
Abstract
Abstract
Metagenomics is a next-generation omics field currently impacting postgenomic life sciences and medicine. Binning metagenomic reads is essential for the understanding of microbial function, compositions, and interactions in given environments. Despite the existence of dozens of computational methods for metagenomic read binning, it is still very challenging to bin reads. This is especially true for reads from unknown species, from species with similar abundance, and/or from low-abundance species in environmental samples. In this study, we developed a novel taxonomy-dependent and alignment-free approach called MBMC (Metagenomic Binning by Markov Chains). Different from all existing methods, MBMC bins reads by measuring the similarity of reads to the trained Markov chains for different taxa instead of directly comparing reads with known genomic sequences. By testing on more than 24 simulated and experimental datasets with species of similar abundance, species of low abundance, and/or unknown species, we report here that MBMC reliably grouped reads from different species into separate bins. Compared with four existing approaches, we demonstrated that the performance of MBMC was comparable with existing approaches when binning reads from sequenced species, and superior to existing approaches when binning reads from unknown species. MBMC is a pivotal tool for binning metagenomic reads in the current era of Big Data and postgenomic integrative biology. The MBMC software can be freely downloaded at http://hulab.ucf.edu/research/projects/metagenomics/MBMC.html.
Introduction
M
Many computational methods have been developed to bin reads in metagenomic projects. According to Mande et al. (2012), these methods can be classified into two categories. One category is the taxonomy-dependent methods (Brady and Salzberg, 2009; Haque et al., 2009; Huson et al., 2007; Krause et al., 2008; Matsen et al., 2010; McHardy et al., 2007; Meyer et al., 2008; Mohammed et al., 2011; Stark et al., 2010; Wood and Salzberg, 2014; Wu and Eisen, 2008), which compare reads with sequences in public databases or models inferred from public databases to group reads and determine which known species are present. The other category is the taxonomy-independent methods (Chatterji et al., 2008; Diaz et al., 2009; Wang et al., 2012, 2015; Wu and Ye, 2011), which employ the difference of GC content (guanine-cytosine content), k-mer (a k base pairs long DNA segment) frequencies, etc., of different microbes in the same samples to bin reads.
Despite the existence of dozens of methods for read binning, it is still challenging to bin reads from metagenomic projects accurately and efficiently. The difficulty stems from the fact that the majority of microbial species are still not sequenced, therefore making read binning from these species problematic by the developed taxonomy-dependent approaches (Albertsen et al., 2013; Mande et al., 2012). The difficulty also commonly arises when the genome coverage of different microbial species is similar (<2 fold difference), which renders it unlikely to separate reads from species with similar abundance by taxonomy-independent methods such as AbundanceBin (Wu and Ye, 2011) and MetaCluster (Wang et al., 2012). In this study, the genome coverage (or sequence depth) of a genome in a dataset under consideration is defined as the sum of the length of all reads from this genome in this dataset divided by the length of the genome. As a result of these limitations, more powerful read binning methods are required.
In this study, we developed a novel taxonomy-dependent and alignment-free approach called MBMC (Metagenomic Binning by Markov Chains). Different from all existing methods, MBMC bins reads by measuring the similarity of reads to the trained ninth-order Markov chains for different taxa instead of directly comparing reads with known genomic sequences. We showed that MBMC reliably determined the species number and binned reads with an average accuracy of more than 91% when tested on 10 simulated datasets with similar species abundance. Additionally, we demonstrated that MBMC reliably binned reads for both known and unknown species when tested on 10 additional simulated datasets and 6 experimental datasets. Compared with existing approaches, MBMC had comparable or better performance than existing approaches on reads from known species and superior performance when dealing with reads from unknown species.
Materials and Methods
Known species used and their Markov chain representation
We downloaded all 2773 completely sequenced microbial genomes from ftp://ftp.ncbi.nlm.nih.gov/genomes/Bacteria/. We also downloaded their taxonomical information from GOLD (https://gold.jgi-psf.org/) (Reddy et al., 2015), including the species, genus, family, order, class, and phylum IDs of these species. Only 2592 genomes had all six-level taxonomical information and were used in the training of MBMC. In total, we had 32 phyla, 56 classes, 110 orders, 239 families, 608 genera, and 1330 different species (multiple genomes may be from the same species).
