
Abstract
Abstract
The annual economic burden of visual disorders in the United States was estimated at $139 billion. Ophthalmology is therefore one of the salient application fields of postgenomics biotechnologies such as proteomics in the pursuit of global precision medicine. Interestingly, the protein composition of the human iris tissue still remains largely unexplored. In this context, the uveal tract constitutes the vascular middle coat of the eye and is formed by the choroid, ciliary body, and iris. The iris forms the anterior most part of the uvea. It is a thin muscular diaphragm with a central perforation called pupil. Inflammation of the uvea is termed uveitis and causes reduced vision or blindness. However, the pathogenesis of the spectrum of diseases causing uveitis is still not very well understood. We investigated the proteome of the iris tissue harvested from healthy donor eyes that were enucleated within 6 h of death using high-resolution Fourier transform mass spectrometry. A total of 4959 nonredundant proteins were identified in the human iris, which included proteins involved in signaling, cell communication, metabolism, immune response, and transport. This study is the first attempt to comprehensively profile the global proteome of the human iris tissue and, thus, offers the potential to facilitate biomedical research into pathological diseases of the uvea such as Behcet's disease, Vogt Koyonagi Harada's disease, and juvenile rheumatoid arthritis. Finally, we make a call to the broader visual health and ophthalmology community that proteomics offers a veritable prospect to obtain a systems scale, functional, and dynamic picture of the eye tissue in health and disease. This knowledge is ultimately pertinent for precision medicine diagnostics and therapeutics innovation to address the pressing needs of the 21st century visual health.
Introduction
T
Mapping the potential tissue-based applications of proteomics in visual health requires a basic understanding of the anatomy of the eye tissue. In this context, the iris is a muscular diaphragm located behind the cornea and in front of the crystalline lens. The space between the cornea and the iris is called the anterior chamber and it is filled with aqueous humor. Anatomically, the iris is composed of two layers of tissues with dissimilar embryological origin. The iris stroma is the anterior layer, which is derived from the mesoderm and consists of a loose collagenous tissue (Rennie, 2012). The muscles of the iris, the sphincter iridis and dilator pupillae, are derived from the neural ectoderm, which forms the other layer.
During embryonic development, by the 4th month of gestation, the rim of the optic cup differentiates into a circular band of muscle, the sphincter iridis. The dilator pupillae arises from the outer layer of the primitive optic cup. The muscles of the iris are innervated by the autonomic nervous system. The sphincter iridis is supplied by the parasympathetic nervous system and its contraction leads to constriction of the pupil. The dilator pupillae receives sympathetic innervations and its contraction leads to pupillary dilatation. These two muscles together control the size of the pupil and thus titrate the amount of light entering the eye (Flom, 1987).
Several attempts have been made to characterize the molecular profiles of various ocular tissues in the recent times using RNA-Seq and proteomic approaches with an aim to understand pathological conditions at the molecular level. Proteomic profiling of ciliary body, vitreous humor, aqueous humor, tears, retina, drusen, and choroid—Bruch's membrane complex (Crabb, 2014; Goel et al., 2013; Keenan et al., 2012; Murthy et al., 2014, 2015), has been recently carried out. Interestingly, mass spectrometry (MS)-based proteomics technologies have not been used yet to identify the proteins expressed in the iris tissue.
The availability of proteome profile of the iris tissue will provide a platform for future research on molecular aspects of uveitic diseases and provide new insights into the factors involved in their pathology. We subjected the iris tissue for proteomic analysis using a nanoflow liquid chromatography coupled with a high-resolution tandem mass spectrometer (nano-LC-ESI-MS/MS) to identify the proteins expressed in human iris tissue. The data arising from this study serve as a baseline to compare the changes in protein expression patterns occurring in other sight-threatening pathological diseases of the uvea such as sarcoidosis, Behcet's disease, Vogt Koyonagi Harada's disease, and juvenile rheumatoid arthritis.
Materials and Methods
Sample preparation and fractionation
The iris tissues for the experiments were acquired from healthy donor eyes after obtaining approval from the institutional review board of Vittala International Institute of Ophthalmology (Bangalore, India). The study had institutional ethic board approval and adhered to the tenets of declaration of Helsinki. The iris tissue was dissected from three healthy donor eyes that were enucleated within 6 h of death. The specimen was frozen and stored at −80°C until further analysis. The details of the tissues used have been provided in the Supplementary Table S1.
