
Abstract
Abstract
Brucella is an intracellular bacterium that causes the zoonotic infectious disease, brucellosis. Brucella species are currently intensively studied with a view to developing novel global health diagnostics and therapeutics. In this context, small RNAs (sRNAs) are one of the emerging topical areas; they play significant roles in regulating gene expression and cellular processes in bacteria. In the present study, we forecast sRNAs in three Brucella species that infect humans, namely Brucella melitensis, Brucella abortus, and Brucella suis, using a computational biology analysis. We combined two bioinformatic algorithms, SIPHT and sRNAscanner. In B. melitensis 16M, 21 sRNA candidates were identified, of which 14 were novel. Similarly, 14 sRNAs were identified in B. abortus, of which four were novel. In B. suis, 16 sRNAs were identified, and five of them were novel. TargetRNA2 software predicted the putative target genes that could be regulated by the identified sRNAs. The identified mRNA targets are involved in carbohydrate, amino acid, lipid, nucleotide, and coenzyme metabolism and transport, energy production and conversion, replication, recombination, repair, and transcription. Additionally, the Gene Ontology (GO) network analysis revealed the species-specific, sRNA-based regulatory networks in B. melitensis, B. abortus, and B. suis. Taken together, although sRNAs are veritable modulators of gene expression in prokaryotes, there are few reports on the significance of sRNAs in Brucella. This report begins to address this literature gap by offering a series of initial observations based on computational biology to pave the way for future experimental analysis of sRNAs and their targets to explain the complex pathogenesis of Brucella.
Introduction
B
sRNAs are present in all life forms. Bacterial sRNAs are typically 50–500 nucleotides (nt) long and regulate diverse cellular functions, such as bacterial virulence, secretion, stress response, and quorum sensing. (Gottesman, 2005; Storz et al., 2005). sRNAs regulate the translation (inhibition/activation) by interacting with specific mRNA targets through base pairing. sRNAs downregulate the gene expression either by interfering with ribosome binding or degrading mRNA target (Storz et al., 2011). Very few sRNAs were shown to positively regulate the gene expression by exposing the ribosome binding site and stabilizing the mRNA (Papenfort et al., 2013). The majority of the sRNAs identified are trans-encoded, these sRNA genes are located far away from their mRNA targets, and their hybridization occurs through short imperfect base pairing (Waters and Storz, 2009). These regulatory RNA molecules are situated in the intergenic regions between protein-coding genes (Atluvia, 2007). Another type of sRNA is cis-encoded, which is cotranscribed with its mRNA targets to regulate its transcription or translation. As the number of available whole-genome sequences is increasing, many computational methods have been developed to identify sRNAs from genome sequences. To date, several sRNAs have been identified in pathogenic bacteria, such as Mycobacterium tuberculosis (Arnvig and Young, 2009), Salmonella spp. (Altier et al., 2000), Listeria monocytogenes (Behrens et al., 2014; Mraheil et al., 2011), and Staphylococcus spp. (Bohn et al., 2010; Pichon and Feldon, 2005). Several computational methods have been developed for the identification of sRNAs in bacterial genome sequences.
Brucella spp. are the etiological agents of the zoonotic disease, brucellosis. Brucella is gram-negative, facultative intracellular bacterium and belongs to the class Alphaproteobacteria (Whatmore, 2009). The genus Brucella is classified into 10 species: Brucella melitensis (sheep and goat), Brucella abortus (cattle), Brucella suis (pig), Brucella canis (dog), Brucella ovis (sheep), Brucella pinnipedialis (seal), Brucella microti (common vole), Brucella neotomae (desert wood rat), Brucella ceti (dolphin, whale), and Brucella inopinata (human), based on the host preference and antigenic and biochemical characteristics (O'Callaghan and Whatmore, 2011). Brucellosis is characterized by sterility, infections, and abortion in animals (Corbel, 1997) and, in humans, it causes undulant fever, osteoarthritis, and several neurological disorders (Young, 1995). Brucella spp. are highly conserved at the nucleotide level and do not show many genetic variations among different species (Paulsen et al., 2002). Brucella does not have classical virulence factors, such as fimbria, toxins, and plasmids. (Moreno and Moriyon, 2002). The virulence of Brucella relies on its ability to survive and replicate in the host phagocytes (Kaufmann, 2011). However, the complex mechanism of intracellular survival of Brucella remains poorly understood. Recently, few sRNAs were identified in Brucella and their roles in intracellular survival and pathogenicity were reported (Caswell et al., 2012; Wang et al., 2015). In this study, we report the computational prediction of sRNAs and their mRNA targets in B. melitensis, B. abortus, and B. suis. We also report the functional enrichment analysis of the predicted mRNA targets and the associated pathways.
