μ 100 Å Magic C18, Michrom Bioresources) and analytical column (5 μ 100 Å Magic C18, Michrom Bioresources) were connected to a Proxeon Easy nLC system (Thermo Scientific, Bremen, Germany). The RP-LC was connected on-line with LTQ-Orbitrap Velos ETD mass spectrometer (Thermo Electron, Bremen, Germany). The nanospray source was fitted with an 8 μm emitter tip (New Objective, Woburn, MA) and a voltage of 2 kV was applied. Peptide samples reconstituted in HPLC solvent A (0.1% formic acid) were loaded onto the trap column and washed for 5 min with 97% HPLC solvent A and 3% solvent B (90% ACN in 0.1% formic acid). The peptide separation was carried out using a linear gradient of 7%–30% solvent B for 53 min. at a constant flow rate of 0.4 μL/min. The data were acquired using Xcalibur 1.2 (Thermo Electron). In the scan range of m/z 350 to 1800 Da, 20 most abundant ions were selected for fragmentation. The acquired ions were excluded for 30 sec. Target ion quantity for FT full MS scan was 5×105 and for MSn was 2×105. FT analyzer was used to acquire MS at the resolving power of 60,000 (for precursor). HCD mode was used to carry out MS/MS with resolving power of 15,000 at 400 m/z (for fragment ions) and lock mass option was enabled for accurate mass measurements. Polydimethylcyclosiloxane ions were used for internal calibration.
Mass spectrometric data searches and protein identification
A nonredundant (nr) protein database of Leishmania major strain Fredlin (n=8412) available from TriTrypDB (http://tritrypdb.org/common/downloads/release-4.1/Lmajor/) kinetoplatid genomics resource, as of July 20, 2012 was used for our analysis. Database dependent searches were submitted to Mascot (version 2.2) and Sequest search engines using Proteome Discoverer (Thermo Scientific, version 1.2) and the data was analyzed. The search parameters used were as follows: a) Proteolytic enzyme used was trypsin (with up to one missed cleavage); b) 20 ppm was set as the peptide mass error tolerance; c) 0.1 Da was set as the fragment mass error tolerance; d) Fixed modification was cysteine carbamindomethylation; e) Oxidation of methionine and protein N-terminal acetylation were included as variable modifications. Peptide data from Mascot and Sequest search algorithms were extracted with 1% false discovery rate (FDR) as threshold using Thermo Proteome Discoverer. In the first pass search, the proteomic data were searched against the L. major protein database, and unassigned spectra not mapping to the protein database were filtered using a spectrum confidence filter. These unassigned spectra were searched in the second pass search against the six-frame translated L. major genome database, which resulted in the identification of GSSPs. Only Rank 1 peptides with high confidence were extracted and used for further analysis. Unique peptide data from the both search algorithms (Mascot and Sequest) after protein database dependent searches were used for further analysis.
Proteogenomics data analysis
The Leishmania major strain Fredlin genome sequence was downloaded from TriTrypDB (http://tritrypdb.org/common/downloads/release-4.1/Lmajor/) and a six-frame translated database (n=1,117,604) was created using in-house python programs, as described previously (Nirujogi et al., 2013). Briefly, the six-frame translated genome database of Leishmania major was created by masking the gaps in the genome. The frequently encountered contaminants such as trypsin, keratins, and BSA were added to both the protein and six-frame translated genome databases that were used for MS/MS ion search. Searches were submitted through Proteome Discoverer console (Thermo Scientific, version 1.2) to Sequest and Mascot (version 2.2) search engines as described above.
Mass spectrometric data were searched against the L. major protein database in the first pass search. The unassigned spectra were searched using a L. major six-frame translated genome database in the second pass search. These peptides that map uniquely to the six-frame translated genome database but not to the protein database were referred to as GSSPs. We carried out further analysis of the genomic regions where these GSSPs map to, to identify novel genes or corrections to existing annotations. Alternative gene models were searched using two different gene prediction programs—FgeneSH and GeneMark for eukaryotes. Thus extensions in gene models, as well as novel genes obtained using peptide evidence and gene prediction tools, were checked for their conservation across Kinetoplast family (includes genus Leishmania and Trypanosoma). In addition, we checked other well-curated Leishmania databases such as TriTrypDB and ensemble L. major genome browser to see if the N-terminal extension and novel genes identified in our analysis have been incorporated in the current gene builds in these databases.