The iris tissues were lysed in lysis buffer containing 4% sodium dodecyl sulfate (SDS), 100 mM dithiothreitol (DTT), and 100 mM Tris pH 7.5. The tissue lysates were then homogenized and sonicated in lysis buffer followed by heating for 10–15 min at 75°C. The lysates were cooled to room temperature and centrifuged for 10 min at 16,000 rpm, and buffer exchange was carried out. The concentration of proteins in the lysate was estimated using bicinchoninic acid assay, and equal amounts of protein from each of the specimens were pooled for further fractionation. In-solution trypsin digestion was carried out using Filter Aided Sample Preparation (FASP) method (Wisniewski et al., 2009). Briefly, 450 μg proteins from the pooled samples were buffer exchanged with 8 M urea to reduce the SDS concentration using 30 kDa MWCO spin filters (EMD Millipore, Billerica, MA, USA), alkylated with 10 mM iodoacetamide, and digested using sequencing grade modified trypsin (Cat. No. V5111; Promega, Madison, WI. USA) overnight at 37°C. The peptide digest was desalted using Sep-Pak C18 columns (Waters Corporation, Milford, MA, USA), and the cleaned digest was lyophilized.
The lyophilized samples were reconstituted in basic reverse phase liquid chromatography (bRPLC) solvent A (10 mM triethyl ammonium bicarbonate [TEABC] in water at pH 8.5), loaded on XBridge C18, 5 μm 250 × 4.6 mm column (Waters, Milford, MA, USA) connected to Agilent 1100 series high-performance liquid chromatography (HPLC) system. The peptide digest was resolved using a gradient of 0 − 100% solvent B (10 mM TEABC in Acetonitrile, pH 8.5) in 50 minutes. The total fractionation time was 60 min. Ninety-six fractions were collected and concatenated to 24 fractions, which were then vacuum dried and stored at −80°C until further liquid chromatography–mass spectrometry analysis.
Liquid chromatography–tandem mass spectrometry analysis
The peptide digest from bRPLC fractionation was analyzed on LTQ-OrbitrapVelos mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) interfaced with Easy-nLC II nanoflow liquid chromatography system (Thermo Scientific, Odense, Southern Denmark). The peptide digests from each fraction were reconstituted in Solvent A (0.1% Formic acid) and loaded onto trap column (75 μm × 2 cm) packed in-house with Magic C18 AQ (Michrom Bioresources, Inc., Auburn, CA, USA) (5 μm particle size, pore size 100 Å) at a flow rate of 5 μL/min with solvent A (0.1% formic acid in water). The peptides were resolved on an analytical column (75 μm × 15 cm) using a linear gradient of 7–30% solvent B (0.1% formic acid in 95% acetonitrile) over 60 min at a flow rate of 300 nL/min. Data-dependent MS acquisition was carried out with full scans (350–1800 m/z) acquired using an Orbitrap mass analyzer at a mass resolution of 60,000 at 400 m/z.
For MS/MS, top eight intense precursor ions from a survey scan were selected from each duty cycle and subjected to higher-energy collision dissociation with 35% normalized collision energy. The fragment ions were detected at a mass resolution of 15,000 at m/z of 400. Dynamic exclusion was set for 30 sec. Lock mass from ambient air (m/z 445.1200025) was enabled for internal calibration as described previously (Murthy et al., 2014).
Data analysis
MS-derived data were searched against Human RefSeq 59 database (consisting of 33,947 entries along with common contaminants) through Proteome Discoverer 1.4 (Thermo Scientific, Bremen, Germany) using Sequest and Mascot (version 2.2) search algorithms. The parameters used for data analysis included trypsin as protease with maximum two missed cleavages allowed. Cysteine carbamidomethylation was specified as fixed modification, and acetylation of protein N-termini and oxidation of methionine were included as variable modifications. Six amino acids were specified as minimum peptide length. The precursor mass tolerance was set to 10 ppm, whereas for fragment ions it was set to 0.05 Da. The data were searched against decoy database, and the false discovery rate was set to 1% at the peptide level.