We represented each genome by a Markov chain. A Markov chain describes a random process that transits from one state to another state in a state space. In this study, we applied Markov chains to model how an N-mer (an N nucleotide long DNA segment) starting from a position in a microbial genome transited to the N-mer starting from the next position in the same genome, where N is called the order of the Markov chain and all 4 N N-mers are the different states of this Markov chain. To determine the proper order of the Markov chain, we tried N from 5 to 10 (Results section). We did not consider N larger than 10 or smaller than 5 because Markov chains with larger orders had too many parameters to be modeled effectively and Markov chains with smaller order did not have enough parameters to distinguish different genomes. We finally chose the ninth-order Markov chains because they were much better than the Markov chains with smaller orders and relatively comparable with the 10th-order Markov chains, which required much smaller number of parameters (Results section).
For a microbial genome and a given order, say N, we estimated two important sets of parameters in a Markov chain, the stationary probabilities and the transition matrix, as follows. We counted the N-mer and (N + 1)-mer frequencies on both the positive and negative strands of the genome under consideration. We then added a pseudocount, 0.0001, to the count of each N-mer frequency and the count of each (N + 1)-mer frequency to avoid any frequency to be zero. We then normalized these obtained frequencies to calculate the stationary probabilities and the transition probability matrix according to their definitions. For a taxon with multiple sequenced genomes, such as a phylum, we obtained its stationary and transition probabilities by averaging the corresponding stationary and transition probabilities of the sequenced genomes that belong to this taxon.
Simulated and experimental datasets
To compare the different order of Markov chains, we generated 10 random datasets (Supplementary Table S1). In each dataset, paired-end reads were simulated with MetaSim (Richter et al., 2008) to cover the genomes of three species from the same genus three times. To determine whether the eighth-order or ninth-order Markov chains should be used in MBMC, we generated 25 groups of species, each of which contained three species. We then generated 50 simulated datasets using the 25 groups of species, with the genome coverage as either 3 or 0.5 (Supplementary Table S2). As mentioned above, the genome coverage measures the sequencing depth of a genome.
To compare MBMC with existing methods, we simulated 10 more complicated datasets. At each taxonomical level (phylum, class, order, family, or genus), two datasets were generated. The first one contained five species from the same taxon with the corresponding genome coverage as 1, 1, 1, 2, and 2. The second one contained six species from the same taxon with the corresponding genome coverage as 2, 3, 4, 5, 6, and 7 (Supplementary Table S3). Species from the same taxonomical level with similar coverage in these datasets made it challenging to bin reads by taxonomy-independent methods (Wang et al., 2015; Wu and Ye, 2011). For each species, Illumina paired-end reads were simulated using MetaSim (Richter et al., 2008) with the read length as 75 base pairs and with the empirical error model in MetaSim. The 75 base pair long reads were used because many benchmark datasets contained reads of such a length, and it was much more challenging to bin shorter reads than to bin longer reads (Wang et al., 2012; Wu and Ye, 2011). We also simulated datasets with longer reads, and in general, MBMC worked even better on these datasets.
To study whether MBMC and other methods could work on datasets comprising reads from unknown species, we generated 10 additional simulated datasets (Supplementary Table S4). Each dataset consisted of reads from three species with the coverage for each genome as 3 or 0.5. The three species were randomly selected from the 181 of the 2773 completed sequenced microbial genomes that did not have complete taxonomical information and were not used to train MBMC.
For experimental datasets, we randomly selected two datasets from the Human Microbiome Project (HMP, http://hmpdacc.org/HMSCP/), SRS017080 and SRS013705. These two datasets contained mapped paired-end reads. We selected four species for the dataset SRS017080, only one of which was known to MBMC and the other three were unknown species. However, these unknown species in the dataset SRS017080 had known sequenced genomes from the same species, but different strains. For the dataset SRS013705, we selected five species, one of which was known and the remaining four were unknown. One of these four species had a known genome from a different strain (Supplementary Table S5).