Primer designing and RT-PCR validation
Specific primers were designed for gene models (novel genes and extensions) using Gene Runner (Version 3.05) software. The designing of the primer was based on annotation of gene models as predicted by gene prediction algorithms or homology to other Leishmania species. The size of the amplicon was chosen to be 300 to 500 bp to ensure good sequencing quality. Total RNA was isolated from promastigote stage of L. major using Qiagen RNeasy kit (Qiagen, Netherlands) and the yield of the RNA was estimated using Nanodrop 2000 (Thermo Scientific, DE). DNase I treatment was given to the RNA. Preparation of cDNA was carried out by reverse transcription of 1 μg RNA using ABI high capacity cDNA reverse transcription kit. The PCR reaction mixture consisted of approximately 1 μL cDNA obtained from protmastigote lifestage of L. major, 10 nM of forward and reverse primers, 1.5 mM MgCl2, 0.2 mM dNTP mix, 1.5 U of Taq polymerase and Taq PCR buffer in 25 μL reaction volumes. Amplification of the targets was achieved by the following PCR cycle: 95°C for 5 min, 35 cycles of 94°C for 60 sec, 50°C–60°C for 45 sec, 72°C for 60 sec, and final extension at 72°C for 10 min. PCR reaction carried out without cDNA from promastigote stage served as negative control. Amplicon size was checked using DNA ladder on 1.5% agarose gel. Specific amplicons were purified by Qiagen gel extraction kit and subjected to sequencing by Sanger's method. The cDNA sequences obtained have been submitted to GenBank.
Availability of proteomic data
The raw mass spectrometry data (.raw files) generated from this study have been made publically available to other researchers through the Tranche server (http://proteomecommons.org/tranche). The Tranche Hash is: cONyF1wG0tlabIAwSiSrzrcdNDiXbqaoZfRQoDHu8Z+0OQck IyvAGp 7Rv3kQ3U5pZHDUIDhDcqGsMcjQlA6SGIDJm5AAAAAAAAAYvg==
Results
Summary of proteomic data
We carried out extensive proteomic profiling of L. major promastigotes to map its proteome and annotate its genome. This study generated one of the largest protein catalogs of L. major to date. The workflow used for proteomic analysis and genome annotation of L. major is outlined in Figure 1. Peptide fractions obtained from two different fractionation methods (i.e., in-gel and SCX) were analyzed on an LTQ-Orbitrap Velos ETD mass spectrometer, and 49 LC-MS/MS runs were carried out. We used high resolution settings for both MS and MS/MS fragmentation. The mass spectrometry data were searched using Sequest and Mascot search algorithms. Approximately 285,651 MS/MS spectra were acquired in this study, which resulted in 172,513 peptide spectrum matches (PSMs). In total, 23,333 unique peptide sequences that passed the 1% FDR threshold were identified from protein and genome database searches. Thus the high resolution mass spectrometry data yielded more identification of proteins compared to previously published studies characterizing the proteome of L. major. The complete list of proteins and peptide identified in protein database searches is shown in Supplementary Tables S1 and S2 (supplementary material is available online at www.liebertonline.com/omi).

Work flow for sample processing, fractionation, and proteomic analysis of L. major promastigote. Leishmania major promastigotes were used for the proteomic analysis as indicated. Proteins from lysates of promastigote were extracted using SDS and subjected to SDS-PAGE or digested with trypsin and subjected to SCX (strong cation exchange) chromatography. LC-MS/MS analysis of digested gel bands or SCX fractions was carried out on a high resolution mass spectrometer. The mass spectrometry data was searched against a protein database and a six-frame translated genome database of L. major.