Bioinformatics analysis
Proteins identified in our current analysis were categorized based on the biological process, molecular function, and subcellular localization by comparing the list of identified proteins against the manually curated Human Protein Reference Database (HPRD; http://hprd.org) and Human Proteinpedia (Goel et al., 2012; Prasad et al., 2009) using in-house scripts. Data from various sources, including neXtProt (Gaudet et al., 2013), Human Proteome Map (Kim et al., 2014), and proteome data sets of ciliary body, aqueous humor, and vitreous humor (Goel et al., 2013; Murthy et al., 2014, 2015), were also used to compare with the data obtained from this study.
Data availability
Raw MS files used in this experiment have been uploaded to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) through the PRIDE partner repository with the data set identifier PXD001299.
Results and Discussion
A global unbiased proteomic approach was followed to identify the proteins expressed in human iris tissue. The overall experimental scheme of the study is depicted in Figure 1. A total of 162,700 MS/MS spectra were acquired in the Fourier Transform mode, which resulted in the identification of 27,466 peptides corresponding to 4959 proteins. A complete list of proteins and nonredundant list of peptides identified in the human iris are provided in Supplementary Tables S2 and S3, respectively. To the best of our knowledge, this is the first study to establish the proteome of human iris tissue, and these results will provide novel insights into the molecular composition of the human iris tissue.
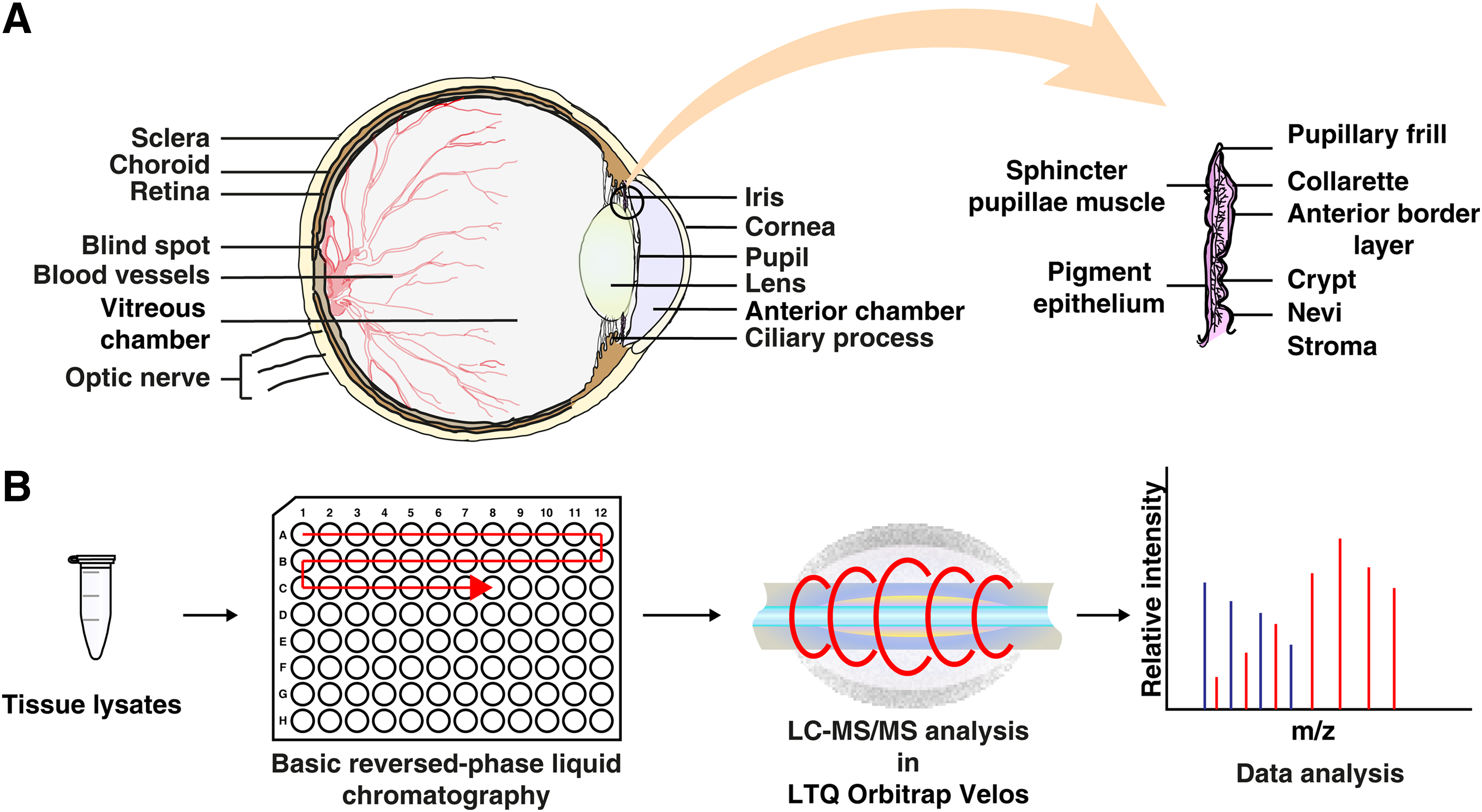
Schematic representation of anatomical structure and proteomic analysis of human iris tissue.