Moreover, we generated an HMP mock dataset that contained single-end reads from SRR172902 and SRR172903. This mock dataset consisted of reads from 22 microbial genomes. All reads were mapped to the 22 genomes by SOAPdenovo2 (Luo et al., 2012) with default parameters. To be consistent with our species abundance cutoff in the default version of MBMC, 5%, we selected all top abundant species that contained more than 5% of total reads in this HMP mock dataset. There were six such species. Reads from these six species accounted for 71.62% of total mapped reads.
In addition, we used a human gut dataset from 15 randomly selected samples from ftp://public.genomics.org.cn/BGI/gutmeta/High_quality_reads/ (Qin et al., 2010). There were 257,158,754 paired-end reads in this dataset, each of which was 75 base pairs long. These reads were mapped to the following three species using the software SOAPdenovo2 (Luo et al., 2012), Bacteroides uniformis, Alistipes putredinis, and Ruminococcus bromii L2-63, because they were the most abundant species and/or had more complete genome sequences in the gut datasets (Qin et al., 2010; Wang et al., 2015). In total, 4,684,098 reads were mapped to the three genomes and used to test MBMC and other methods.
Besides this, we generated two additional datasets, HiSeq dataset comprised totally 9,000,000 HiSeq Illumina reads from nine species, one of them is unknown, and MiSeq dataset contained 5,000,000 MiSeq Illumina reads from five species, one of them is unknown. For each genome, we selected 1,000,000 reads. Reads from known species were obtained from GAGE-B project (Magoc et al., 2013) and reads from two unknown species were from the NCBI Sequence Read Archive (SRA run SRR515982 and SRR493654).
MBMC: a novel taxonomy-dependent approach to bin metagenomic reads
We developed MBMC, a novel taxonomy-dependent approach, to bin metagenomic reads. For a given dataset, MBMC determines and selects potential taxa with the ordinary least squares (OLS) method, and then assigns reads to the selected taxa based on the similarity score of the reads to the Markov chains corresponding to the selected taxa. The details are in the following.
To determine potential taxa, MBMC models all input reads by a mixture of ninth-order Markov chains. That is, the frequency of 9-mers and 10-mers in all input reads and their reverse complement reads is counted to calculate the stationary and the transition probabilities of the Markov chain mixture. Correspondingly, MBMC models each taxon at every taxonomical level by a ninth-order Markov chain. MBMC then applies the following OLS to identify potential taxa:
where,
In the above formula, n =
MBMC only keeps taxa whose relative abundance β is larger than a prespecified cutoff, say 0.01 or 0.05. Since there are much more taxa at lower levels such as at the species level than those at higher levels such as at the phylum level, to speed up the prediction process, MBMC iteratively chooses potential taxa from high levels to low levels (Figure 1).

Flowchart of MBMC. The cutoff to select potential taxa was defined as 0.01 at the phylum to genus levels and 0.05 at the species level.
In brief, MBMC first infers potential phyla. There are 32 known phyla in total and X is thus initially a 410 by 32 matrix. By the above OLS, MBMC keeps all potential phyla whose corresponding relative abundance
For instance, assume
Next, MBMC infers the potential classes. Note that although there are 56 classes in our training datasets, X in the above OLS only considers classes that belong to the selected potential phyla and thus the known p classes in the Equation 1 may be much smaller than 56. Next, MBMC similarly predicts potential orders, families, and genera, respectively. Finally, MBMC similarly predicts potential species by requiring
With the
in which,
Comparisons with other methods
We compared MBMC with two taxonomy-independent methods, AbundanceBin (Wu and Ye, 2011) and MetaCluster 5.1 (Wang et al., 2012). We ran MBMC without the information of the number of species present. We ran AbundanceBin and MetaCluster with the actual species number m as input since they hardly predicted the correct species number. We ran AbundanceBin using the command “./abundancebin–input reads_file–output output_file -bin_num m.” We ran MetaCluster by using the command “./MetaCluster5_2 reads_file—Species m”. Because the genome coverage was low for all simulated species, we only ran the second command of MetaCluster 5.1, which was used to process low-abundance species.