Materials and Methods
Prediction of sRNAs in three Brucella species
sRNA candidates were predicted by integrating the outputs of two programs, SIPHT and sRNAscanner. SIPHT (sRNA Identification Protocol using High-throughput Technologies) identifies candidate sRNAs by searching for putative Rho-independent terminators downstream of conserved intergenic sequences. Each predicted locus is then annotated with several features, including its conservation in other species, its association with one of several transcription factor binding sites, its position relative to flanking genes, and its homology and conserved genomic position with previously identified sRNAs (Livny et al., 2008). SIPHT searches on Web server (http://newbio.cs.wisc.edu/sRNA/) were used to detect sRNAs with following parameters: sRNA length −50–550 nt, maximum blast E value –1e-15, minimum TransTerm confidence value −87, maximum FindTerm score −10, and maximum RNAMotif score—9. Other parameters were kept at their default values. To ensure the accuracy of computational prediction, we considered only the candidates denoted as RNA by the QRNA (Rivas and Eddy, 2001) analyses of SIPHT.
sRNAscanner (Sridhar et al., 2010) was the other program used to screen sRNA candidates in three Brucella genomes. sRNAscanner identifies intergenic sRNA transcriptional units based on the transcriptional signals. This computational program uses position weight matrices of sRNA promoter and rho-independent terminator signals derived from experimentally defined Escherichia coli K-12 MG1655 to identify sRNAs through sliding window-based genome scans. The searches were restricted to intergenic regions, and the sRNA length for prediction was set to 50–550 nt. All other parameters were kept at default values.
Target prediction
The mRNA targets of the sRNA candidates were predicted using TargetRNA2 (Kery et al., 2014) (http://cs.wellesley.edu/∼btjaden/TargetRNA2) with default parameters.
Functional enrichment analysis
Functional categorization of predicted target genes was done using COG (cluster of orthologous groups) analysis and Gene Ontology (GO) annotations. COG classification was done using COG database (Tatusov et al., 2003) and Comparative GO was used for GO annotations (Fruzangohar et al., 2013). Pathway annotations were done using KEGG pathway database with default parameters.
GO regulatory network
Regulatory relationships among the biological processes were analyzed using the Comparative GO Web server (Fruzangohar et al., 2013). GO regulatory network (GRN) provides information on (1) regulatory relationships between GO terms and their associated genes, (2) enrichment levels of GO terms, and (3) genes associated with each GO term. We constructed GRN for the predicted sRNA target genes in three Brucella species.
Results
In-silico prediction of sRNA candidates in three Brucella species
sRNAs in three genomes of Brucella were predicted using SIPHT and sRNAscanner. We considered only the candidates predicted as RNA by QRNA algorithm of SIPHT as sRNAs. The number of sRNAs predicted for each Brucella genome is shown in Figure 1 and the details of predicted sRNAs are given in Tables 1–3. sRNAs predicted by both methods were considered as potential sRNA candidates. In B. melitensis, 21 sRNAs were consistently predicted by both QRNA and sRNAscanner. Similarly, in B. abortus and B. suis, 13 and 16 consensus RNAs, respectively, were predicted. sRNA candidates identified varied in length between 62 and 487 nt (Fig. 2). The majority of the candidates were between 51 and 150 nt in length. GC content of sRNA candidates ranged from 34% to 65%. The consensus sRNAs predicted were searched against Rfam database (http://rfam.sanger.ac.uk/) and Bacterial Small Regulatory RNA Database (http://bac-srna.org/BSRD/index.jsp#) to eliminate the duplicates if present. Two sRNA candidates in B. melitensis, one in B. abortus, and two in B. suis were showing homology with already identified sRNAs in Rfam database (Table 4).

sRNA candidates predicted in

Length distribution of sRNAs identified in B. melitensis, B. abortus, and B. suis.
Novel sRNAs in the major range (51 to 150 nt) identified in this study.
sRNAs having homologs in Rfam.
sRNAs, small RNAs.
Novel sRNAs in the major range (51 to 150 nt) identified in this study.
sRNAs having homologs in Rfam.
Novel sRNAs in the major range (51 to 150 nt) identified in this study.
sRNAs having homologs in Rfam.
Comparative analysis
The novel sRNAs identified were searched against the nonredundant database of NCBI using blastn algorithm with the parameter, exclude organism Brucella (taxid: 234), to identify the conservation of sRNA sequences in other organisms. In B. melitensis 16M, 15 sRNA candidates were found to be Brucella specific. Similarly, 12 and 13 candidate sRNAs of B. abortus 2308 and B. suis 1330, respectively, were Brucella specific. sRNA candidates were also searched against all other Brucella species to see the conservation in the genus. sRNA sequences were conserved in the genus except in nonzoonotic species B. neotomae and B. inopinata. In addition, we compared the sRNAs predicted in one species with other species to identify the homologous sRNAs in three Brucella species (Table 5). One sRNA of B. melitensis (BmsRNAs19) was found in other two species also. Similarly, seven sRNAs of B. abortus were found in B. suis, but not in B. melitensis, and one sRNA of B. melitensis was found in B. suis, but not in B. abortus (Table 5). In total, 14 sRNA candidates in B. melitensis were identified as novel and Brucella specific. Similarly, three sRNAs in B. abortus and five sRNAs in B. suis were identified as novel and Brucella specific.
sRNAs having homologs in other organisms also (Table 4).