Functional classification of proteins identified in iris
To acquire a functional overview of identified proteins in the human iris tissue, the list of identified proteins was categorized by their biological processes, molecular functions, and subcellular localization. Of the total 4959 identified proteins, 3968 proteins could be mapped to specific biological processes, 1888 proteins had a wide variety of molecular functions, and 3868 proteins were associated with diverse cellular components.
Classification based on biological processes revealed that the highest number of proteins was involved in cell signal transduction (14%), followed by those associated with cell communication (13%), metabolism (12%), energy pathways (12%), protein metabolism (9%), regulation of nucleic acid metabolism (7%), cell growth and maintenance (7%), transport (6%), immune response (2%), and other processes (6%). A large group of proteins had no associated biological processes (12%) as seen in Figure 2A.

Functional characterization of proteins identified in iris tissue by
Molecular function-based classification showed that the identified proteins were involved in various functions, including cell transporter activity (8%), catalytic activity (8%), RNA binding activity (4%), ubiquitin-specific protease activity (4%), transcription regulator activity (4%), and receptor signaling complex scaffold activity (3%). However, molecular functions were not known for a large percentage of proteins (28%). The remaining proteins were classified into enzyme activity (26%) and other functions (15%).
Classification of proteins by their subcellular localization revealed that the majority of proteins were localized to the cytoplasm (31%), followed by nucleus (18%), plasma membrane (13%), mitochondrion (10%), extracellular region (6%), endoplasmic reticulum (4%), integral membrane (3%), Golgi apparatus (3%), ribosome (2%), lysosome (2%), and others (8%) as seen in Figure 2B.
Proteins previously reported in other studies
Our study is the first to attempt MS-based proteomics to study the proteome of human iris. Through this analysis, we were able to identify several proteins previously reported to be expressed in the iris, confirming the validity of our proteomic approach. The list of proteins and the approach used by other groups (Bystrom et al., 2006; Chandra et al., 2013; Hamann et al., 1998; Kim et al., 2012; Silverman et al., 2001; Tran et al., 2013; Van Haeringen et al., 2000) are summarized in Table 1. Representative MS/MS spectra for two of the previously studied proteins, aquaporin 1 (AQP1) and intercellular adhesion molecule 1 (ICAM1), are shown in Figure 3A and B.
