
Abstract
Abstract
Travelers' diarrhea (TD) is one of the most important global health issues among international travelers, especially those traveling to developing countries. Enterotoxigenic Escherichia coli (ETEC) is the most frequently isolated pathogen causing TD. Although TD is self-limiting, it is also severely incapacitating. Ciprofloxacin is one of the standard quinolone antibiotics used in patients with TD. However, the alarming levels of antibiotic resistance have necessitated the urgent need to identify new drugs. The drug target of ciprofloxacin is gyrA, which is exclusively present in prokaryotes and essential for bacterial survival. Several mutations in the quinolone resistance-determining region of gyrA are associated with increased quinolone resistance in vivo, which accounts for reduced affinity toward quinolones. To understand the molecular events underlying the mechanism of drug resistance, we report, for the first time to the best of our knowledge, the structural and dynamic effects of mutations in ETEC gyrA on ciprofloxacin affinity in comparison with the wild-type protein. Our simulations reveal that mutations significantly alter the gyrA residue interaction network and overall pattern of global dominant motions in major distinctive domains of N-terminal regions of gyrA. This study therefore provides important insights for the design of more potent antibacterial agents with high ligand efficacy for treating drug-resistant bacterial infections, including the TD that continues to adversely impact global health and international travel.
Introduction
T
Fluoroquinolones are potent antimicrobial agents for treatment of severe cases of TD. Increased use of quinolones led to an escalating rate of antibiotic resistance against these antibiotics in E. coli. Increased quinolone resistance is related to (1) decreased influx of antibiotic, (2) plasmid-mediated resistance, and (3) acquisition of mutation in genes encoding gyrase. Among these, mutations are reckoned to be most clinically relevant (Fabrega et al., 2009; Hooper and Jacoby, 2015; Jacoby, 2005). Such mutations can cause structural modifications of the target and ultimately affect binding affinity. Thus, it becomes imperative to explore the structural impact of these mutations to significantly advance our knowledge of associated molecular changes in the functionally significant regions of the target.
E. coli gyrA, a type II topoisomerase, is an attractive drug target for antibacterials. gyrA is exploited as a drug target with high therapeutic potential due to its ability to introduce negative supercoils in DNA in an ATP-dependent reaction. It is one of the two essential subunits of DNA gyrase. gyrA facilitates DNA unwinding at replication forks and is involved in the regulation of DNA supercoiling (Champoux, 2001). DNA gyrase is a heterotetramer (A2B2), which comprises two subunits of GyrA (Mol Wt.: 97 kDa) and GyrB (Mol Wt.: 90 kDa) encoded by genes, gyrA and gyrB, respectively (Reece et al., 1991). A 59 kDa (GyrA59) N-terminal domain (NTD) and a 38 kDa C-terminal domain (GyrA-CTD) associate to form GyrA subunit. Currently no high-resolution structure for the gyrase holoenzyme (A2B2) exists. However, X-ray crystal structure of the GyrA59 fragment from E. coli (PDB ID: 1ab4) was solved with a resolution of 2.8 Å, which consists of residues 30–522 (Cabral et al., 1997). gyrA N-terminal region, which cleaves single-stranded DNA, has three distinctive domains (Fig. 1).
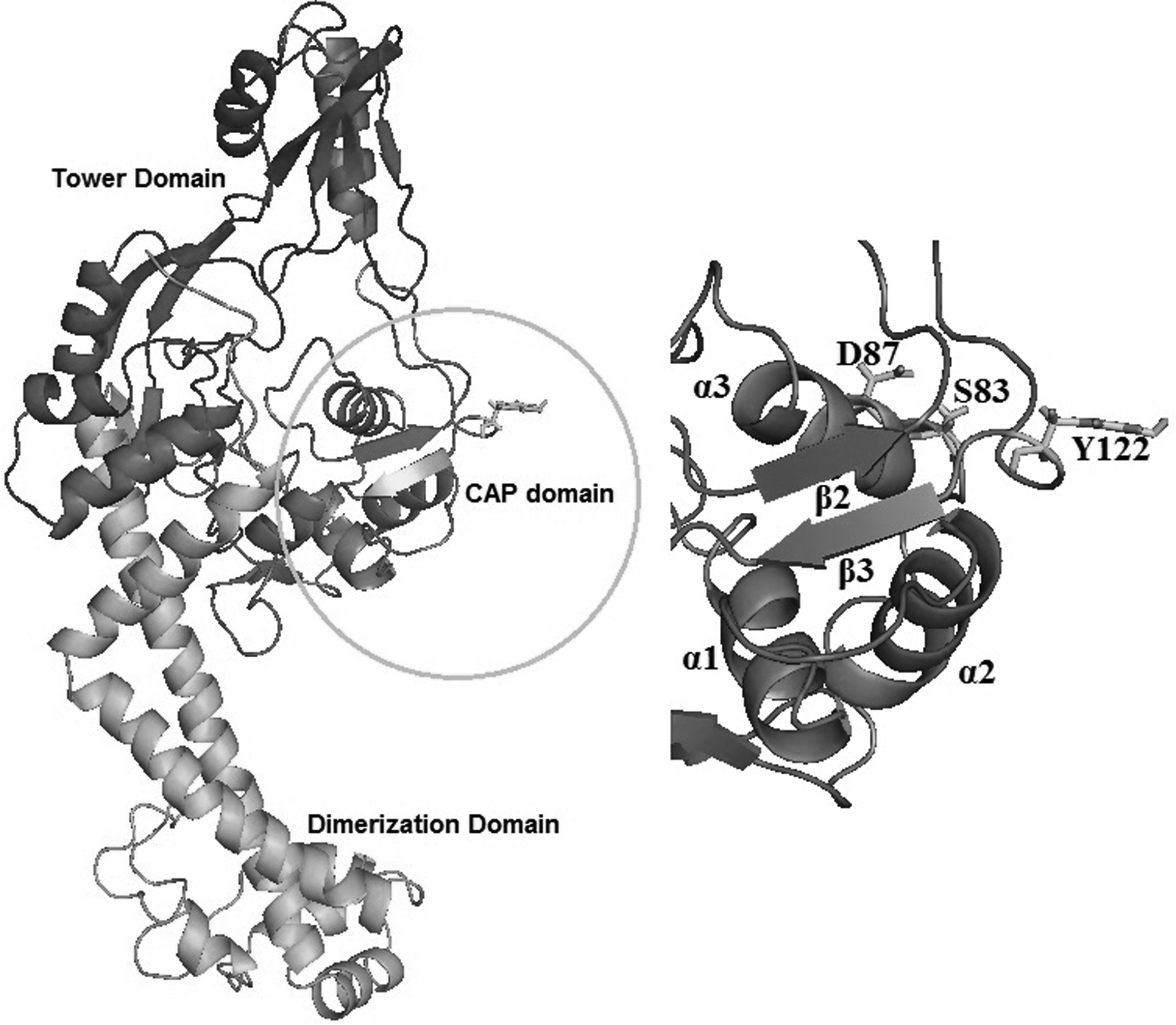
Cartoon representation of ETEC gyrA showing its distinctive domains, catalytic site, and mutations associated with quinolone resistance reported in diarrheal patients. QRDR mutations and the catalytic site are in stick representation and shown in yellow and cyan color, respectively. The CAP domain is colored violet purple, the tower blue, and the dimerization domain and connecting a-helices colored orange. Zoomed in is a view of CAP domain showing HTH motif with three helices and two beta-sheets with corresponding residue ranges as α1 (43–56), α2 (66–77), α3 (81–92), β2 (101–106), and β3 (124–128). CAP, Escherichia coli catabolite activator protein; HTH, helix-turn-helix; QRDR, quinolone resistance-determining region.