Confirmation of annotated protein coding genes in L. major genome
The majority of the annotated proteins coding genes in L. major genome were identified based on in silico gene prediction algorithms, and most of the proteins are hypothetical proteins with no known assigned function. We wanted to carry out in-depth proteomic profile of L. major promastigotes that would directly identify protein-coding genes. L. major has 8412 protein coding genes and, in our study, we identified 23,307 unique peptides derived from 3613 proteins. Of the 3613 proteins identified in L. major promastigote, 58% (2082) proteins were identified by three or more peptides per protein, 14% (508) proteins by two peptides, and 28% (1023) proteins by single peptide. Of the 8412 proteins in L. major, the majority of the proteins are uncharacterized hypothetical proteins (5336), and we have confirmed the existence of 1999 of these hypothetical proteins (24% of L. major proteome). Most of these hypothetical proteins have no known biological function assigned and their role in L. major cellular physiology remains unexplored. Figure 2 illustrates an example that shows 51 unique peptides (2036 PSMs) mapping to LmjF.28.2770 protein coding gene which codes for 658 amino acid long heat-shock protein 70 (HSP70). A representative MS/MS spectrum of HSP70 peptide SVHDVVLVGGSTR identified in L. major has been provided. HSP70 has been shown to play a crucial role in Leishmania stage differentiation (Louw et al., 2010), virulence (Folgueira et al., 2008), and cell cycle (Raina and Kaur, 2012). In addition to HSP70, we also identified proteins belonging to different categories to be abundantly expressed in L. major promastigotes such as calpain-like cysteine peptidases (LmjF.27.0500, 278 unique peptides), glyceraldehyde 3-phosphate dehydrogenase (LmjF.30.2970, 38 unique peptides), pyruvate phosphate dikinase (LmjF.11.1000, 72 unique peptides) elongation factor 1-alpha, and paraflagellar rod proteins, among others. These proteins had multiple peptides mapping to them with hundreds of PSMs. These proteins were abundantly expressed and seem to play an important role in parasite survival.

Identification of heat shock protein 70 (hsp70).
N-terminal acetylation and confirmation of translational start sites
Protein translational start site (TSS) identification and its correct assignment in predicted transcripts has always been a challenging task. The traditional approach for TSS determination relied on annotating the longest open reading frame (ORF) in a given nucleotide sequence under study. Hence, other methods such as mass spectrometry-based approach (Gevaert et al., 2003; Molina et al., 2005) and homology-based bioinformatics approach (Peri and Pandey, 2001) have been used as an additional line of evidence to determine N-terminal acetylation of annotated proteins. N-terminal acetylation reaction is catalyzed by N-acetyltransferase by transferring acetyl group from acetyl-CoA to the first amino acid post excision of initiator methionine. The excision of N-terminal initiator methionine is catalyzed by methionine aminopeptidases (Frottin et al., 2006). N-terminal acetylation occurs in the majority of eukaryotic proteins. There are five major groups of N-terminal acetyl transferases (A to E) that catalyze N-terminal acetylation (Arnesen et al., 2009; Hollebeke et al., 2012). Mass spectrometry-based approaches can be used to ascertain the exact TSSs using N-terminal acetylation of peptides (Helbig et al., 2010). N-terminal acetylated peptides can be determined by searching mass spectrometry data using N-terminal acetylation as a modification during database searches. In our present analysis, we identified 266 N-terminal acetylated peptides; among these, 21% of initiator methionine residues were found to be acetylated. In addition, other amino acids at second position were found to be acetylated. Among these, serine (47%), alanine (25%), and threonine (7%) residues were also found to be acetylated. This approach can be used to correctly annotate TSS for protein coding genes. The complete list of N-terminal acetylated peptides and the corresponding proteins is shown in Supplementary Table S2.
Proteogenomics analysis of L. major
Identifying novel genes and corrections to existing gene models can be achieved by applying a proteogenomics approach. The majority of genome annotations are carried out using in silico approaches. Proteomics can provide a direct evidence for existence of a protein coding gene in a given genome understudy. We carried out a proteogenomics analysis of L. major, which resulted in identification of genome search specific peptides (GSSPs). These GSSPs mapped uniquely to the L. major genome and were identified in our study by searching unassigned spectra that were filtered out post L. major protein database searches. These unassigned spectra were searched using a L. major six-frame translated genome database in the second pass search. This proteogenomics study resulted in identification of 15 N-terminal extensions and three novel genes in L. major. A complete overview of the proteogenomics workflow adopted for the L. major promastigote MS/MS data is shown in Figure 3. The details of the novel genes and N-terminal extensions along with the corresponding GSSPs are provided in the Supplementary Table S3.

Proteogenomics workflow used to analyze L. major promastigote proteomic data. L. major mass spectrometry data was first searched against a L. major protein database resulting in the identification of 3613 proteins in L. major. The unmatched spectra were searched in the second pass search against the six-frame translated L. major genome database, which resulted in identification of GSSPs. These GSSPs, in turn, provided evidence for existence of novel protein coding genes and also resulted in correction to existing annotations in L. major upon further analysis of genomic regions.