Target prediction
To understand the role of sRNAs in biological functions, an in silico analysis was done for the prediction of mRNA targets of the predicted sRNAs, using the Web server TargetRNA2. Target genes predicted for each sRNA of all three species are listed in Supplementary Tables S1–S3. In B. suis, no targets were predicted for BssRNA7. Five candidate targets with high scores for each candidate sRNA were considered for further analysis.
Functional categorization of target genes
To gain more insights on the role of mRNA targets predicted, target genes were functionally classified based on COG analysis and GO annotations. In all the three species, mRNA targets were enriched in COG categories of transport and metabolism of carbohydrates, amino acids, lipids, nucleotides, and coenzyme, energy production and conversion, replication, recombination and repair, transcription, etc. (Fig. 3). Enriched GO terms for the mRNA targets are described in Supplementary Tables S4–S6. GO terms were distributed widely with regard to their respective biological processes. The target genes were enriched in GO terms, such as metabolic pathways, transport pathways, transcription, response to stress, DNA replication, and pathogenesis (Fig. 4A, D, G) (Supplementary Tables S4–S6). When the targets are categorized as molecular function, a majority of the genes were related to GO terms, catalytic activity and binding. Particularly, the genes were enriched in nucleic acid binding, DNA binding, ATP binding, GTP binding, ion binding, and transporter activity, etc. (Fig. 4B, E, H) (Supplementary Tables S4–S6).
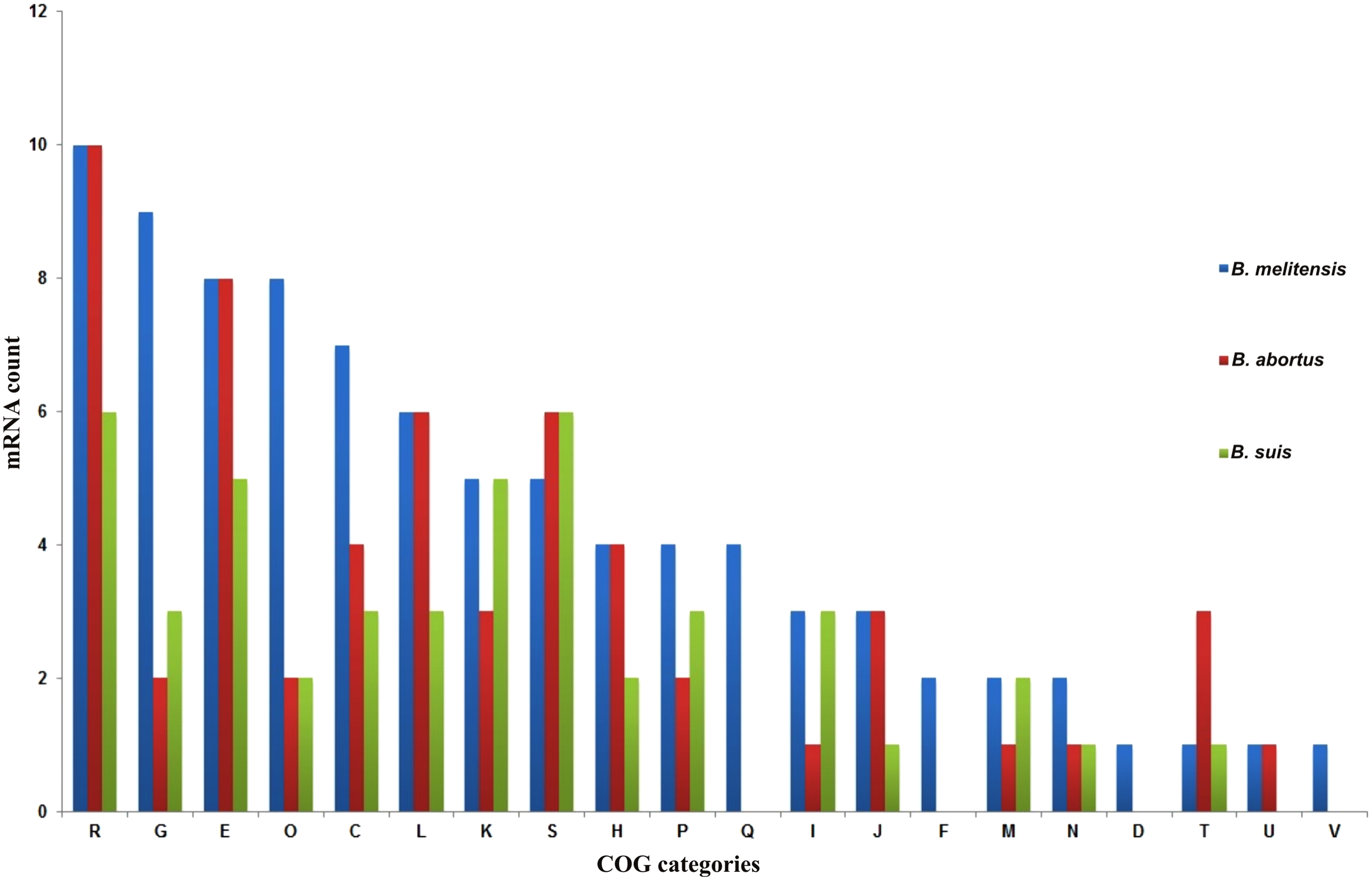
COG classification of target genes in B. melitensis, B. abortus, and B. suis. The COG (cluster of orthologous groups) categories are coded as follows: C, energy production and conversion; D, cell division and chromosome partitioning; E, amino acid transport and metabolism; F, nucleotide transport and metabolism; G, carbohydrate transport and metabolism; H, coenzyme metabolism; I, lipid metabolism; J, translation; K, transcription; L, DNA replication, recombination, and repair; M, cell wall/membrane biogenesis; O, post-translational modification, protein turnover, and chaperones; P, inorganic ion transport and metabolism; Q, secondary metabolite biosynthesis, transport, and catabolism; R, general functional prediction only; S, function-unassigned conserved proteins; T, signal transduction; and V, defense mechanisms.