The first domain contains a helix-turn-helix (HTH) motif similar to that of the E. coli catabolite activator protein (CAP). It contains the quinolone resistance-determining region (QRDR) and active site tyrosine, which is responsible for breakage and religation of the DNA (Cabral et al., 1997; Horowitz and Wang, 1987). The second domain is termed as the tower and adopts an extended bilobed α/β structure. The third domain, termed as dimerization domain, is a dense bundle of small α-helices connected to the tower domain and the CTD through long α-helices (Cabral et al., 1997).
It is noteworthy that the Ser83Leu and double mutation Ser83Leu/Asp87Asn in gyrA have been widely associated with quinolone resistance in ETEC isolates from diarrheal patients of diverse geographic regions (Daniel et al., 2002; Mendez et al., 2009). These mutations have been mapped to the QRDR of E. coli gyrase subunit A (residues 67–106) (Yoshida et al., 1990).
Molecular dynamics (MD) simulation studies play an important role in delivering more potent broad-spectrum antibiotics (Fjell et al., 2012) by using a modified virtual screening protocol wherein the library of compounds is docked against multiple target conformations. This approach has been successfully applied in designing inhibitors against many microbial targets (Durrant et al., 2011; Schames et al., 2004; Wang et al., 2011). MD simulations complement experimental approaches such as X-ray and nuclear magnetic resonance where thermodynamic properties of complexes are ignored. MD simulations provide insights into such dynamic movements at the atomistic level. Such extreme molecular details further help in describing ligand affinities and selectivities and thus help in design of potent inhibitors.
The present study was undertaken to better understand the dynamic behavior of the gyrA in ETEC and to decipher the structural changes associated with mutations, Ser83Leu and Ser83Leu/Asp87Asn, leading to ciprofloxacin antibiotic resistance in ETEC gyrA.
Materials and Methods
Preparation of starting structures
The full-length canonical structure of gyrA is encoded by a polypeptide of 875 amino acids with a molecular weight of about 97 kDa. The structure of gyrA from the ETEC strain, E24377, using E. coli gyrA (PDB code: 1AB4) as template was homology modeled in Discovery Studio version 4.1 (DS) (Mehla and Ramana, 2016). Heteroatoms, crystallographic waters, and cofactor were removed from the structure before docking. In DS, the starting structures of the mutants were modeled in silico by mutating the target residues with the desired amino acids, keeping the secondary structure intact. The complex structures of gyrA with ciprofloxacin were predicted by computational docking for the wild-type (WT) and mutant (MT) structures in DS. The initial constructed models were further optimized and docked structures from the study by Mehla and Ramana (2016) were used as the starting structures for MD simulations.
MD simulations
The structures of gyrA WT and MT complexes were simulated using the Amber 11.0 (Case et al., 2010) and Ambertools 1.5 package. Hydrogen atoms were added using the tLeap program implemented in Ambertools and parameters were assigned according to AMBER FF99SB force field (Hornak et al., 2006). The system was solvated with TIP3P water model (Jorgensen et al., 1983) in an octahedral box with 15 Å buffer around the complex. Twelve Na+ counterions were added to maintain neutrality of the systems.
Each molecular system, that is, (1) the native modeled complex of 1AB4, (2) the modeled complex of 1AB4 with substitution of Ser to Leu at position 83, and (3) with substitution of Ser to Leu at position 83 and substitution of Asp to Asn at position 87, was energy minimized through three rounds of minimizations of 1000 steps each.
In each set of minimization, protein was energy minimized for 500 steps of steepest descent, followed by 500 steps of conjugate gradient with a time step of 2 fs to eliminate close van der Waals contacts. In the first and second minimization rounds, position restraints of 10 kcal−1Å−2 and 2 kcal−1Å−2, respectively, were imposed. In the third minimization, the entire system was minimized without any restraints. Each system was then gradually heated from 0 to 300 K, followed by constant temperature equilibration at 300 K and 1 atm pressure for 100 ps. Following the stabilization of thermodynamic properties, a 10-ns MD simulation was performed for each system with an integration step of 2 fs in an isothermal isobaric ensemble (NPT, T = 300 K and P = 1 atm) with periodic boundary conditions. NPT conditions were maintained with Langevin thermostat (Davidchack et al., 2009) and Berendsen barostat (Berendsen et al., 1984). SHAKE algorithm (Ryckaert et al., 1977) was applied to constrain all covalent bonds involving hydrogen atoms.
Long-range electrostatic forces were treated using the particle mesh Ewald method (Darden et al., 1993) with a charge grid spacing of ∼1.0 Å and the charge grid interpolation on a cubic grid by setting the direct sum tolerance to 4.0 × 10−6. Short-range electrostatics and van der Waals interactions were evaluated using a 9.0 Å atom-based cutoff. Trajectory coordinates were saved at every 2 ps for further analysis. After performing MD simulations, the root mean square deviations (RMSDs) and root mean square fluctuations (RMSFs) of the MD trajectories were analyzed with respect to initial set of coordinates. MD simulation protocol has been validated based on the modeled structure of WT-gyrA. The convergence of simulations was analyzed by monitoring parameters such as the energy components, RMSD from initial modeled structure, and RMSF for WT-gyrA, and each of the mutants (S83L, S83L/D87N).