Identification of novel genes in L. major
We identified GSSPs mapping to the six-frame translated L. major genome. These were categorized as intergenic (i.e., mapping to particular genomic regions that have no known protein coding genes). The identification of these novel protein coding genes in L. major shows that these genes are not a part of the current L. major genome annotation. In the current study, three unique peptides were identified mapping to the intergenic region of L. major genome that resulted in identification of three novel protein coding genes in L. major. An illustrative example of a novel gene identified in L. major coding for a conserved hypothetical protein is shown in Figure 4. We identified a unique GSSP mapping to intergenic region on chromosome 32 on the negative strand in the third frame. Upon gene prediction analysis, we identified a protein that is 125 amino acids long in this region. This is a novel gene in L. major and there is no conserved annotated ortholog in other Leishmania species, namely L. donovani, L. braziliensis, and L. infantum. However, Trypanosoma cruzi has a gene Tc00.1047053511707.20 that codes for this hypothetical protein. Upon further analysis of the genomic sequences from different Leishmania species, we identified similar conserved ORFs in other Leishmania species that are not yet identified and annotated as protein-coding genes. Our proteogenomics data show that these novel genes have protein-coding potential. Further investigations need to be carried out to determine whether they are also expressed in other Leishmania species that have similar conserved genomic sequences. Another example of a novel gene in L. major is one which codes for a hypothetical protein. We identified a unique GSSP mapping to the intergenic region on chromosome 9 on the negative strand in the first frame. Gene prediction analysis resulted in identification of 140 amino acid long protein. We have not identified any conserved orthologs in other protozoans.
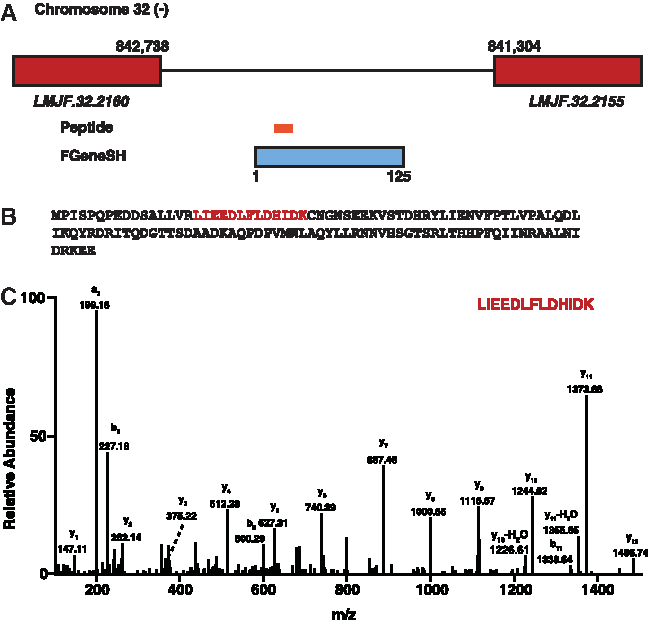
Identification of a novel protein coding genes in L. major based on peptide evidence.
Identification of N-terminal extension in L. major
In the present study, we have corrected the annotation of existing gene models in L. major. Towards this end we identified GSSPs that mapped to 5′ boundary of existing genes in L. major. Hence, we identified 23 unique peptides that resulted in N-terminal extension of 15 proteins in L. major. An example of N-terminal extension of an existing L. major gene is of leucine-rich repeat coding gene (LmjF.30.0150), which codes for a 216 amino acid long protein and is present on chromosome 30 negative strand first frame. We identified 3 GSSPs mapping to upstream of this gene. This resulted in extension of this protein by 140 amino acids. Upon gene prediction analysis, we identified a longer protein which contained the 3 GSSPs. It had a conserved ortholog in L. braziliensis which was coded by the gene LBRM_30_0160 which codes for a 359 amino acid longer protein (Fig. 5). Thus we corrected the annotation of the existing gene in L. major and identified an N-terminal extended protein product in L. major. Another example of N-terminal extension of the gene LmjF.26.1550, which codes for mitochondrial tri-functional enzyme alpha subunit of length 726 amino acids, is present on chromosome 26 on the positive strand in the second frame. We identified a single GSSP mapping upstream of this gene. This resulted in extension of this protein by 88 amino acids. Upon gene prediction analysis, we identified a longer protein that contained the GSSP. It had a conserved ortholog in L. braziliensis that was coded by the gene LBRM_26_1570, which codes for an 804 amino acid long protein.