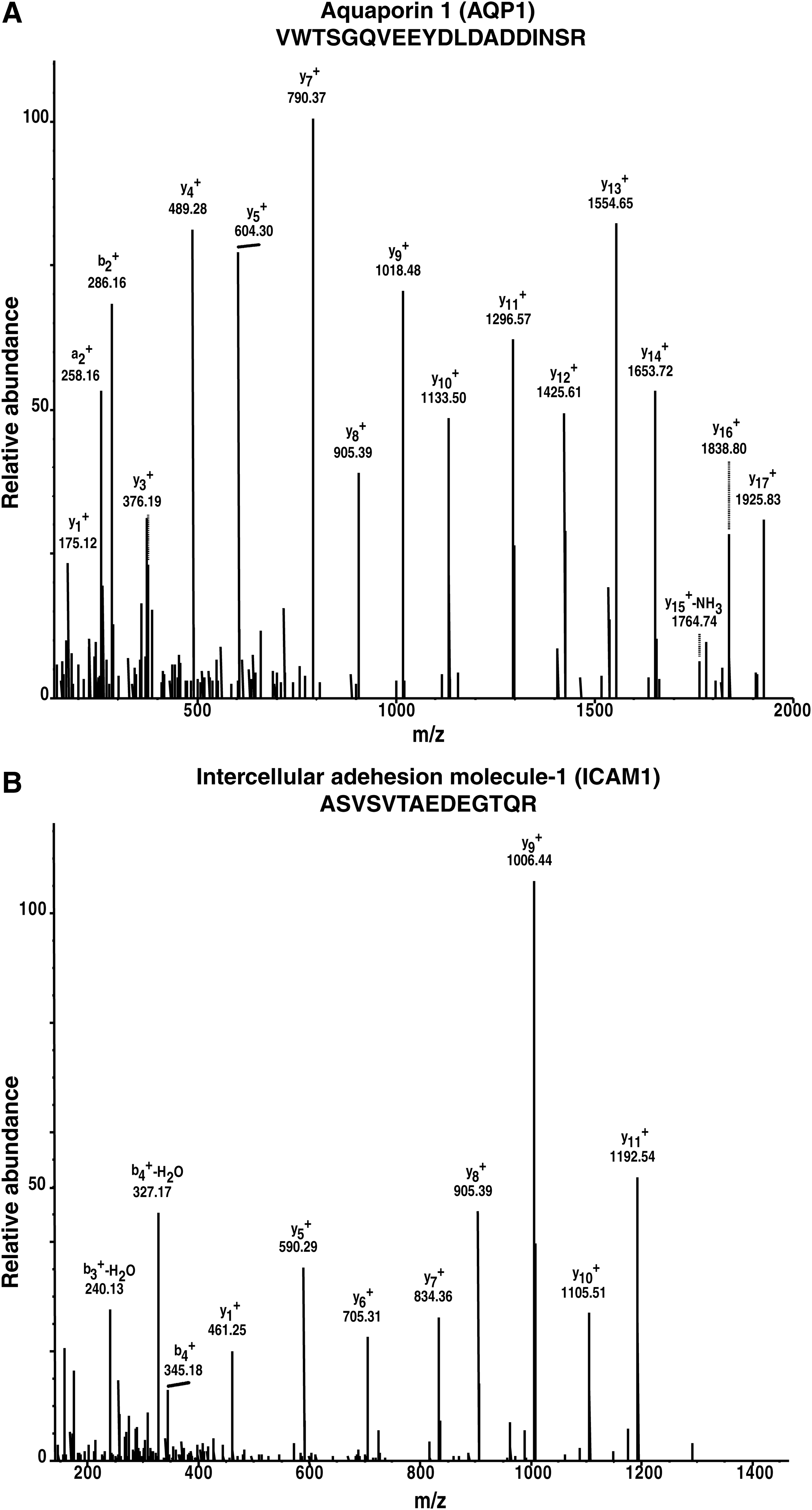
Representative MS/MS spectra for two of the previously studied proteins.
ADAMTSL4, ADAMTS-like protein 4; COX1, cyclooxygenase-1; COX2, cyclooxygenase-2; ICAM1, intercellular adhesion molecule 1; ICAM2, intercellular adhesion molecule 2; LAMB1, laminin subunit beta-1; OPTC, opticin.
The aquaporins (AQPs) are a family of small (∼30 kDa/monomer) and specialized transmembrane water channels (Papadopoulos et al., 2008). They are expressed in diverse cell types and are hydrophobic in nature. AQPs are mainly involved in the transportation of water and glycerol (Gomes et al., 2009). AQP1 is a hexa-transmembrane domain protein (Yool et al., 1996). It is a member of the membrane intrinsic protein gene family and is well known to provide pathways for water flux across cell membranes (Anthony et al., 2000). The expression of AQP1 is limited to fluid-secreting and absorbing tissues in the human body such as the brain, kidney, and eye (Stamer et al., 2003; Yin et al., 2008). In the human eye, it is known to be present in the iris epithelium, the nonpigmented ciliary epithelium, the corneal endothelium, and the epithelium of lens (Stamer et al., 1994). It has been reported to play a role in the production of aqueous humor in the ciliary body epithelia, corneal endothelial fluid transport (Verkman et al., 2008), and also implicated in the development of cataract (Verkman et al., 2014).
ICAM1 is an inducible cell surface glycoprotein belonging to the immunoglobulin super family. ICAM1 has a role in inflammation, immune responses, intracellular signaling events, and migration of activated leukocytes to sites of inflammation (Silverman et al., 2001). Expression of ICAM1 in ocular tissues and its upregulation during inflammatory eye diseases have been reported (Xu et al., 1994). It is also a key mediator of acute ocular inflammation (Mammari et al., 2014). ICAM1 is studied for its potential involvement in diabetic retinopathy (Kamiuchi et al., 2002; Kociok et al., 2009), macular degeneration (Mullins et al., 2006), uveitis (Cimino et al., 2010), and Grave's disease (Kretowski et al., 2003; Zhao et al., 2010).