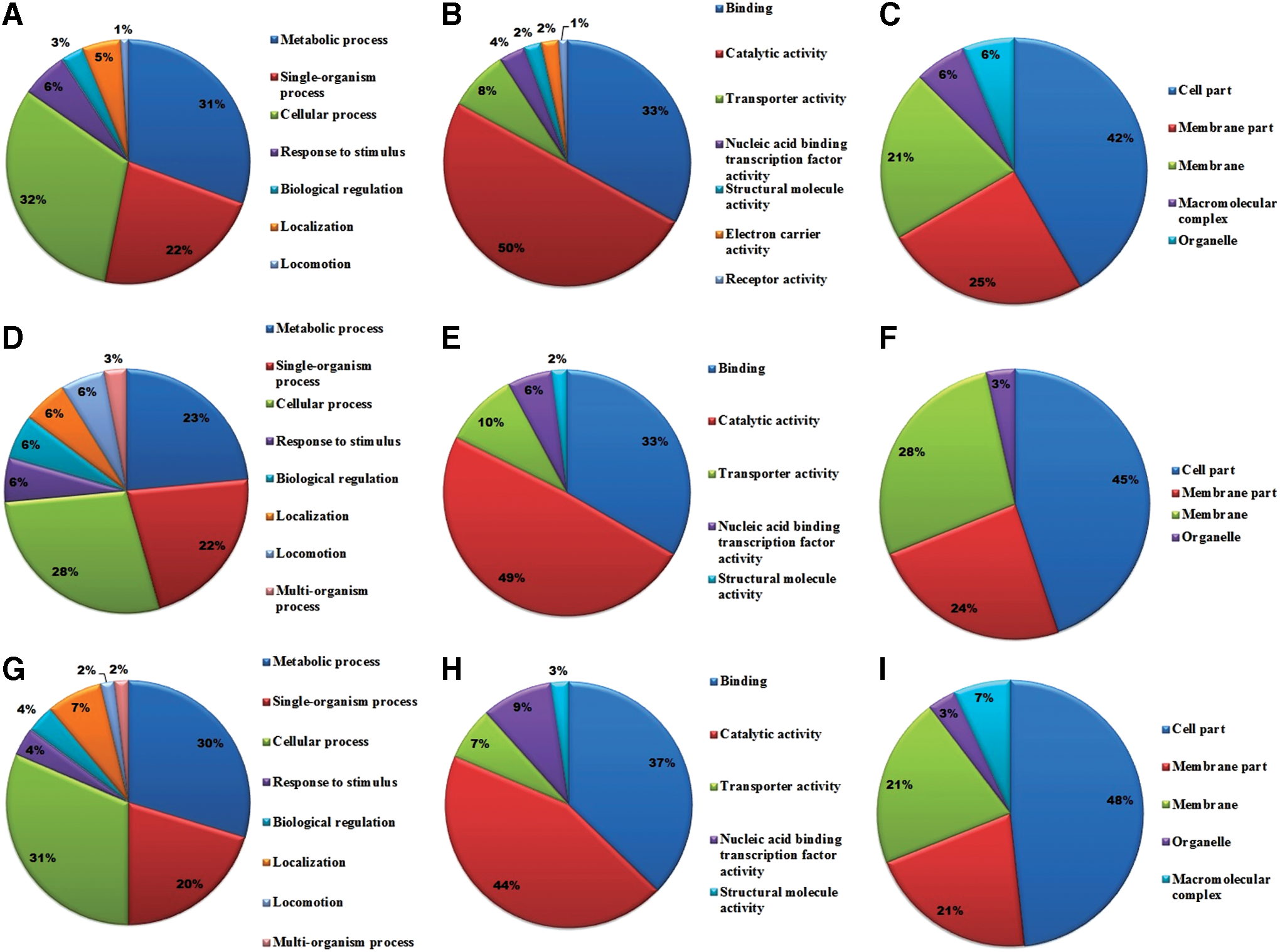
Gene Ontology (GO) analyses of predicted target genes in B. melitensis, B. abortus, and B. suis. GO analysis of target genes that are predicted to be involved in
To further understand the role of the target genes in biological pathways, the predicted target genes were mapped to KEGG database using KEGG mapper. In B. melitensis, the target genes were mapped onto 43 known pathways with a p-value <0.05. The targets were predominantly enriched in metabolic pathways, microbial metabolism in diverse environments, ABC transporters, and biosynthesis of secondary metabolites. In B. abortus, the target genes were mapped onto 27 known pathways with a majority of the genes mapped on to metabolic pathways, ABC transporters, biosynthesis of secondary metabolites, and microbial metabolism in diverse environments. In B. suis, a majority of the genes mapped on to metabolic pathways, biosynthesis of secondary metabolites, and carbon metabolism.