MM-PBSA calculations
Free energies were calculated using the conventional MM-PBSA and MM-GBSA approaches in AMBER 11. Free energies were estimated by averaging the configurations that were extracted from a total of 2500 frames at every 20 ps giving a total of 125 frames for energetic analysis during the last 5 ns of the MD simulation. The binding free energy profiles were calculated for WT and each of the mutants (S83L, S83L/D87N) using the following equations:
where T and S correspond to the temperature and the total solute entropy, respectively; Egas signifies gas-phase energy, which is the sum of internal energy (Eint), electrostatic (Eele), and van der Waals contributions (Evdw). Egas is evaluated using parameters from the FF99SB force field terms. The solvation free energy (Gsol) can be further decomposed into polar and nonpolar solvation states. The polar solvation contribution (GGB and GPB) is determined by solving Poisson Boltzmann (PB) and Generalized Borne (GB) equations. The nonpolar solvation contribution (GSA) is estimated using 0.0072 kcal mol−1Å−2 as the value for constant γ and the solvent-accessible surface area (SASA) determined using a water probe radius of 1.4 Å. Dielectric constant values for solute and solvent were set to 1 and 80, respectively.
To determine the contribution of individual amino acids to free energy, a per residue decomposition analysis was performed using the GB model, implemented in Amber11. Decomposition analysis was performed on l25 frames obtained during last 5 ns of MD simulation. The amino acids, which contributed more than 0.5 kcal/mol to the binding energy, were identified as the hotspot amino acids. These hotspot amino acids add up most to the complex stability and are crucial for the gyrA-ciprofloxacin interaction.
Principal component analysis
To investigate the direction and amplitude of the dominant motions of MD trajectories, essential dynamics calculations were performed. Principal component analysis (PCA) was performed to reduce the dimensionality of the MD simulation data. PCA can help in identifying an essential subspace that probes pronounced motions corresponding to large-scale vibrational modes of groups of atoms in normal mode analysis (Amadei et al., 1993). Before analysis, the overall translational and rotational motions were excluded by translating MD trajectory to the geometrical center of the molecule and superimposing onto a reference structure (Amadei et al., 1996).
PCA constructs the configurational space by orthogonal linear transformation in a new coordinate system to generate a covariance matrix (C). Associated eigenvectors (Vi), namely principal components (PC) and eigenvalues (λi), are generated by diagonalization of the covariance matrix. Vi give a vectorial description of each component of the motion by indicating the direction of the motion and λi represent the amplitude of the eigenvectors along the multidimensional space. Projection Proj[M,PCi] of any frame M onto ith PC is calculated by Equation (6):
where Mα is the Cα atom of every structure after overlaying M with the reference structure. Projection highlights the time-dependent motions that the components perform in the particular vibrational mode.
PCA was carried out using the PCAsuite software (http://mmb.pcb.ub.edu/software/pcasuite/) on i), WT, S83L, and S83L/D87N mutant models. MD trajectories of corresponding atoms were extracted using the PTRAJ program and analysis carried out for the last 5 ns using 63 frames.
Residue interaction network Analysis
The average structure derived from the last 5 ns trajectory of each system, wild and S83L, S83L/D87N mutants, was used for constructing the residue interaction networks (RINs). Reduce program (Word et al., 1999b) adds hydrogen atoms to the input structure using local geometry. PROBE (Word et al., 1999a) identifies interacting amino acids by evaluating their atomic packing using small-probe contact dot surfaces. PROBE uses several scoring functions to quantify noncovalent interactions such as interatomic contact, hydrogen bonds, salt bridges, and pi–pi interaction. RINs generated from the averaged MD structures were visualized using RINalyzer (Doncheva et al., 2012). Analyzing different RINs helps us to extract changes in the networks that reflect alterations in physicochemical characteristics of the structures.
Results and Discussion
Characterization of structural stability
To understand the effect of substitutions at amino acid positions, 83 and 87, on quinolone susceptibility, we carried out MD simulations. To evaluate the conformational flexibility of the WT and MT structures, time-dependent RMSDs of the backbone atoms were monitored relative to the corresponding energy-minimized structure. Figure 2 depicts time-dependent RMSDs of WT and MT models compared with the reference structure. RMSDs were calculated from the last 5 ns of the 10-ns MD simulations. The trajectories for all the simulation systems were stabilized over the time course, with moderately small fluctuations in the models. The final RMSD values range from 1.2 to 1.3 Å for all three systems.
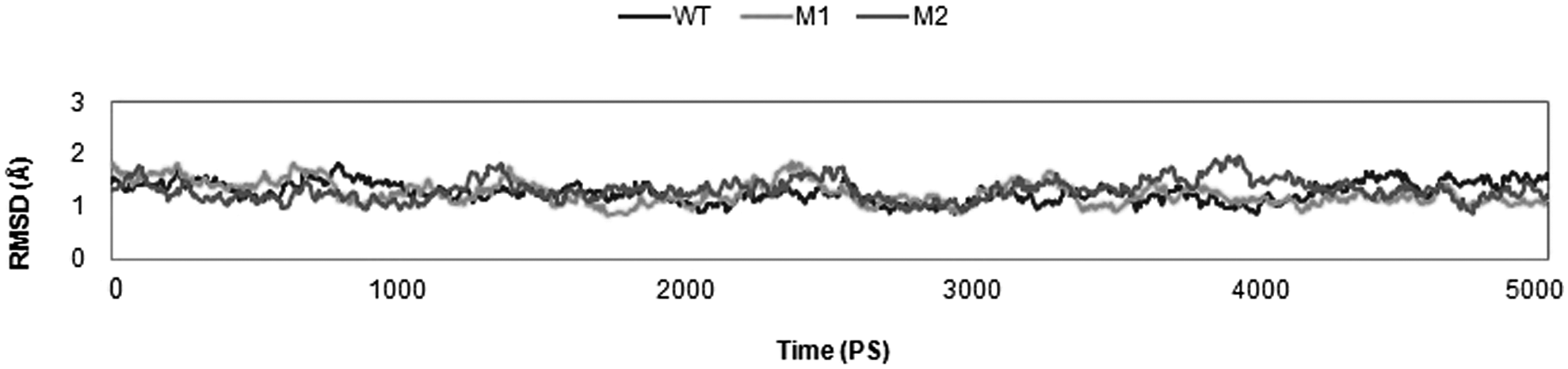
Time evolution RMSD of gyrA backbone throughout the 10-ns MD simulation time for WT (blue), S83L mutant (yellow), and S83L/D87N mutant (red). WT, wild-type.