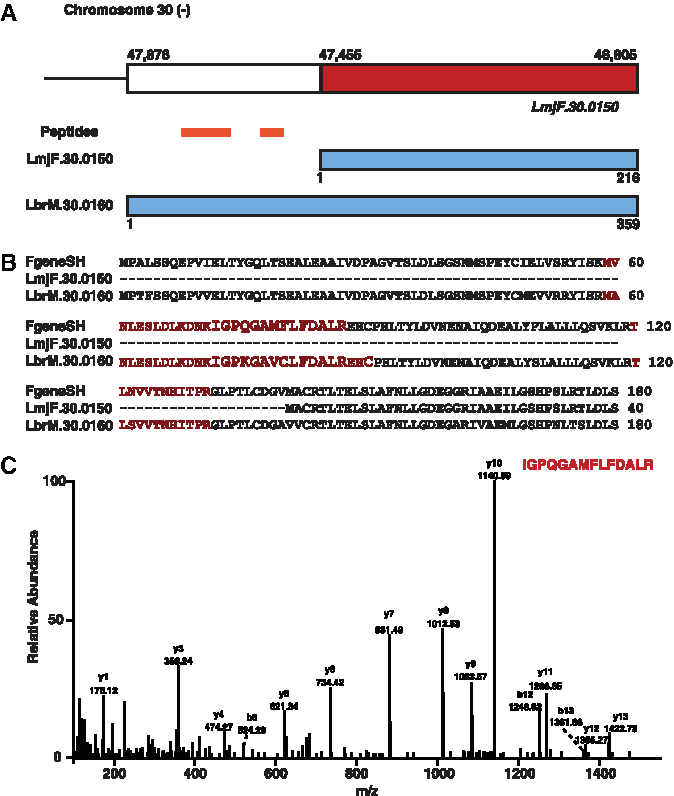
Identification of N-terminal extended proteins in L. major.
Bioinformatics analysis
Identified proteins were classified into various groups based on their primary subcellular localization (e.g., membrane, cytoplasm). Additionally, proteins were also grouped on the basis of biological process (e.g., metabolism). The analysis was carried out in accordance with Gene Ontology (GO) standards. The 3613 proteins identified in the current proteogenomics analysis of L. major promastigote were categorized on the basis of biological processes (e.g., cell signaling and communication). This resulted in the identification of 1418 proteins (39%), which were grouped into one of biological processes (Fig. 6a). The majority of the grouped proteins play a role in cellular metabolism, protein synthesis, degradation, and transport. However, only a small number of the identified proteins are known to be associated with pathogenesis. This points towards the fact that many of the virulence factors associated with infection and survival of L. major in infected host cells (i.e., neutrophils and macrophages) are expressed constantly at a certain basal level, even in axenic L. major cultures. Expression of these pathogenicity-associated factors probably increases post exposure of these pathogens to the host immune system. Additionally, proteins were classified on the basis of primary subcellular locations (Fig. 6b). We have also analyzed biological domains and motifs of the proteins identified in the current proteogenomics analysis of L. major. The details of the corresponding domains and motifs can be found in Supplementary Table S1. Many of the identified proteins could not be classified in any known category, and they remained unclassified. Many of these proteins could be potential drug targets or vaccine candidates.

Gene ontology based bioinformatics analysis of proteins identified in L. major.
A bioinformatics analysis of L. major promastigote proteins was carried out by searching TriTrypDB for identification of signal peptide and transmembrane domain information. Of the 3613 proteins identified in L. major, 399 proteins contained transmembrane (TM) domain, and 366 proteins contained only signal peptides (SP). Additionally, 296 proteins contained both a transmembrane domain and a signal peptide (Fig. 6c). Identification of secreted and transmembrane proteins in L. major can provide valuable information regarding the role of these membrane/extracellular proteins in virulence and pathogenicity of L. major during cutaneous leishmaniasis.