GO regulatory network
We determined GO term enrichments for the mRNA targets identified for all the sRNAs from B. melitensis 16M, B. abortus 2308, and B. suis 1330. We describe the implementation of GO-based gene selection and GO network discovery. We constructed a GO interaction network between biological process GO terms for the mRNA targets identified in all the three genomes. The GO network of mRNA targets of B. melitensis 16M sRNAs is shown in Figure 5. Transcription antitermination (GO ID: 31564) was the central node in the GRN. Transcription antitermination protein NusG (BMEI0744) showed interaction with other GO terms, such as response to stress, transport, DNA replication, transcription DNA-templated, cell redox homeostasis, DNA repair, protein folding, cytolysis, cell cycle, nitrogen compound metabolic process, and ATP-coupled electron transport. (Fig. 5). In the case of B. abortus 2308, DNA-templated negative regulation of transcription (GO ID: 45892) was the central node in the network (Fig. 6) governed by BAB10578, which codes for a transcriptional regulator, betI. BAB10578 governed GO has interactions with many other GO terms, such as response to stress, pathogenesis, gluconeogenesis, transport, nucleoside metabolic process, DNA replication, lipopolysaccharide biosynthesis process, DNA repair, and protein folding. (Fig. 6). Cell redox homeostasis (GO ID: 45892) was the central node of the GRN of B. suis 1330 with the involvement of dihydrolipoamide dehydrogenase (BR1126). Cell redox homeostasis showed interaction with pathogenesis, DNA repair, transport, DNA recombination, DNA template transcription, metabolic process, and rRNA processing, etc. (Fig. 7).
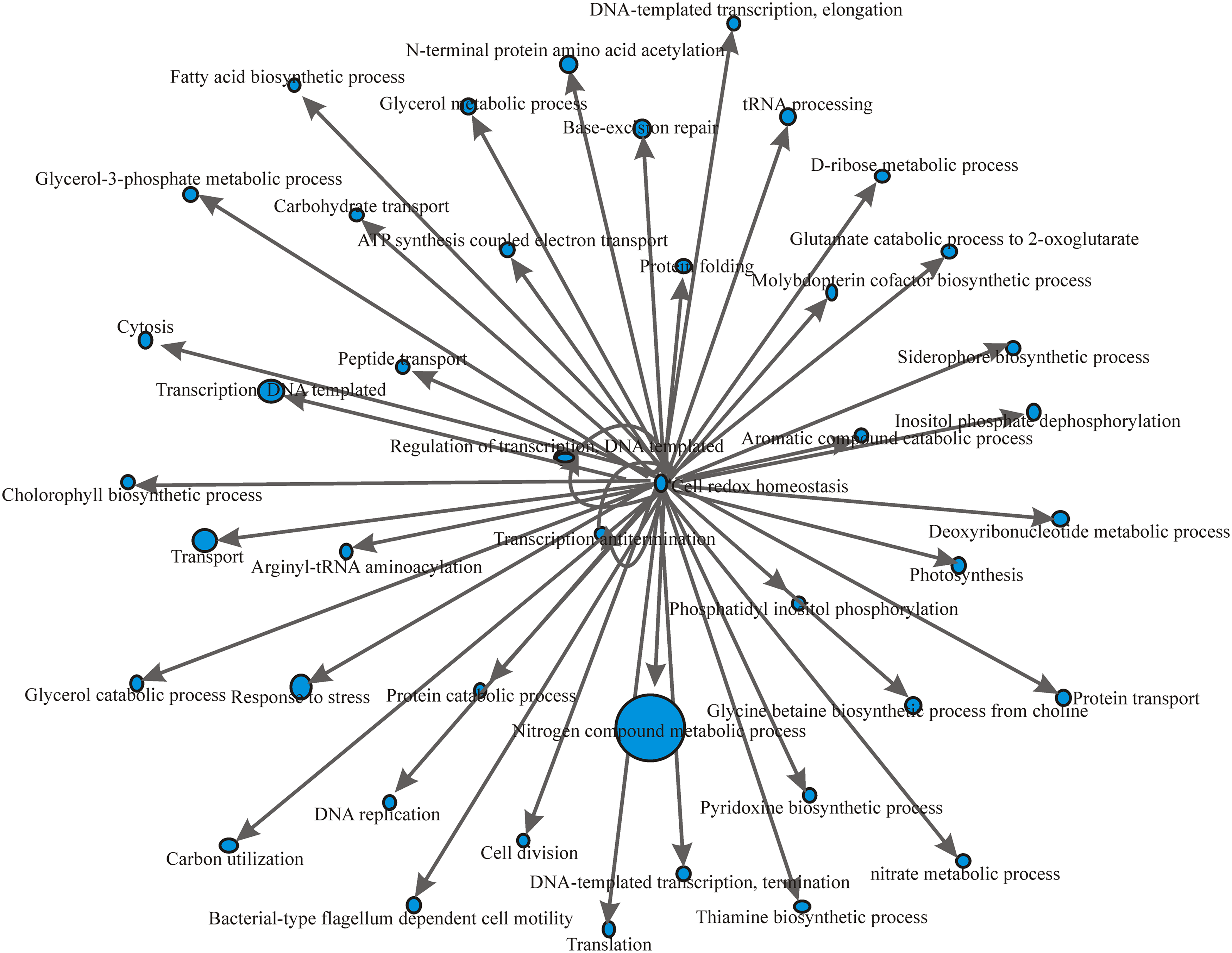
GO regulatory network (GRN) based on the mRNA target genes of B. melitensis 16M.