A narrow range of time-averaged RMSD values reflects that deviations in the native and mutant structures were relatively constant. It was proposed that these mutations do not disrupt the backbone conformation or cause significant global change, although they could affect the dynamic behavior of mutant complexes, thus leading to reduced binding affinity. From their initial conformations, S83L/D87N showed maximum deviation. It is evident from the RMSD plot that RMSDs of the both MT models are comparatively higher than that of the WT.
Furthermore, to assess the effect of these mutations on dynamic behavior of residues, RMSFs between native, S83L, and S83L/D87N were calculated and plotted in Figure 3. RMSFs were calculated from the average structures generated during the last 5 ns of the 10-ns MD simulations, which were also used as a reference to calculate the RMS deviations in the last 5 ns. The apex points of RMSFs were different for all the trajectories. Hence, regardless of the structural similarities between WT and MT models of gyrA (RMSDs ∼1.2 Å), mutant forms considerably deviate from the WT forms in their conformational flexibility. The deviations in residue fluctuations may influence the interaction with ligand and thus the ligand binding efficiency. The regions of high RMSFs were not confined to the vicinity of the mutated residues only, but were distributed across the entire protein backbone.
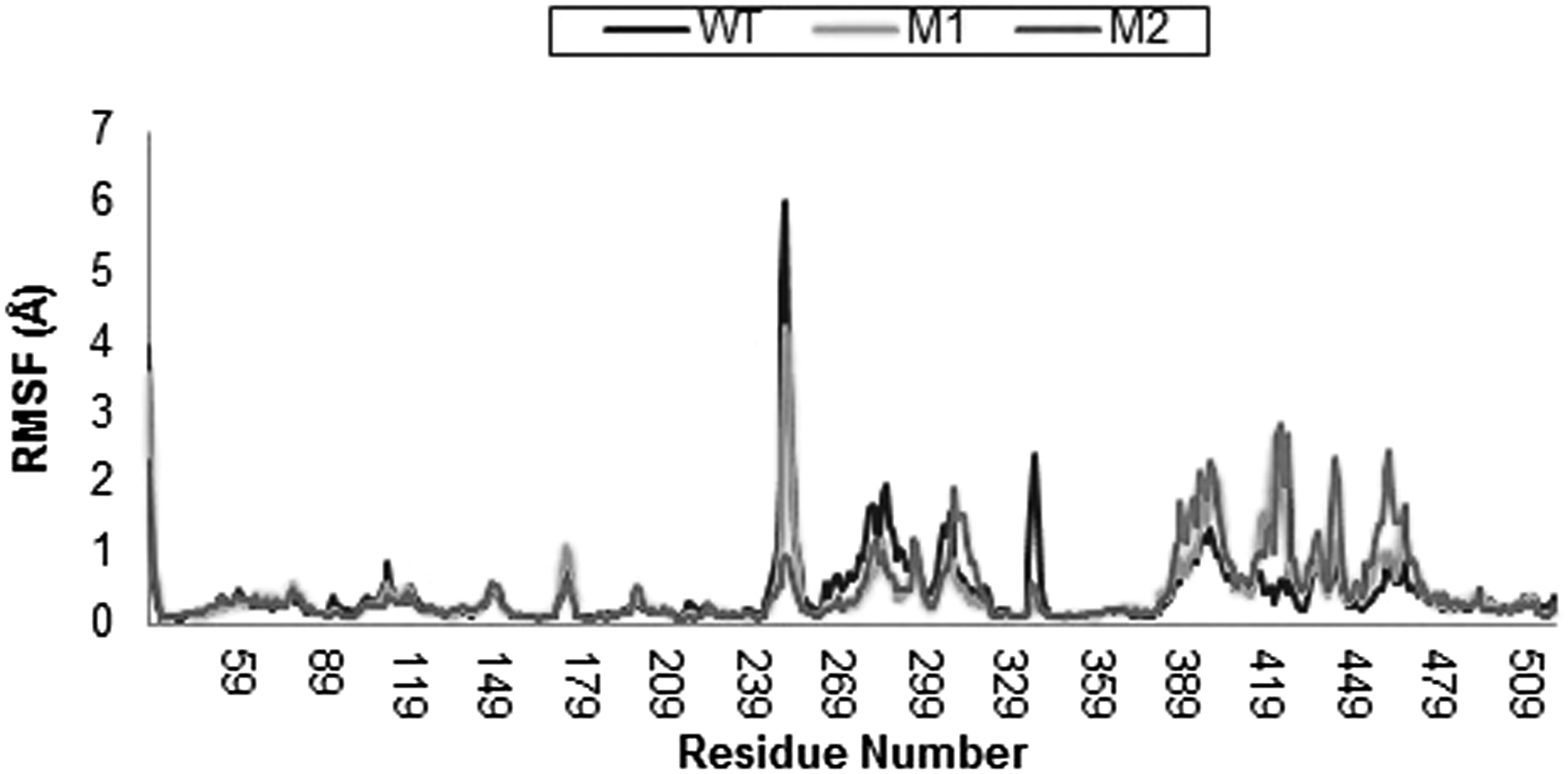
Time evolution RMSF of gyrA backbone atoms throughout the 10-ns MD simulation time for WT (blue), S83L mutant (yellow), and S83L/D87N mutant (red). RMSF, root mean square fluctuation.
For example, RMSFs of the residues ranging from 240 to 270 show large fluctuations in WT compared with both MT models, which suggests that these mutations affect the entire protein structure and not just adjacent residues. The difference in RMSDs of WT and MT models at several apex points was higher than 1 Å, which further strengthens our argument that such mutations introduced in native structure influence the dynamic properties of the protein. Flexibility of active site residue Tyr 122 is reduced in MT compared with WT models, indicating the relative rigidity of this region in MT models. Tyr 122 lies in domain III, which has folds similar to CAP and is responsible for DNA cleavage (Berger et al., 1998).