We also compared MBMC with two taxonomy-dependent methods, MEGAN5 (Huson et al., 2007) and Kraken (Wood and Salzberg, 2014). It must be noted that we used the same 2592 genomes to train MEGAN5 and Kraken. MEGAN5 is an alignment-based method. We ran MEGAN5, by first mapping reads to the 2592 reference genomes using BLASTN with default parameters, and then ran it by inputting mapped reads to its GUI mode program. Kraken is a composition-based method. We first generated a reference database using the 2592 genomes. Because of the limited memory-computing environment, we used the same command as they generated their MiniKraken database. To increase Kraken's accuracy, we concatenated paired-end reads together with a single N between the sequences, as suggested by Kraken. With the built reference database and concatenated reads, we ran Kraken by the command “./kraken—preload—db my_own_db reads_file >>output_file.”
Results
The ninth Markov chain models are effective in representing microbial genomes
We represented each completed microbial genome by an N-th-order Markov chain, where N was from 5 to 10. Intuitively, the higher order a Markov chain has, the better it represents a microbial genome. However, a Markov chain with a higher order has at least four times more parameters, which demands higher memory storage and more computation time. Therefore, it is necessary to investigate which order gives more accurate read assignment with reasonable memory usage.
To see which order of Markov chains was better, we studied read assignment in 10 random datasets (Supplementary Table S1). For a given order, a read was assigned to one of the three Markov chains for which this read had the largest similarity score. The read assignment accuracy in a dataset was defined as the number of correctly assigned reads divided by the total number of reads from the three species. Figure 2 showed the read assignment accuracy in each dataset when using different orders of Markov chains. From the 5th to 10th-order Markov chains, the average accuracy increased. The largest increment, 19.05%, happened when the order was changed from seven to eight. The second largest increment, 10.57%, occurred from order eight to order nine. The increment from the 9th order to the 10th order was 2.78%, which together with the above observations suggested that the eighth- or ninth-order Markov chains likely are the most effective ones to represent a microbial genome.
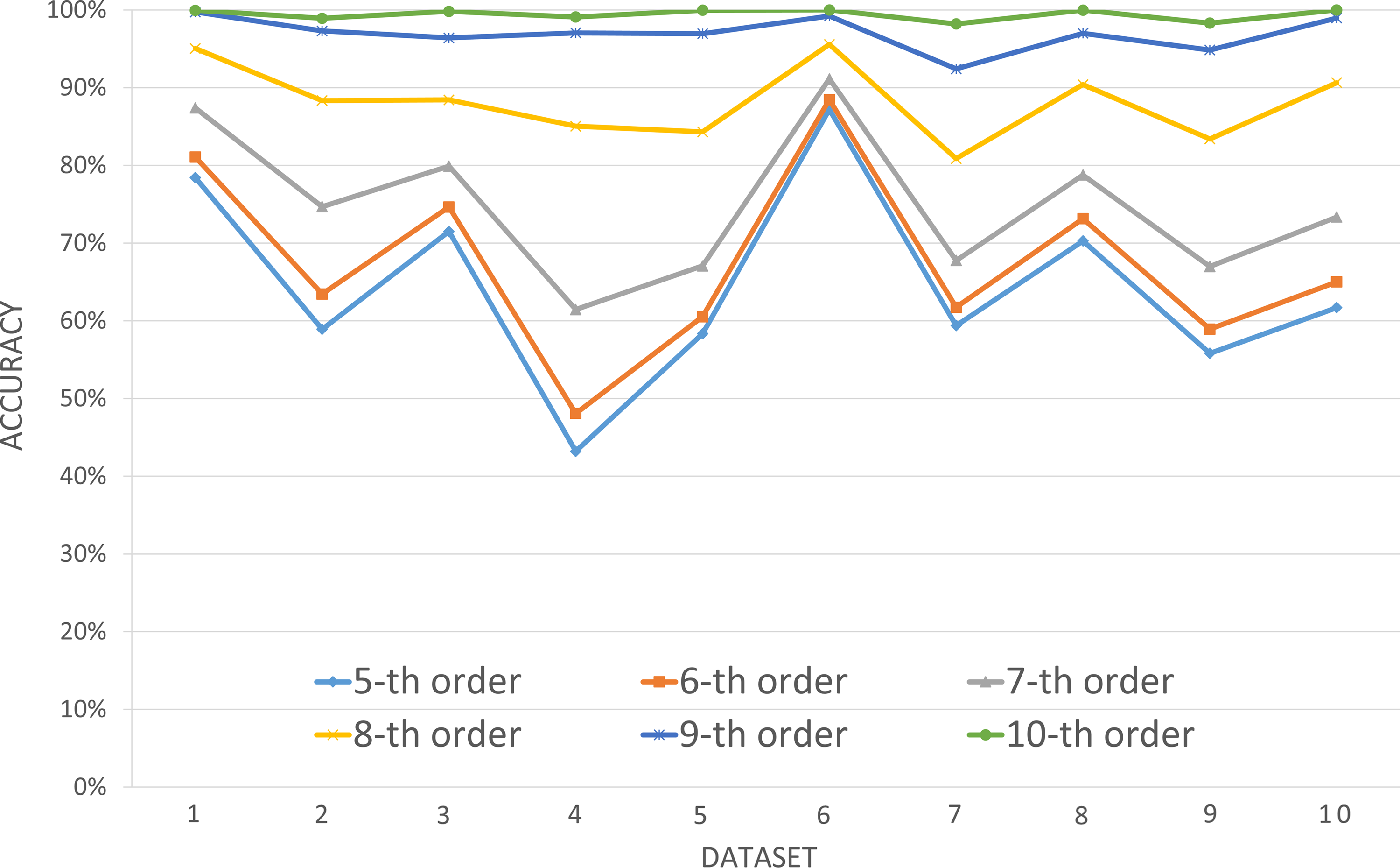
Comparisons of read assignment accuracy using different orders of Markov Chains.
To compare the representation of the eighth Markov chains and the ninth-order Markov chains, we tested them on 50 simulated datasets (Supplementary Table S2). In this study, although reads in each dataset were still from three randomly chosen species, every read was scored with m instead of three Markov chains to calculate the similarity scores of the read to the Markov chains and to determine the assigned species (m was the number of potential species predicted by MBMC). The assignment accuracy was correspondingly defined as the percentage of reads that were correctly assigned to the correct species.