Some of the interesting surface proteins that are shown to be associated with virulence are as follows: Tuzins are surface proteins, which have been studied in relation to Trypanosoma cruzi and its expression, was shown to be regulated post transcriptionally (Teixeira et al., 1999). Kinetoplastid membrane protein-11 (KMP-11) is a surface protein that is expressed in the promastigote stage of Leishmania. KMP-11 expression is severely downregulated in the amastigote stage and is localized to flagellar basal body and flagella. KMP-11 is present either as membrane bound or soluble protein (Berberich et al., 1998). It is highly antigenic and stimulates a variety of immune cells such as B-cells and dendritic cells. Additionally, circulating anti-KMP-11 antibodies have been found in patients with leishmaniasis (Trujillo et al., 1999). The partial list of transmembrane and secreted proteins identified in L. major promastigote is shown in Table 1.
RT-PCR validation of mass spectrometry derived data
To validate our finding in the current study, a subset of novel identifications were validated using RT-PCR. These included 5 N-terminal extensions and 3 novel genes. In total 8 RT-PCR reactions were performed to validate the existence of transcripts for novel genes and N-terminal extension events. The cDNA sequences have been submitted to NCBI Genbank. Figure 7 shows the RT-PCR amplification products for the novel genes and N-terminal extensions identified in our study. The details of validated novel genes and N-terminal extensions with the corresponding GenBank accession numbers are provided in the Supplementary Table S4.
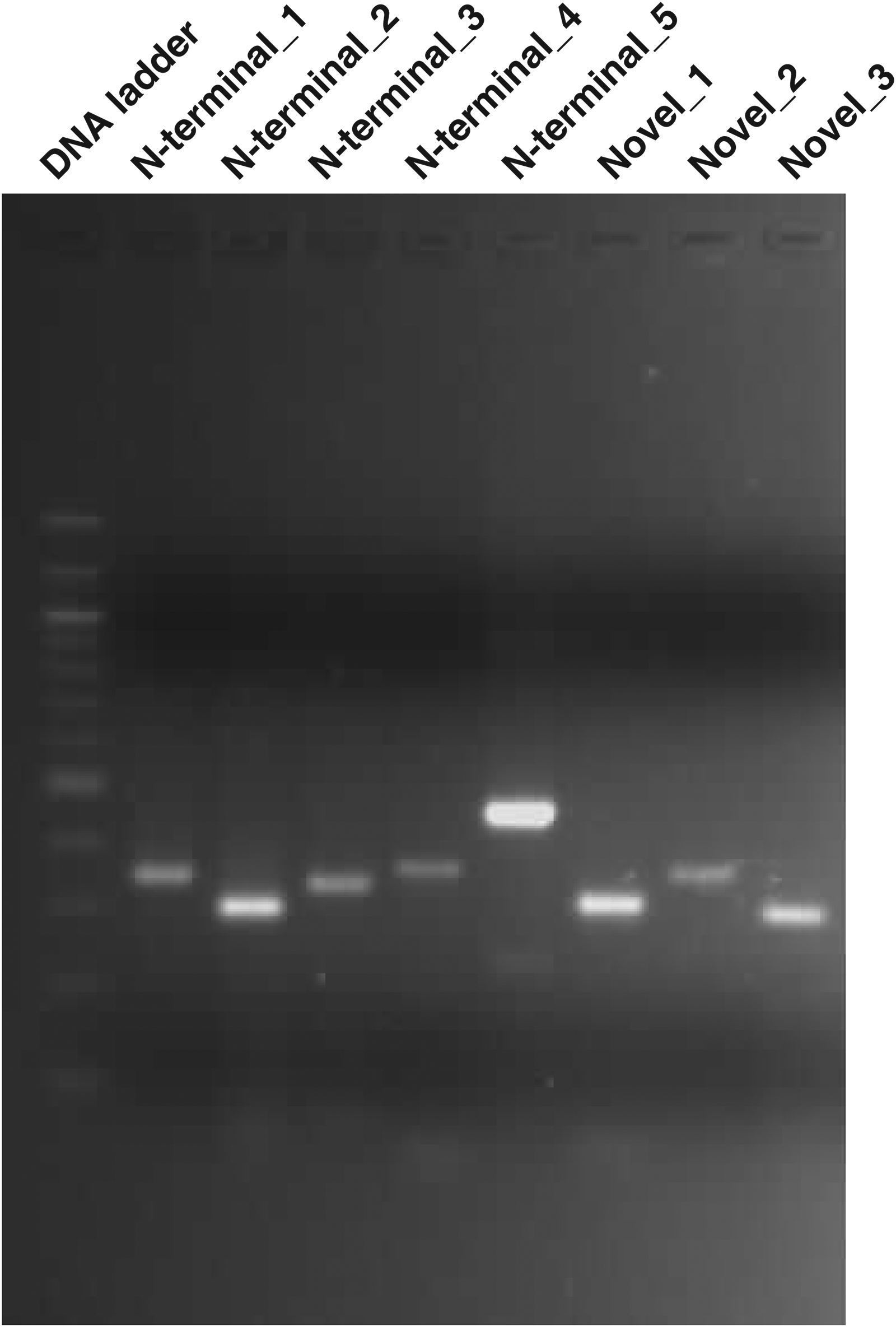
RT-PCR based validation of novel genes and N-terminal extension identified in L. major. We validated the results from our proteogenomics study using RT-PCR and sequencing it. We carried out agarose gel electrophoresis of the PCR products of N-terminal extensions and novel genes identified in L. major proteogenomics study.