Secondary structure content
The ensemble-averaged secondary structure populations for each amino acid residue are depicted in Figure 4. Although WT and MT variant conformations appear to be dominated by turns and α-helices, regular secondary structures (helical and β-sheet structures) in small fractions are clearly traceable. The population of the pi-helix and parallel sheet is almost negligible for all the systems investigated. The helical content is conspicuously higher than the β-sheet and -turn contents in all molecular systems. No significant changes were observed in the secondary structure content of ensemble-averaged structures from MT and WT models. This finding ideates that the backbone conformations of the WT and MT complexes do not change significantly upon mutations.
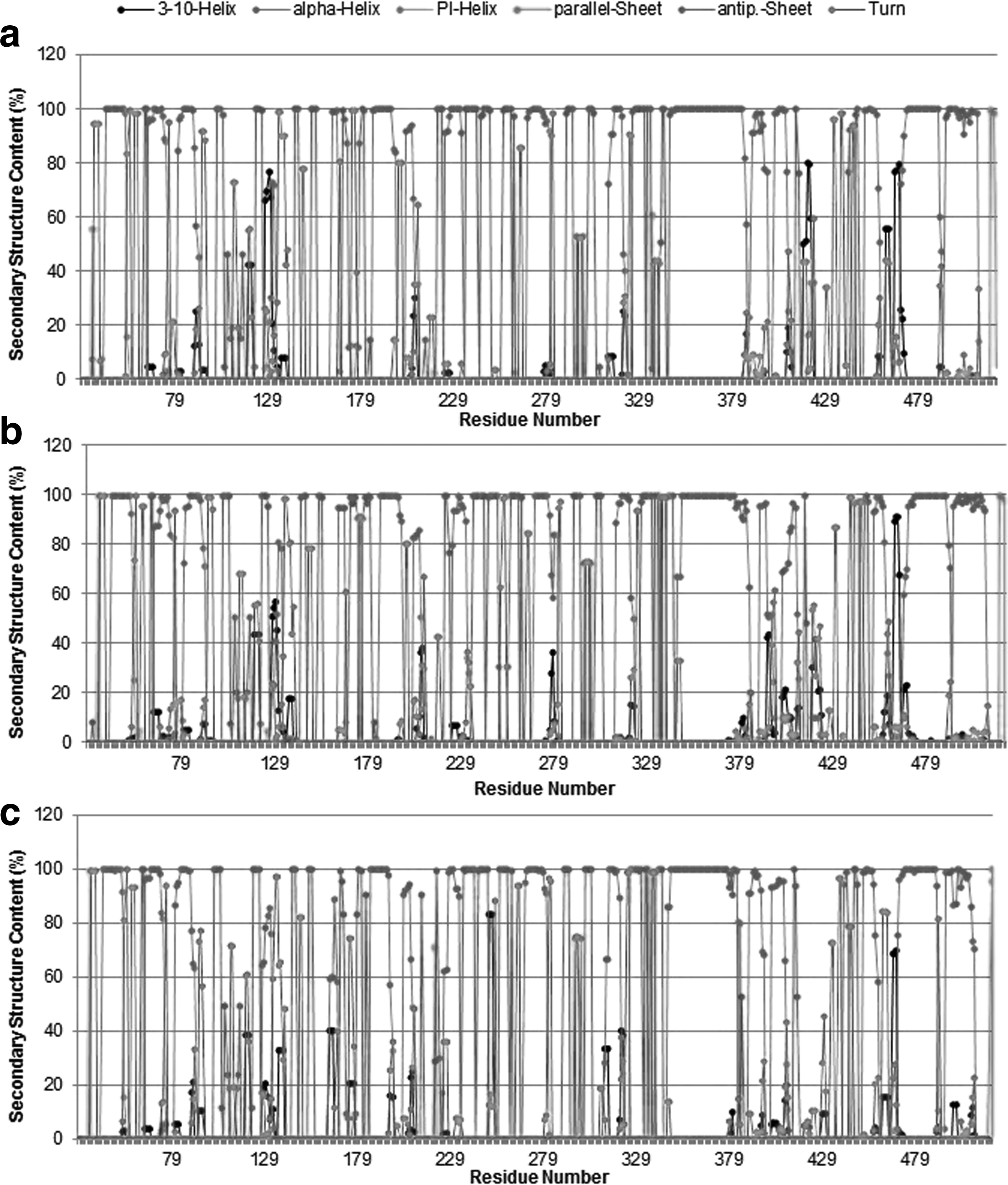
Average secondary structure contents of protein backbone residues in
DNA-dependent ATPase activity of gyrA is governed by the enzymatic site comprising residue Tyr122. To investigate the dynamic structural changes of the catalytic residue and nearby active site residues, we visualized the enzymatic site using visual MD (Humphrey et al., 1996). It was observed that the conformation of catalytic residue, Tyr122, fluctuates in WT, S83L, and S83L/D87N mutants during the course of simulation. Three snapshots per mutant at 0, 5, and 10 ns of the MD trajectories were saved reflecting conformational changes of Tyr122 (Fig. 5). Conformational changes of Tyr122 were quantified by computing the phi-psi dihedral angles across WT and MT structures (Fig. 6). Mean dihedral angles in WT gyrA, S83L, and S83L/D87N varied from −115.06 to −122.91 and −118.32 for phi; 4.47 to 0.73 and 5.84 for psi; respectively. It is noteworthy that neither RMSD/RMSF comparisons nor secondary structure assessment provides any evidence for decreased structural stability of mutated structures compared with WT structures.