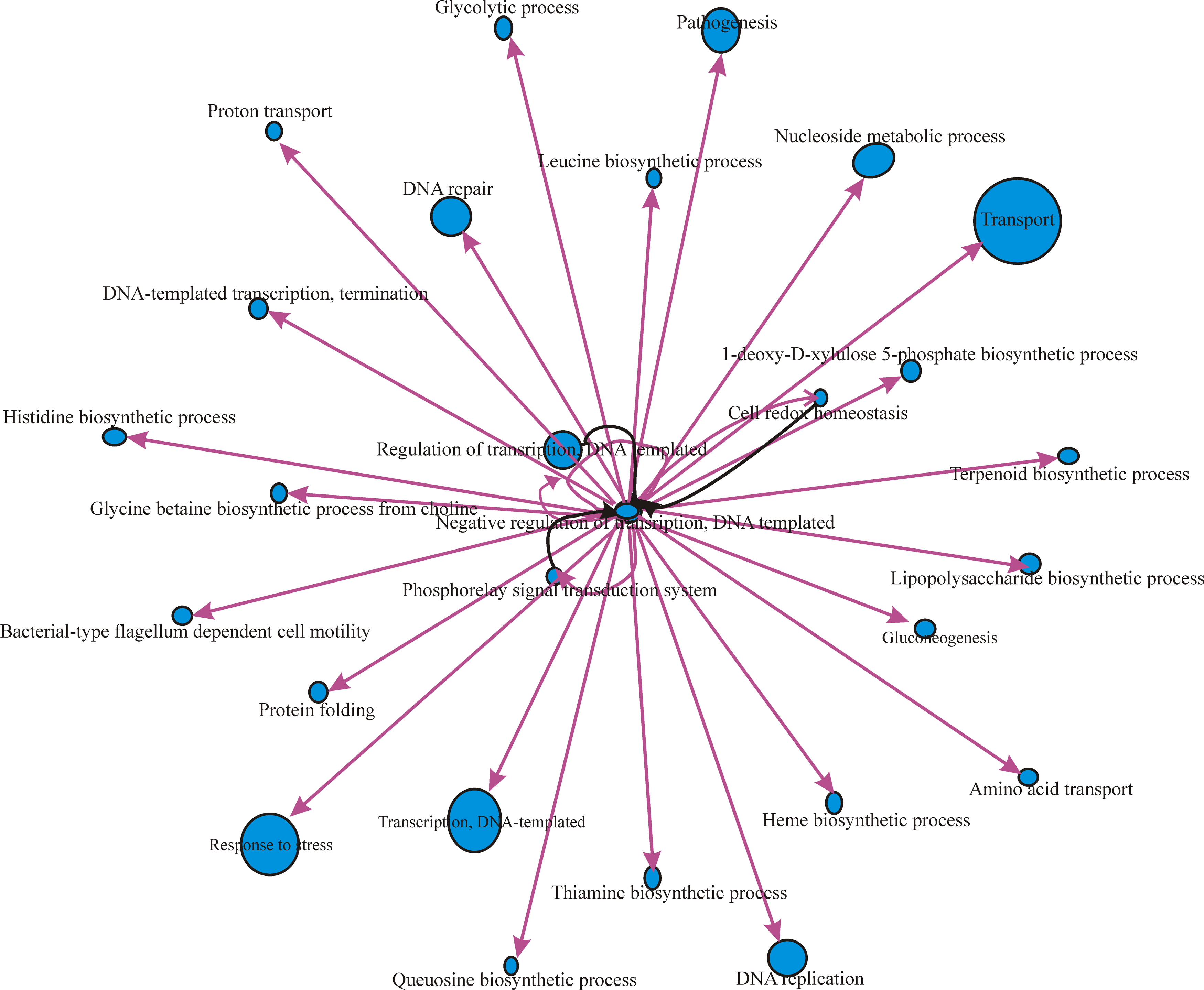
GRN based on the mRNA target genes of B. abortus 2308.