From the comparisons, it was evident that the accuracy was improved by 2.9% for the ninth-order Markov chains (Supplementary Table S6). For one of the datasets, dataset 3_5, because of the inaccurate representation of the genome sequences by the eighth-order Markov chains, the obtained potential species even did not include the real ones. The assignment accuracy was only 71.40% for the eighth-order Markov chain on this dataset, while the ninth-order Markov chains achieved more than 89% accuracy on every dataset. We therefore chose the ninth-order Markov chain representation in MBMC in the following analyses.
It was worth mentioning that the ninth-order Markov chains representing species from the same taxa were in general more similar to each other than those representing species from different taxa. For instance, the ninth-order Markov chains representing species from the same genus, Spiroplasma, had an average distance of 212147, while the ninth-order Markov chains representing species from Spiroplasma and from any other genus (such as Genus Aquifex) had an average much larger distance (such as 246549). The distance of two Markov chains was calculated as the sum of the absolute difference of the corresponding entries in the two transition matrices. The more different the taxonomical information of two species was, in general, the larger distance the two corresponding Markov chains had. This implied that the Markov chain representation of an unsequenced species may be approximated by the Markov chain representation of a sequenced species that had similar high-level taxa as this unsequenced species. In other words, one may group reads from unsequenced species by using the Markov representation of sequenced species.
MBMC reliably predicted the species number and accurately grouped reads in simulated datasets
With the ninth-order Markov chain representation of microbial genomes, we developed a novel taxonomy-dependent binning method called MBMC (Materials and Methods section). To show whether MBMC reliably binned reads on more complicated datasets, we tested MBMC on 10 simulated datasets (Supplementary Table S3).
MBMC performed well on these datasets (Table 1). In all 8 of 10 datasets, MBMC correctly predicted the actual species number. The binning accuracy in a dataset, which was defined as the percentage of correctly binned reads divided by the total number of reads, varied from 69.39% to 99.04%, with an average accuracy as 91.71%. It was also evident that the binning accuracy was relatively higher at high taxonomical levels and lower at low taxonomical levels. For instance, the average binning accuracy at the phylum level was 96.61%, while it was 81.38% at the genus level (Table 1).