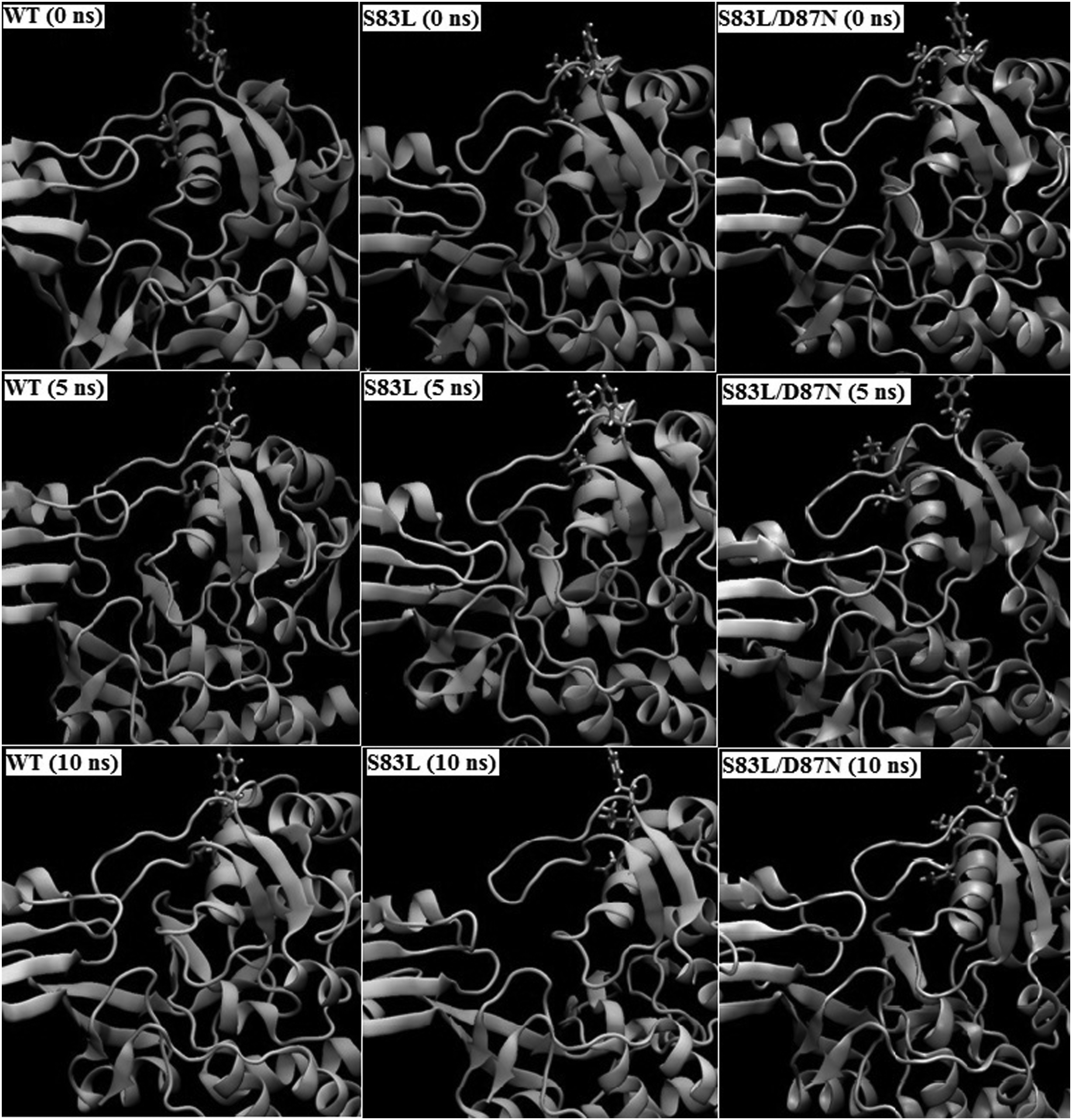
Observed conformational changes of catalytic residue, Tyr 122, and the mutated residues, Ser 83 and Asp 87, during the course of MD simulation in WT (Column 1), S83L mutant (Column 2), and S83L/D87N mutant (Column 3) at 0 ns (start of production phase), 5 ns, and 10 ns (end of production phase).
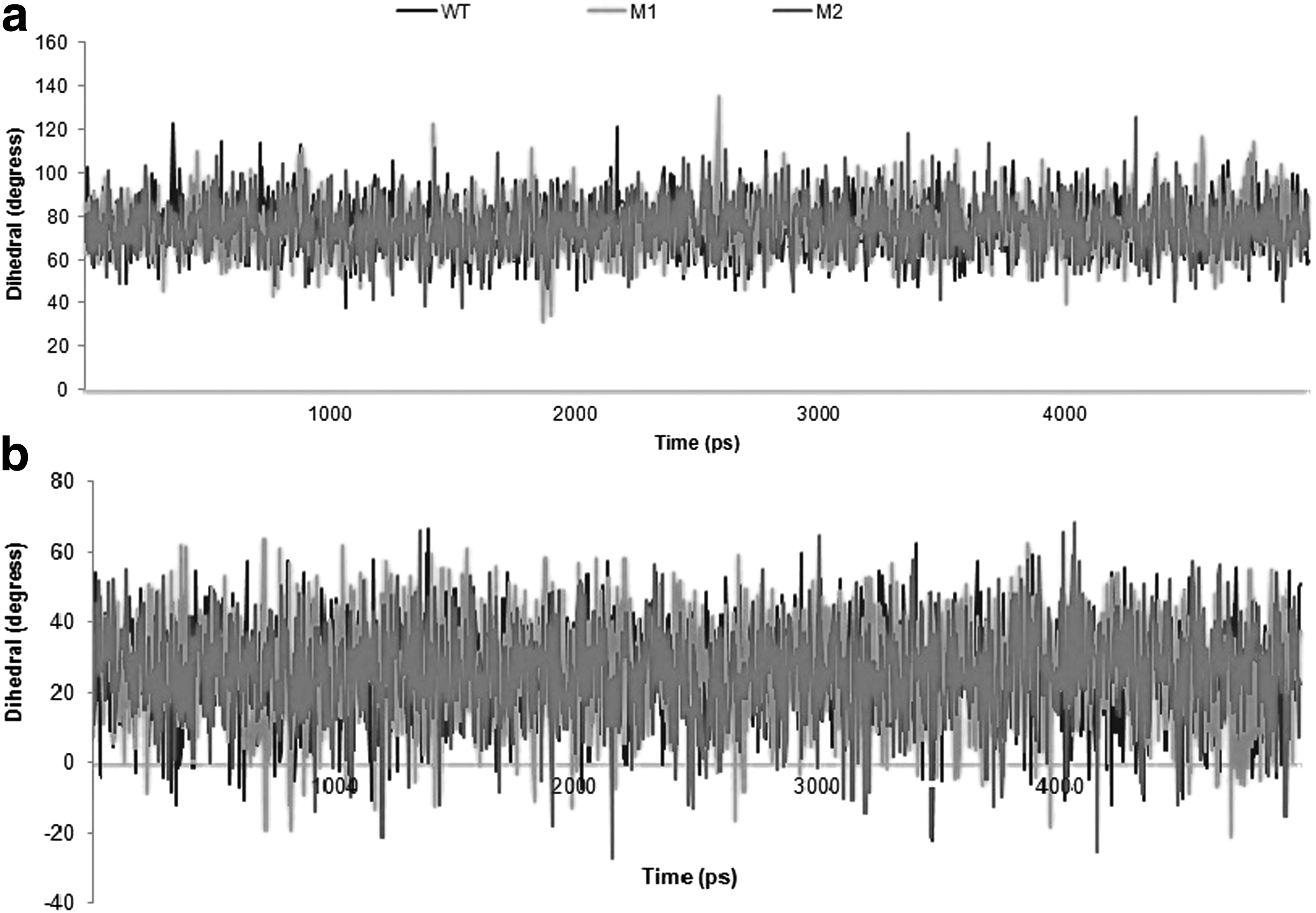
Time evolution of rotation of dihedral bond angles
MM-PB(GB)/SA binding free energy calculations
To evaluate the free energy differences between WT and MT models, MM-PB(GB)/SA methods were employed. For calculating binding energy, 125 frames from the last 5-ns simulation were analyzed. Predicted binding energies and energy component breakdown for different simulation systems are summarized in Table 1. The total binding free energies using both methods follow the same trend of reduced binding affinity in MT relative to WT models. In the double mutant S83L/D87N, a significant drop in binding energy (ΔG binding) was observed using both methods. Reduced binding affinities imply that the mutations influence the efficiency of binding of ciprofloxacin to gyrA. Computationally predicted binding affinities are in accordance with the experimental study, which reports increased minimum inhibitory concentration values for ciprofloxacin in S83L and S83L/D87N variants of ETEC gyrA (Mendez et al., 2009). The predicted van der Waals (ΔEVDW) and electrostatic contribution (ΔEELE) are significantly higher in WT gyrA than both MTs (Table 1). It is evident from Table 1 that majority of favorable contributions are due to ΔEVDW and ΔEELE terms.