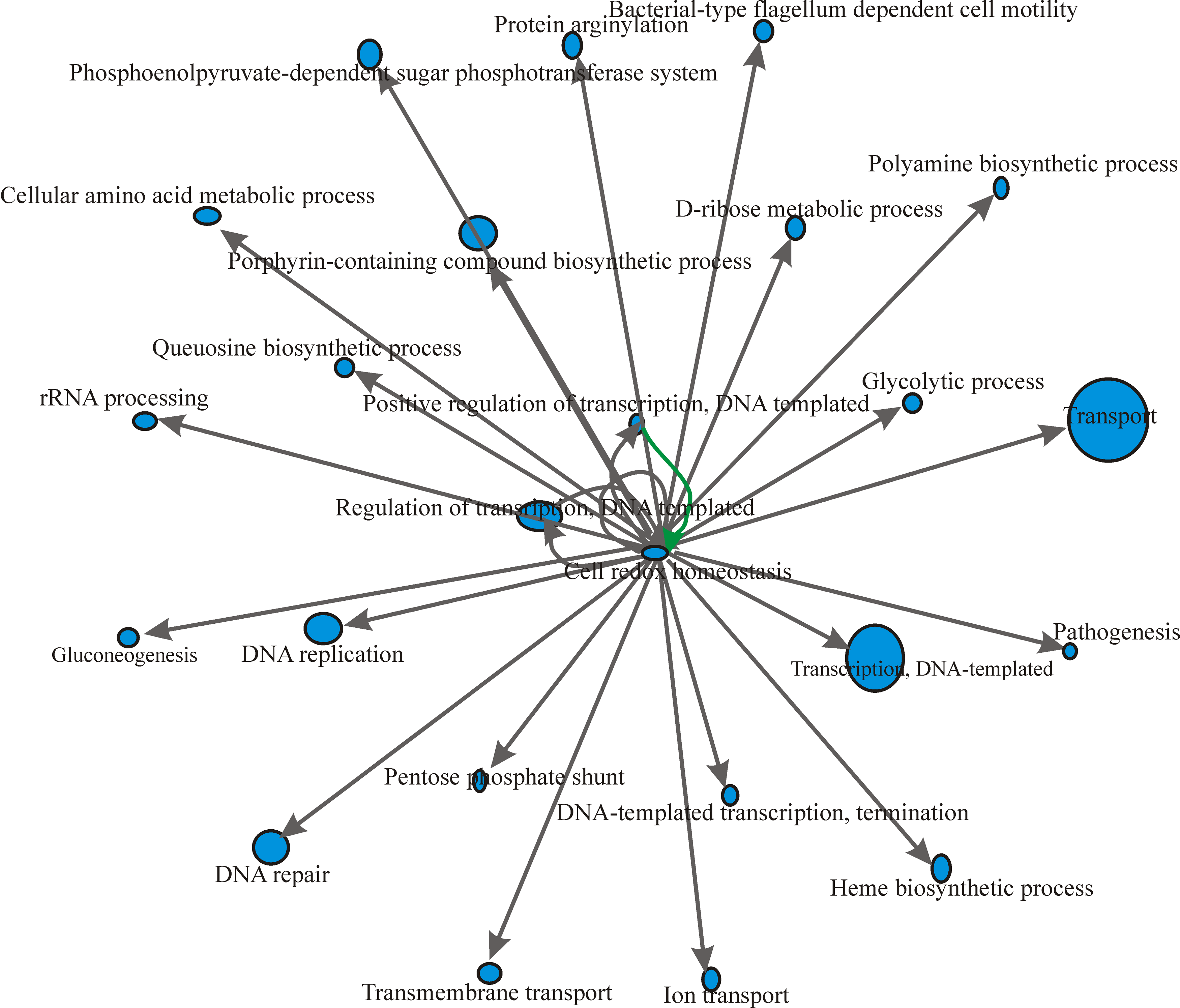
GRN based on the mRNA target genes of B. suis 1330.
Discussion
sRNAs are known modulators of gene expression in prokaryotes (Papenfort et al., 2015; Prevost et al., 2011; Sakurai et al., 2012). However, very few reports are available on the roles of sRNAs in Brucella. Caswell et al. (2012) have reported two sRNAs and their roles in virulence of Brucella. Recently, sRNA responsible for Brucella adaptation to stress conditions and intracellular survival has been reported (Wang et al., 2015). In this study, we have used the computational approach to predict the sRNA candidates in three Brucella species.