Comparison with ciliary body, aqueous humor, and vitreous humor proteome
The proteomic data from the present study were compared with our previously published studies on other tissues of the eye, including ciliary body, aqueous humor, and vitreous humor (Goel et al., 2013; Murthy et al., 2014, 2015), to analyze proteomic differences among various components of the human eye. A total of 2793 ciliary body proteins were compared with the iris tissue proteome data set, and we observed that 2535 proteins were unique to the iris tissue, while 2388 proteins were common between the data sets. Comparison of the aqueous humor proteome with the iris tissue proteome data set revealed 463 common proteins, whereas 4455 proteins were unique to the iris tissue. Similarly, comparison with vitreous humor resulted in the identification of 668 common proteins and 4250 proteins unique to iris tissue. Interestingly, 2302 proteins were found to be unique to iris tissue and were not identified from ciliary body, aqueous humor, and vitreous humor.
Some of the proteins identified in the present study include optineurin (OPTN), toll-like receptor 7 (TLR7), adenosine deaminase (ADA), angiotensin-converting enzyme isoform 1 (ACE), double-stranded RNA-specific adenosine deaminase (ADAR), ancient ubiquitous protein 1 (AUP1), developmentally regulated GTP-binding protein 2 (DRG2), and H/ACA ribonucleoprotein complex subunit 1 (GAR1). The proteins OPTN and AUP1 are discussed in the subsequent sections and their respective MS/MS spectra are shown in Figure 4A and B.
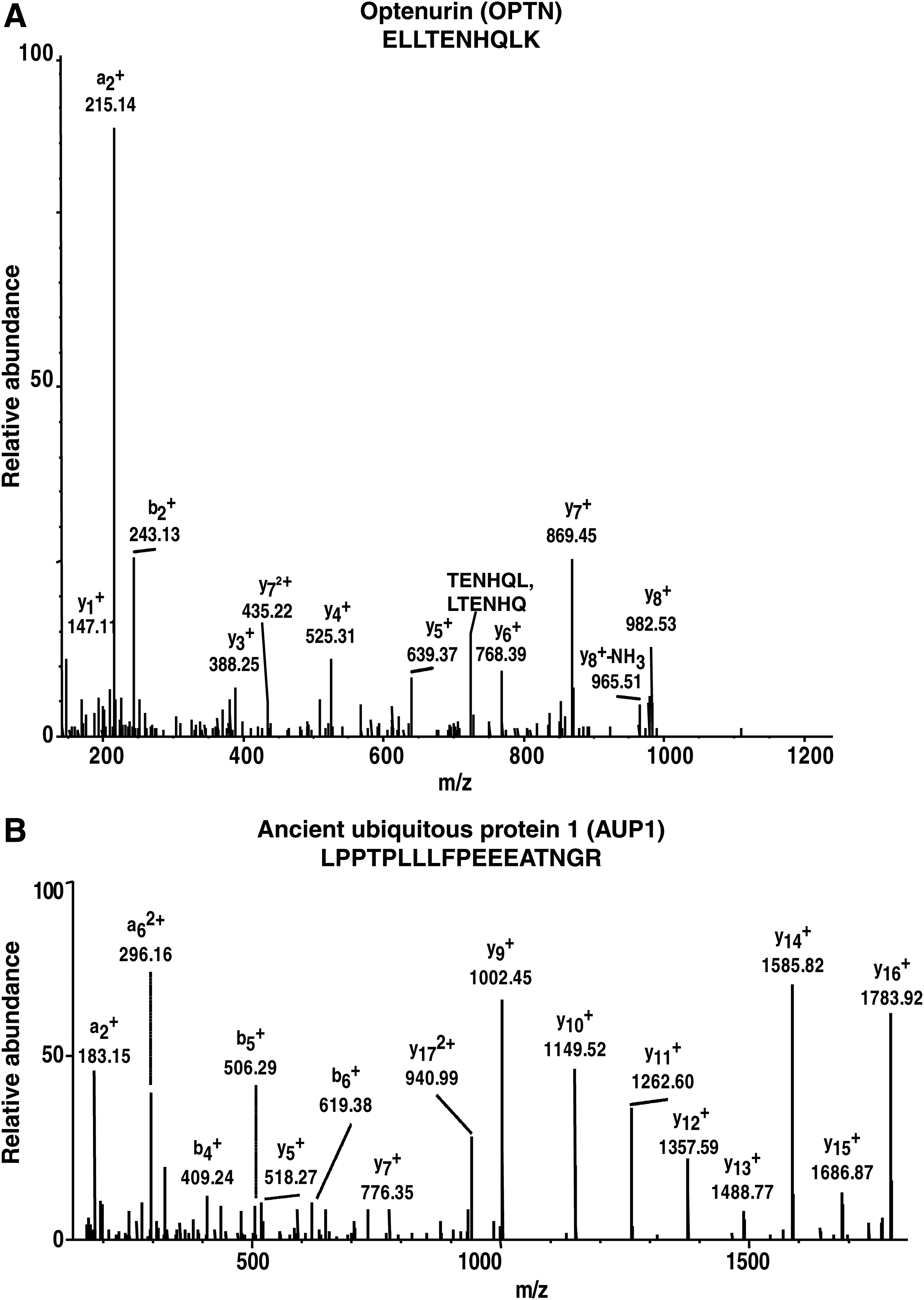
Representative MS/MS spectra for two of the proteins identified in this study.