At each taxonomical level (phylum, class, order, family, and genus), two datasets were generated. The first one contained five species from the same taxon with the corresponding genome coverage as 1, 1, 1, 2, and 2, respectively. The second one contained six species from the same taxon with the corresponding genome coverage as 2, 3, 4, 5, 6, and 7, respectively.
We noticed that for the last dataset, the dataset 5_2, although MBMC correctly predicted the species number, it had a low accuracy. This was caused by the similarity among the six Markov chains corresponding to these species. For instance, the smallest distance between two Markov chains in the dataset 5_2 was 102014, which was much smaller than that in all other datasets (111182 in the dataset 3_2). Recall that the distance of two Markov chains was calculated as the sum of the absolute difference of the corresponding entries in the two transition matrices. It was worth pointing out that the average distance of two ninth-order Markov chains for two species from the same genus was 160615, which was much larger than the smallest distance of Markov chains in the dataset 5_2.
We also applied four existing methods to the same 10 simulated datasets (Materials and Methods section). The two taxonomy-independent methods, AbundanceBin and MetaCluster, could not predict the correct species number in any dataset. Therefore, we specified the actual species number in each dataset when we ran them. On average, the binning accuracy of AbundanceBin and MetaCluster was 31.49% and 61.32%, respectively (Table 1). We observed that at every taxonomical level, the accuracy of MetaCluster was much higher in the second dataset than in the first one. This may be because the coverage in the first dataset was smaller than in the second dataset and MetaCluster could not work well with species of low coverage. This also implied that MBMC worked well for datasets with low-abundance species.
The binning accuracy of the two taxonomy-dependent methods, MEGAN5 (Huson et al., 2007) and Kraken (Wood and Salzberg, 2014), was high because the species in these datasets were known to the two methods (Table 1). In addition, as reads were usually mapped to many different species by the two taxonomy-dependent methods, when we calculated their binning accuracy, we considered only groups containing more than 5% of total reads and assumed groups with most reads from a species as the correctly predicted groups for that species. Even with such a treatment, both MEGAN5 and Kraken incorrectly predicted the species number in three datasets. Overall, MBMC had slightly lower average accuracy than MEGAN5 and Kraken at lower taxonomical levels (order, family, genus), but higher accuracy in higher taxonomical levels (phylum, class), suggesting that the Markov chains for species from different higher level taxa were more different than those from lower level taxa.
MBMC worked well on datasets with unknown species
In contrast to the existing taxonomy-dependent approaches, MBMC compares reads with the Markov representation of the sequenced microbial genomes instead of the microbial genome sequences themselves. We hypothesize that this unique aspect may enable MBMC to reliably bin reads from unknown species. The rationale was that an unknown species may be from the same phylum, class, order, family, or genus as certain sequenced species, whose Markov representation may thus aid the accurate binning of reads from this unknown species.
To test this hypothesis, we applied MBMC to 10 additional datasets (Supplementary Table S4). Each dataset comprised reads from three randomly selected unknown species that were not used to train MBMC. The genome coverage was set as 3 or 0.5 for each species in each dataset. The binning accuracy of MBMC together with other four existing methods is summarized in Table 2.
Each dataset contained three unknown species with the corresponding genome coverage as 3, 3, 3, or 0.5, 0.5, and 0.5 (datasets marked with*). Here m represents the number of the predicted species, and na means no meaningful output.
MBMC had an average accuracy of 70.30% on these datasets, which was at least 10.13% higher than any of the four methods. Among the four methods, the two taxonomy-independent methods had much higher accuracy than the two taxonomy-dependent methods. We again input the actual species number for the two taxonomy-independent methods to improve their accuracy. As for the two taxonomy-dependent methods, they were barely able to bin any read in any dataset (Table 2) because reads from unknown species unlikely satisfied the required high similarity to the genome sequences used to train the two methods. For instance, Kraken did not group more than 5% of the total reads to any bin, although three such groups of reads existed in each of the 10 datasets. It was also evident that although MetaCluster performed better than AbundanceBin on datasets with high coverage (coverage ≥3), it could not work on any dataset with coverage as 0.5.