Discussion
Leishmania major genome was the first genome whose cosmid contig map was constructed amongst Leishmania species. Also, the genome of L. major was the first genome to be completely sequenced and annotated. It is at present the best annotated genome amongst Leishmania parasites. The L. major complete genome sequence has been exploited for comparative genomics with other sequenced genomes of Leishmania that showed conservation of gene content and synteny amongst different species of Leishmania (Peacock et al., 2007). The gene sequences also helped in transcriptomic analysis in different Leishmanial spp. (Cohen-Freue et al., 2007; Depledge et al., 2009; Li et al., 2008). Various in silico analyses have been performed using this genome sequence to yield interesting information such as proteins targeted to glycosomes (Opperdoes and Szikora, 2006). Despite this information, very few studies have been carried out with L. major, especially in the area of transcriptomics and proteomics. Studies reported in these areas used L. donovani, L. mexicana, L. infantum, and L. panamensis, and showed that ∼10%–12% of proteins are differentially expressed in amstigotes (Papadopoulou, 2008). This may be because it is easier to obtain motile promastigotes, infective metacyclic and axenic non-motile amastigotes by in vitro cultures with these parasites. Axenic amastigotes cannot be obtained in vitro from L. major, which make it an unsuitable model for most research of Leishmania. Study of proteomics in Leishmania has also helped reveal post-translational modifications (Rosenzweig et al., 2008), immunodominant antigens (Forgber et al., 2006), and virulence factors (Fasel, 2008). Proteins from the promastigotes stage, which were previously reported to be uniquely expressed in the amastigote stage such as 40S ribosomal S2 protein, β-tubulin, and eukaryotic initiation factor 5a, putative have been reported (Nugent et al., 2004).
Our proteogenomics study is significant because it is the largest catalog of proteins detected by mass spectrometry as compared to other reported studies of different Leishmanial spp. In total, 23,333 unique peptide sequences were identified, which belonged to 3613 proteins in L. major promastigotes. This represents 43% of L. major proteome comprising of a large set of proteins having different cellular locations and function. We compared our L. major promastigote data with gene expression analysis reported previously to identify stage-specific expression in L. major promastigotes (Akopyants et al., 2010). The gene expression data were downloaded from TriTrypDB. We identified various proteins that were reported to be expressed in log phase promastigotes, but also detected proteins that are known to be expressed in the metacyclic stage. We found many proteins in our proteomic data that are reported to be upregulated in metacyclic stage of L. major as seen by the relative increase in their mRNA as compared to log phase promastigotes stage of parasite. These proteins belong to cell surface proteins including amastins, hydrophilic acylated surface protein, and various transporters. Proteins that were upregulated in metacyclics responsible for oxygen radical metabolism such as tryparedoxin and glutathione peroxidase were also detected in our preparation (TritrypDB). In fact, metacyclic promastigotes are enriched from promastigotes culture, and presence of these proteins in our preparation indicates presence of metacyclic forms in the culture. We also compared our L. major promastigote data with a previously published gene expression analysis data on L. major by Rochette et al. (2008), who compared gene expression profiles between mid-log phase promastigotes and lesion-derived amastigotes of L. major. Of the 431 unique genes found to be differentially expressed in L. major promastigotes, we identified 260 proteins in our study. These include proteins again responsible for flagellar components, surface antigen proteins, fatty acid metabolism, and various transporters.