WT, wild-type.
Per residue decomposition of binding energy
Per residue decomposition of binding energy sheds light on the residues, which majorly contribute to gyrA-ciprofloxacin binding in both WT and MT models. We compared the protein–ligand interaction spectra between gyrA WT and MT models and Figure 7 shows contribution of individual hotspot residues to polar and nonpolar interaction terms. It can be observed from the energy decomposition analysis that in the case of ciprofloxacin-bound WT complex, major contributions were from Lys42, His45, Arg91, Leu98, Ser172, Gly173, and Ile174. Decrease in the overall interaction energy in MT models is largely accountable to change in the van der Waals contribution of hotspot residues. Mutation of Ser to Leu at position 83 in double mutant S83L/D87N shows improved binding at mutation site (Total Score: −0.609 kcal/mol; van der Waals Score: −0.88 kcal/mol) likely due to better hydrophobic interactions with the Leu side chain. However, it is accompanied with a reduction in overall binding energy (Table 1) as S83L substitution negatively impacts the binding affinity of the nearby residues.
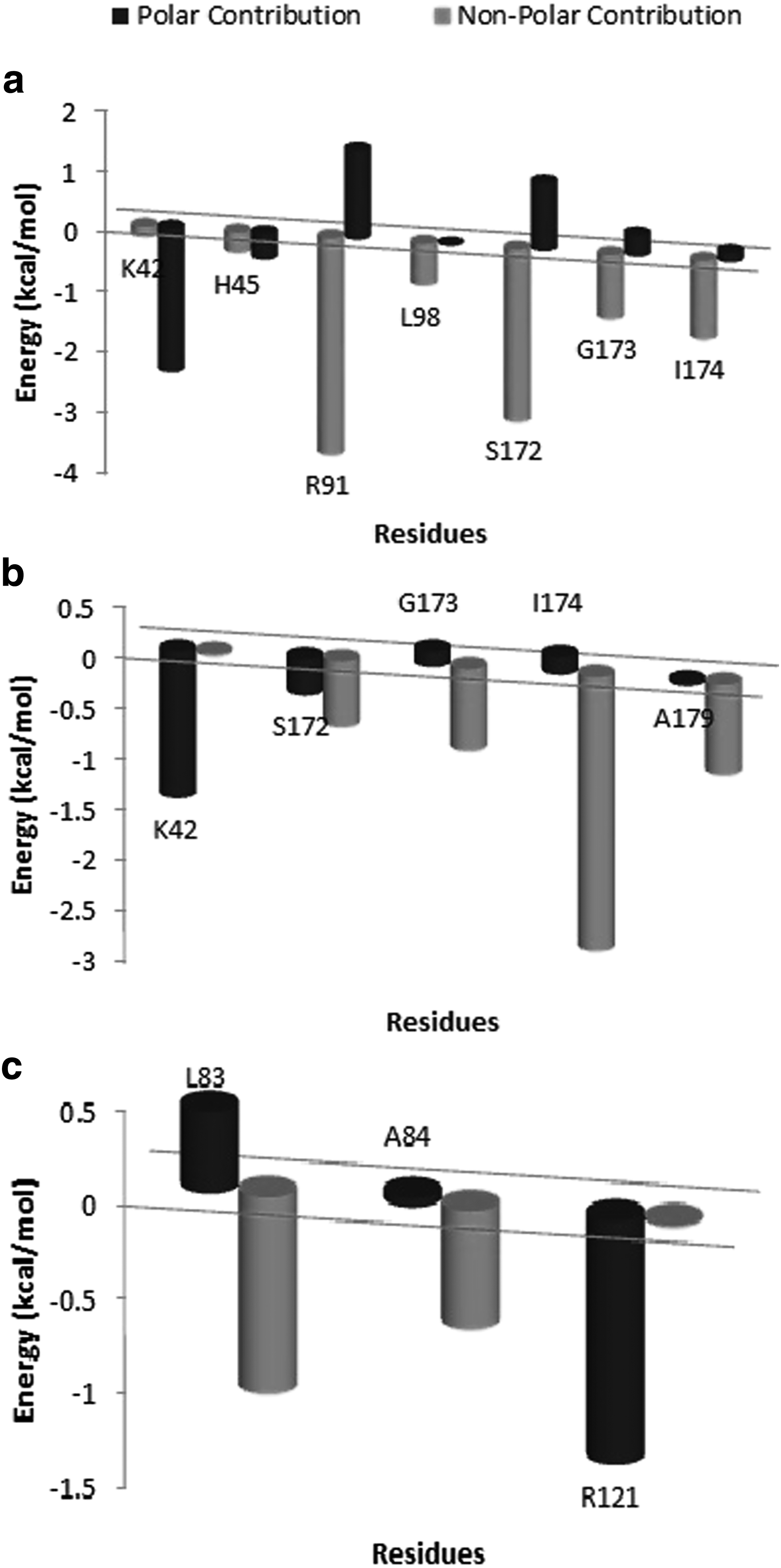
Polar and nonpolar contribution of hotspot residues from MM-PBSA-based energy decomposition analysis of total binding energy (kcal/mol) during last 5 ns of MD simulation in
Principal component analysis
To identify the dominant motions in WT and MT complexes, PCA was carried out on the last 5-ns trajectories of each molecular system. Figure 8 shows the porcupine plot for the ciprofloxacin-bound WT and MT complexes along the direction of first PC. Significant difference in the overall pattern of global motions between three systems can be observed. Dominant motions represented by high magnitude arrows were mostly observed in the three distinctive domains of N-terminal regions. CAP domain contains HTH motif, which have been previously described to be responsible for contacts with DNA targets (Berger et al., 1998).