We also noticed that MBMC did not accurately predict the actual species number in these datasets (Table 2). This was because multiple genome sequences used to train MBMC may be similar to the genome sequences of an unknown species. Reads from the unknown species may thus be divided into several groups, each of which may be more similar to one of the known genomes and predicted as a species.
We scrutinized our prediction in these 10 datasets and confirmed that this was the case. For instance, in the ninth dataset, five predicted species, Bacillus amyloliquefaciens, Bacillus licheniformis, Geobacillus_Y412MC61, Solibacillus silvestris, and Paenibacillus Y412MC10, shared the same order with the actual unknown species Exiguobacterium sibiricum. These five predicted species should be combined into one predicted species as 70.91% of reads from them were from the actual species E. sibiricum, which was not used to train MBMC. In the same dataset, another four predicted species, Sinorhizobium fredii, Agrobacterium H13 3, Agrobacterium fabrum, and Rhizobium leguminosarum, shared the same family as the actual unknown species Rhizobium sp. IRBG74. Again, these four predicted species should be combined into one predicted species as 92.13% of reads from them were from the actual species Rhizobium sp. IRBG74.
A valid question was thus how to tell whether there existed unknown species in a dataset. We observed that the predicted abundance of each bin was relatively small in these datasets. For instance, in the fifth dataset, at the species level, the largest relative abundance was only 0.041. Moreover, as we mentioned above, we also observed that several genome sequences corresponding to these small bins were quite similar to each other. In practice, one may take the above two observations into account when interpreting the predictions by MBMC.
MBMC was superior to other methods on experimental datasets
To assess the practical application of MBMC, we studied the performance of MBMC on six experimental datasets: two HMP real datasets SRS017080 and SRS013705, one HMP mock dataset, one human gut dataset, and two additional datasets, which comprise HiSeq and MiSeq Illumina reads, respectively (denoted as HiSeq and MiSeq). Two HMP real datasets, one human gut dataset, and HiSeq/MiSeq datasets were used because such datasets were widely used in assessing other metagenomic tools (Alneberg et al., 2014; Kultima et al., 2012; Namiki et al., 2012; Wang et al., 2012; Wood and Salzberg, 2014). The HMP mock dataset was used because it was different from other three datasets and contained only single-end read. In each dataset, at least one species was known (Supplementary Table S5).
The read binning accuracy of MBMC together with that of other four existing methods is shown in Table 3. Overall, MBMC had a higher binning accuracy than other methods in five of the six datasets, where there existed unknown species (Table 3). In the three datasets with unknown species, MBMC in general binned reads into more groups, with several groups corresponding to the same unknown species. MEGAN5 and Kraken had relatively poor predictions when unknown species existed in the datasets as many reads from the unknown species were not mapped to the reference genomes. For instance, in the dataset SRS013705, MBMC had more than 28.78% higher accuracy than MEGAN5 and Kraken. It was worth pointing out that MBMC had a similar accuracy as MEGAN5 and Kraken in the dataset SRS017080 because the three unknown species in this dataset had sequenced strains and MEGAN5 and Kraken were able to directly map a large number of reads to these sequenced known strains. However, even in this dataset, MBMC had a more than 8.07% higher accuracy than MEGAN5 and Kraken, supporting the advantage of comparing reads with the Markov representation of the genome sequences instead of the actual genome sequences themselves.
The parentheses in each dataset ID include the number of known species, followed by the number of species in the dataset. Here m is the number of the predicted species in a dataset.
HMP, Human Microbiome Project.
Besides this, in the HiSeq and MiSeq datasets, MBMC was shown to predict the real species more accurately than MEGAN5 and Kraken. For example, MBMC can correctly predict 6 of 8 and 3 of 4 known species in the HiSeq and MiSeq datasets, respectively, while MEGAN5 and Kraken predicted less accurately for known species. As for the remaining dataset with only known species, the HMP mock dataset, MBMC correctly predicted five of the six species. MetaCluster and AbundanceBin did not work out on this mock dataset due to the low genome coverage and low abundance ratio of species.