We identified 266 N-terminal acetylated peptides in our study. This is the largest catalog of acetylated peptides to date in Leishmania. N-terminal acetylation is crucial for stability of proteins. In a proteomic study by Rosenzweig et al. (2008), the authors carried out a time course analysis of L. donovani differentiation in a quantitative proteomic approach. They also tried to identify various post-translational modifications in both life stages of L. donovani. Their study resulted in identification of 16 phosphopeptides, 20 methylated peptides, 10 glycopeptides, and 27 acetylated peptides (Rosenzweig et al. 2008). Of those 26 unique acetylated peptides, which mapped to 26 proteins in L. donovani, we identified 10 N-terminally acetylated peptides in our 266 N-terminal acetylated peptide list (black bold in Supplementary Table S5). While the remaining 16 acetylated peptides identified in L. donovani were not identified in our L. major proteogenomics study, in fact one peptide (MNSNHADAGAPAMEK) was redundant in Rosenzweig's list and it mapped to aspartyl-tRNA synthetase, as can be seen in Supplementary Table S5. However, the remaining 256 N-terminal acetylated peptides identified in L. major promastigotes have not been reported earlier and the complete list of these unique 266 acetylated peptides is provided in Supplementary Table S5.
Our proteogenomics analysis resulted in identification of 26 genome search specific peptides (GSSPs), which map uniquely to the six-frame translated genome of L. major and not to the L. major protein database. In all, 23 peptides mapped to N-termini of 15 genes, thereby extending the boundary of these previously annotated genes. These 15 N-terminal extensions could be identified as the GSSPs mapped 5′ to the existing gene models in L. major. It should be pointed out that alternative coding sequences were shown to exist in TriTrypDB database due to alternative splicing via spliced leader peptides. Many of these gene models that we have corrected in L. major have an alternative start site upstream of the currently annotated gene and GSSPs identified in our study mapped to the alternative CDS. However, these gene models were not updated and our study showed that these alternative CDS lead to generation of longer proteins. The full length orthologs for many of these N-terminally extended genes exist in other related kinetoplastids. Our proteogenomics analysis also identified three GSSPs that map to intergenic region where no known ORF was shown to exist in L. major genome. These three GSSPs resulted in identification of three novel genes in L. major and these genes do not have conserved annotated orthologs in other Leishmania. This is quite novel since the L. major genome was the first to be sequenced, and its genome was used as reference for assembling other Leishmania species genomes. The L. major genome is the best curated genome among the Leishmania species. We could still identify three novel protein coding genes that were missed earlier by using our proteogenomics approach. However, further analysis of the genomic regions (containing novel protein coding genes) in L. major and in other related Leishmania species resulted in identification of conserved ORFs that represent novel protein coding genes not yet identified and annotated in L. major and in other related Leishmania species. Thus at the genomic level, there is conservation of these novel genes in most Leishmania species, but these novel protein coding genes need to be identified and annotated. We have validated the existence of these three novel genes and 5 N-terminal extensions in identified in our proteogenomics study of L. major using RT-PCR and sequencing.
Conclusions
In the current proteogenomics analysis, we mapped 43% of the L. major proteome, illustrating the value of high-resolution mass spectrometry derived data in proteomic profiling and genome annotation of L. major. We have identified expression of many hypothetical proteins in promastigotes. In addition, we identified bona fide virulence factors that have been shown to be associated with pathogenesis and survival of Leishmania in infected hosts. Although L. major genome was the first Leishmania genome to be sequenced and is one of the best annotated Leishmania genomes, we identified 26 GSSPs from six-frame translated genome searches of L. major and this resulted in correction to the 15 gene models (N-terminal extensions). In addition, we identified three novel genes in L. major genome on the basis of GSSP evidence. This study shows that genome annotation is a challenging task, and using proteomic and proteogenomics approaches can assist in identification of novel genes missed in the current annotation. We could also refine existing gene models on the basis of peptide evidence.
Footnotes
Acknowledgments
We thank the Department of Biotechnology (DBT), Government of India, for research support to the Institute of Bioinformatics. Harsha Gowda is a Wellcome Trust/DBT India Alliance Early Career Fellow. Harsh Pawar and Sweta N. Khobragade are recipients of Senior Research Fellowships, Gajanan Sathe and Sandip Chavan are recipients of Junior Research Fellowships from Council of Scientific and Industrial Research (CSIR), Government of India. Santosh Renuse is recipient of Senior Research Fellowship from the University Grants Commission (UGC), Government of India. We would like to thank Deepa Chaphekar for assistance in making of figures.
Author Disclosure Statement
The authors declare no conflicting financial interests.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.