Bioinformatic software for sRNA prediction are based on four major principles: (1) comparative genomics, (2) secondary structure and thermodynamic stability, (3) transcriptional signals, and (4) ab initio methods (Sridhar and Gunasekaran, 2013). A common practice to improve the accuracy is to combine several bioinformatic tools for the prediction of sRNAs (Dong et al., 2014; Khoo et al., 2012; Tesorero et al., 2013). In this study, we combined SIPHT and sRNAscanner to improve accuracy. We also made use of the QRNA analysis feature of SIPHT to predict sRNAs.
The sRNAs identified here vary in length and GC content. The variation in GC content is expected to attain different requirements of stability as sRNAs are diverse in both functions and mechanisms of action. Our results show that the primary sequences of sRNAs identified are conserved only within the genus Brucella. We found two sRNAs in B. melitensis16M to have homologs in Rfam. BmsRNA6 and BmsRNA16 were showing homology with BjrC1505 and ctRNA_p42d, respectively. ctRNA (counter-transcribed RNA) is a noncoding RNA encoded by plasmids, which has roles in rolling circle replication. ctRNA binds to repB and causes translational inhibition (Venkova-Canova et al., 2003). BasRNA13 of B. abortus 2308 showed homology with ar15, which is a noncoding sRNA identified in Sinorhizobium meliloti (del val et al., 2007). BssRNA6 and BssRNA 16 of B. suis 1330 showed homology with Atu_C9 and suhB, respectively.
sRNAs regulate the gene expression by binding to the mRNA targets either by perfect or imperfect sequence complementarity. One sRNA might regulate several mRNA targets, which may result in upregulation or downregulation of a set of genes. By regulating the activities of different genes at one time, sRNAs play a fundamental role in organism's biological and cellular functions (Johansen et al., 2008; Lenz et al., 2004; Wang et al., 2015; Wassarman, 2002). In this study, we have identified potential targets of sRNAs, using the bioinformatic approach. The majority of the target genes were enriched in metabolic and transport pathways. KEGG analysis revealed that the target genes may play a significant role in Brucella biological process, replication, and intracellular survival. These sRNAs could regulate several transcriptional factors. GntR family transcriptional regulator is one such transcriptional regulator, which plays a major role in Brucella virulence (Delrue et al., 2004). An sRNA candidate of B. suis was identified to regulate type IV secretion system protein, VirB7, which is required for intracellular survival and pathogenesis of Brucella (Boschiroli et al., 2002; O'Callaghan et al., 1999). In addition, we could see that the identified sRNAs may regulate several hypothetical proteins. Identifying the functions of these hypothetical proteins may give more insights into the complex intracellular survival and pathogenesis of Brucella.
We further constructed the GRN of predicted mRNA targets using the biological process GO terms. Transcription antitermination protein, NusG, constitutes the central node in B. melitensis 16M. It interacts with the termination factor Rho and RNA polymerase and thereby influences transcription termination and antitermination. In B. abortus 2308, the betI gene was the central node in the regulatory network. This gene acts as a transcriptional repressor of bet genes. These genes are required for regulating the osmotic balance of the bacteria and needed to overcome osmotic stress (Lamark et al., 1996). GO network analysis showed that negative regulation of transcription, DNA-templated (GO ID: 45892) governed by the betI gene has influence in the suppression of genes with which it interacts, opens a new avenue for the treatment of brucellosis. In B. suis 1330, dihydrolipoamide dehydrogenase (lpdA-1) was the central node in the network. Dihydrolipoamide dehydrogenase is involved in several pathways, including glycolysis/gluconeogenesis, citrate cycle (TCA cycle), glycine, serine and threonine metabolism, valine, leucine and isoleucine degradation, pyruvate metabolism, glyoxylate and dicarboxylate metabolism, metabolic pathways, biosynthesis of secondary metabolites, microbial metabolism in diverse environments, biosynthesis of antibiotics, and carbon metabolism.
We have used a bioinformatic strategy to predict sRNA candidates in B. melitensis 16M, B. abortus 2308, and B. suis 1330 and predicted 21, 13, and 16 sRNAs, respectively. Most of these sRNAs were predicted for the first time. The functional categorization and pathway analysis of the target genes revealed that sRNAs are involved in various metabolic pathways. GO network analysis in B. melitensis 16M, B. abortus 2308, and B. suis 1330 revealed new biological insights.
Taken together, we would like to reemphasize that although sRNAs are veritable modulators of gene expression in prokaryotes, the reports on the significance of sRNAs in Brucella are quite limited. This work begins to address this literature gap by offering a series of initial observations based on a genome-wide computational biology analysis for future experimental analysis of sRNAs and their targets to explain the most complex multifactorial basis of Brucella pathogenesis and its intracellular survival.
Footnotes
Acknowledgments
This work was supported by the Department of Biotechnology, New Delhi, through the DBT Network Project on Brucellosis. The UGC-CAS, CEGS, NRCBS, DBT-IPLS, DST-PURSE Programs of School of Biological Sciences, Madurai Kamaraj University, are gratefully acknowledged.
Author Disclosure Statement
The authors declare that no competing financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.