Table 3 again demonstrates the strength of MBMC from two aspects. One aspect is that although MBMC is a taxonomy-dependent method, it works well to bin reads from unknown species. For instance, in the human gut dataset, which had two unknown species, MBMC achieved at least 11.39% higher accuracy than other methods. Second, MBMC works well when species abundance is low or the relative abundance among species is low. In both cases, the existing taxonomy-independent methods have difficulties in binning reads, as illustrated in the HMP mock dataset.
Discussion
We developed a novel, taxonomy-dependent alignment-free approach named MBMC to bin metagenomic reads. In contrast to existing taxonomy-dependent approaches, MBMC bins reads by measuring how well reads are modeled by the learned Markov chains from the sequenced microbial genomes instead of comparing each read with each sequenced microbial genome directly. This unique characteristic enables MBMC to reliably group reads from both known and unknown species. We tested MBMC on 10 simulated datasets with only known species and showed that MBMC had more than 91% accuracy on average. We further tested MBMC on 10 additional simulated datasets containing unknown species and showed that MBMC had more than 70% accuracy on average, which was superior to current conventional approaches. We also tested MBMC on six experimental datasets and demonstrated that MBMC outperformed the available methods, especially when there were unknown species. In summary, the major advantages and difference of MBMC compared with other methods are as follows:
• MBMC assigns reads by comparing reads with learned Markov models instead of direct sequence–sequence comparison; • MBMC is comparable with the state-of-the-art methods when tested on datasets that contain only known species; • MBMC is superior to existing methods when tested on datasets that contain unknown species; • MBMC performed better than existing methods when tested on experimental datasets.
We used only 2592 completed microbial genomes to train MBMC. We could also include draft microbial genomes for the training of MBMC. In the future, with more microbes sequenced, their sequences could be used to train MBMC as well. A critical question remaining is whether the training with more genomes will make the binning accuracy of MBMC decrease instead of increasing. It is possible that the accuracy will decrease because more Markov chains from these genomes may be more similar to each other. To address this question, we randomly selected 1000 draft microbial genome sequences from ftp://ftp.ncbi.nlm.nih.gov/genomes/Bacteria/), 955 of which with all six level taxonomical information were combined with the default 2592 genomes to train MBMC. We then applied the trained MBMC to the simulated datasets mentioned above (Supplementary Table S2). We found that the accuracy was almost the same with the accuracy when using 2592 genomes in MBMC (Supplementary Table S6).
As mentioned in Kraken (Wood and Salzberg, 2014), there exist contaminant and adapter sequences in certain draft genomes (Wood and Salzberg, 2014). In this study, training with draft genomes did not affect the accuracy of MBMC much. This suggests that MBMC is robust for contaminant sequences. It also implies that MBMC may become even better with more training genome sequences.
We implicitly employed the tree structure of taxa in MBMC. Alternatively, we could explicitly utilize the taxonomical trees to assign reads. For instance, we could build taxonomical trees at each taxonomical level, and assign reads to each tree, and then combine trees at all levels to make final decisions about which species a read belongs to. Alternatively, one could use only the species tree to assign reads directly. We tested these two alternative approaches and found neither was as time-efficient as the top-down strategy used in MBMC.
MBMC performed better in binning reads from unknown species than existing approaches. We observed that MBMC tended to divide reads from an unknown species into multiple small bins. How to combine these small bins together to reconstruct a unique bin for an unknown species seems challenging and worth further investigation. In addition, although MBMC showed superior performance than existing approaches on simulated and real experimental datasets with unknown species, in practice, the low abundance of many unknown species in metagenomic projects is still challenging for existing methods, including MBMC, to figure out how to bin reads. It is thus important to be able to deal with species with even lower abundance than the current MBMC can deal with. We are working on these directions together with others to further improve MBMC.
Conclusions
We demonstrated that MBMC binned reads from unknown species much better than existing approaches and had similar or better performance when binning reads from known species. It thus will be a useful method and tool for future metagenomic studies.
Footnotes
Acknowledgments
This work is supported by the National Science Foundation [grants 1356524, 1149955, and 1218275] and National Institutes of Health [grant 2R01HL048044-22]. Funding for open access charge was received from The National Science Foundation grant 1218275.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.