OPTN is a 67-kDa protein encoded by the OPTN gene. The protein contains a number of putative domains such as leucine zipper, coiled-coil motifs, an ubiquitin-binding domain, a microtubule-associated protein 1 light chain 3 interacting motif, and NF-kappaB essential modulator-like domain (Wild et al., 2011). OPTN has been found to interact with a diverse set of proteins and acts as a platform to assemble multimolecular complexes to mediate a variety of cellular and molecular functions (Genin et al., 2015). OPTN has been reported to be associated in various processes, including neuroprotection of the optic nerve, maintenance of the Golgi complex, membrane trafficking, and exocytosis (Osawa et al., 2011). OPTN has been implicated in open-angle glaucoma (Rezaie et al., 2002). Mutations in the OPTN gene have been associated with primary open-angle glaucoma, a leading cause of blindness in the world (Sahlender et al., 2005). Furthermore, OPTN is also reported to have a role in amyotrophic lateral sclerosis, formation of Lewy bodies, generation of neurofibrillary tangles, and dystrophic neurites in Alzheimer's disease (Doshi et al., 2006).
AUP1 is a monotopic membrane protein localizing to lipid droplets and endoplasmic reticulum membranes (Spandl et al., 2011; Stevanovic and Thiele, 2013). AUP1 is a 410 amino acids long integral protein with a single predicted membrane domain. The membrane domain of AUP1 is located internally, and both the N and C termini of AUP1 face the cytoplasm, resulting in monotopic/hairpin topology (Stevanovic and Thiele, 2013). AUP1 has multiple functional domains and is reported to facilitate endoplasmic reticulum-associated protein degradation, inside–out signaling in platelets, and neutral lipid storage (Kato and Oshimi, 2009; Spandl et al., 2011; Stevanovic and Thiele, 2013). AUP1 possesses domains related to signal transduction, and it has also been proposed to be involved in integrin signaling (Kato and Oshimi, 2009). It is reported to play a role in the translocation of terminally misfolded proteins from the endoplasmic reticulum lumen to the cytoplasm and their degradation by the proteasome (Olzmann and Kopito, 2011).
Comparison with human proteome data
To identify proteins uniquely reported in this study, the data obtained in the present study were compared with that of “A draft map of the human proteome” (Kim et al., 2014). A total of 17,294 nonredundant proteins identified by Kim et al. were compared with the iris tissue proteome data, and we observed 4872 proteins common to the human proteome data and 26 proteins unique to the present study (Table 2). A partial list of uniquely identified proteins in this study includes alanyl-tRNA editing protein Aarsd1 (AARSD1), melanoma antigen recognized by T-cells 1 (MLANA), olfactory receptor 5D18 (OR5D18), vesicular acetylcholine transporter (SLC18A3), and 5,6-dihydroxyindole-2-carboxylic acid oxidase (TYRP1). Representative MS/MS spectra for two of uniquely identified proteins, alanyl-tRNA synthetase domain containing 1 (AARSD1) and Tyrosinase-related protein-1 (TYRP1), are shown in Supplementary Figure S1. Biological process, molecular function, and domain analysis of uniquely identified proteins are provided in Table 2. These proteins are mainly found to be associated with biogenesis of melanosomes and may play a role in the regulation of the type of melanin synthesized in the iris tissue.
Protein-coding evidence for “missing proteins”
We compared our data with the list of “missing proteins” provided by Chromosome-Centric Human Proteome Project (neXtProt release: 2015-04-28, application release: 3.0.26) (Gaudet et al., 2013). These “missing proteins” do not have any experimental evidence for their coding potential. However, in our study, we found peptide evidence for 16 such proteins. Apart from providing experimental evidence for their expression, we now also know that these genes get translated in the iris. We also manually inspected MS/MS spectra for these peptides to confirm their quality. We have also found multiple peptide spectrum matches (PSMs) for 9 of these 16 proteins. Some of these proteins include ADP-ribosylation factor-like protein 5C (ARL5C), abhydrolase domain-containing protein 12B isoform c (ABHD12B), ATP synthase lipid-binding protein and mitochondrial isoform B (ATP5G3), and doublecortin domain-containing protein 5 (DCDC5). Our analysis revealed that these proteins contain transmembrane domain (TM), doublecortin (DCX), abhydrolase_5 domain (ABH), ankyrin repeats (ANK), and Fes/CIP4homology (FCH) domains. The representative MS/MS spectra for two of the proteins, including Solute carrier family 18 member 3 (SLC18A3) and SLIT-ROBO Rho GTPase activating protein 2B (SRGAP2B), are provided in Supplementary Figure S2. Biological process, molecular function, and domain analysis of these nine proteins are provided in Table 3.
Conclusions
Proteomic analysis of human iris tissue resulted in the identification of 4959 proteins and provides a broad functional analysis of all the nonredundant human iris tissue proteins, being the first step toward the complete characterization of the iris tissue. The current study has identified proteins previously not known to be associated with the iris such as OPTN and AUP1. The identified proteins are found to be involved in neuroprotection, membrane trafficking, maintenance of Golgi apparatus, and immune response. We were also able to identify a group of proteins associated with biogenesis of melanosomes, which are the intracellular organelles of pigment cells devoted to the synthesis, storage, and transport of melanin pigments.
One of the unique proteins identified in this study, TYRP1, is found to play a role in activating pigmentation, cell proliferation, and differentiation. In this study, we have been able to identify many unique proteins and missing proteins, which can be further investigated to characterize their role in human physiology and disease pathology. This exhaustive list of iris proteins can also serve as a baseline data for future research on iris tissue. The data from this study will significantly contribute to the ambitious Human Eye Proteome Project (HEPP), which aims at characterizing the proteome of all ocular tissues (Semba et al., 2013).
Finally, we make a call to the broader visual health, ophthalmology, and omics community (Dawkar et al., 2015; Reddy et al., 2015) that proteomics offers a veritable prospect to obtain a systems scale, functional and dynamic picture of the eye tissue in health and disease. This knowledge is ultimately pertinent for diagnostics and therapeutics innovation to address the pressing needs of the 21st century visual health.
Footnotes
Acknowledgments
The authors thank the Department of Biotechnology (DBT), Government of India, for research support to the Institute of Bioinformatics. They thank the Infosys Foundation for research support to the Institute of Bioinformatics. M.D. is on deputation from Siddaganga Institute of Technology (Tumkur, India) to pursue his research work at IOB toward a PhD degree under Faculty Improvement Program of Sree Siddaganga Education Society (Tumkur, India). R.S.N. is a recipient of Senior Research Fellowship from the Council of Scientific and Industrial Research (CSIR), Government of India.
G.D. is a recipient of Senior Research Fellowships from University Grants Commission (UGC), Government of India. A.K.M. is the recipient of BINC Senior Research Fellowship from DBT, India. H.G. is a Wellcome Trust/DBT India Alliance Early Career Fellow. Dr. T.S.K.P. and Dr. K.R.M. are the recipients of a research grant on “Proteomics based identification and validation of urinary Biomarkers for Age-related Macular Degeneration” from DBT. Dr. T.S.K.P. is the recipient of a research grant on “Development of Infrastructure and a Computational Framework for Analysis of Proteomic Data (BT/01/COE/08/05)” from DBT. All authors have met the ICMJE criteria for authorship.
Author Disclosure Statement
The authors declare that they have no competing financial interests.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.