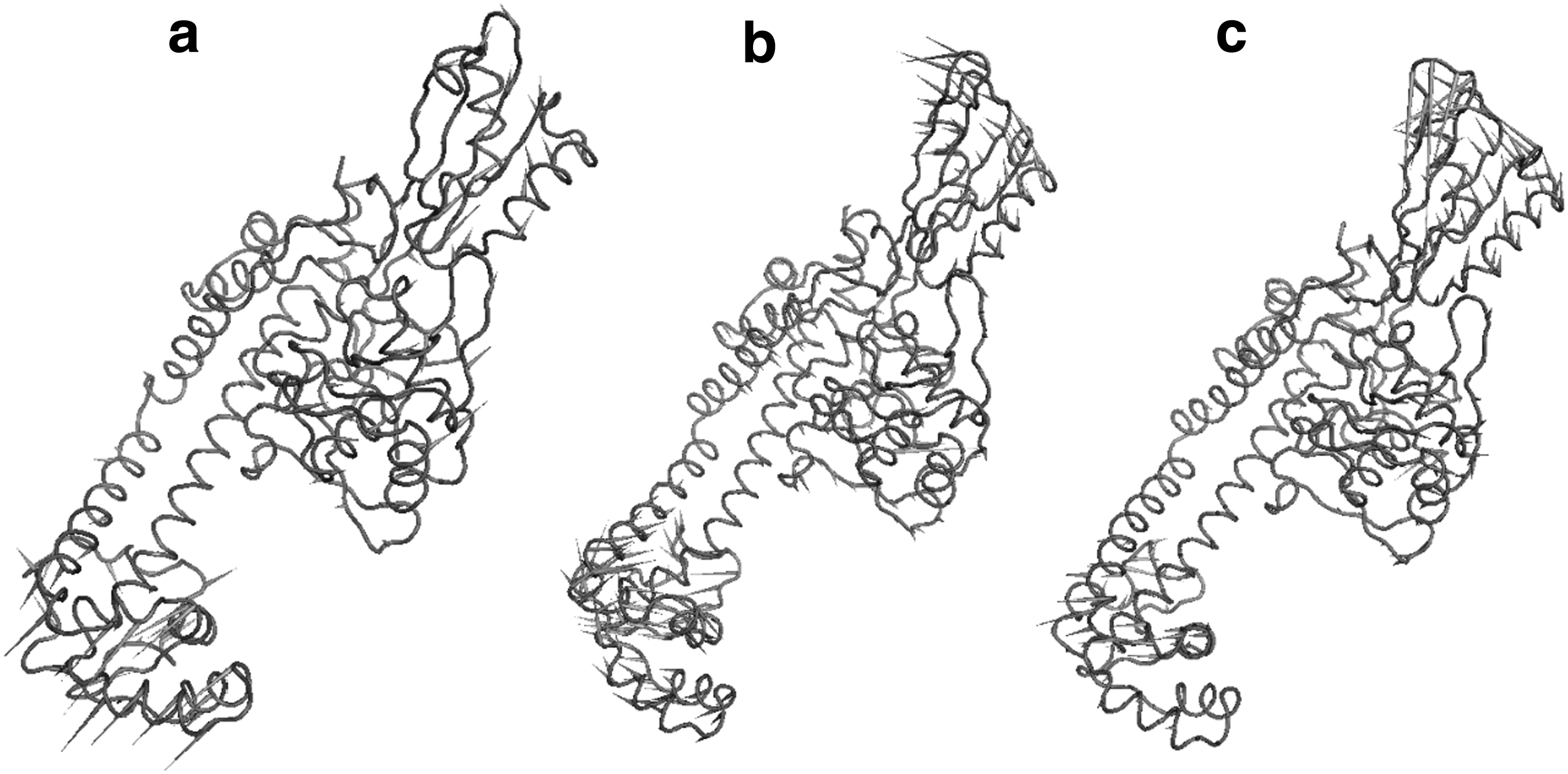
Porcupine plot of the first eigenvector obtained by PCA from simulation ensembles of
Motions in the CAP domain in WT structure were mostly upward, while in S83L, no prominent motions were observed. In double mutant S83L/D87N, random movements were observed in CAP domain. CAP domain in WT structure was observed to be relatively more flexible. Tower domain holds up against CAP domain, providing structural support and DNA binding site (Corbett et al., 2005). Tower domain in WT structure exhibited mostly upward movements, while in S83L and S83L/D87N, leftward and downward/leftward movements were detected. gyrA switches between its open and closed conformations due to the flexion of two connecting α-helices, which connect dimerization domain to CTD and tower domain (Corbett et al., 2005). We observed that prominent motions in these three domains were significantly different in MT variants compared with WT structure, both in direction and magnitude. Consequently, overall conformations of trajectories from the last 5-ns simulation were different across different structures.
RIN analysis
To identify key residue interactions and explore the differences between different WT and MT models, RINs were generated using the representative structures from the last 5-ns trajectories of each molecular system. As evident from the RIN plot (Fig. 9), RINs of MT variants of gyrA vary considerably from each other and WT. Zooming into the RIN plot shows, for instance, the interaction network in the vicinity of mutated residues, Ser 83 and Asp 87, and around catalytic residue, Tyr 122 follows a different trend across WT and MT variants of gyrA. A three-way interaction (peptide bond, close atom interaction, pi-cation interaction) between Thr 123 and Glu 124 in WT gyrA reduces to just a peptide bond interaction in S83L mutant.

Residue interaction network (RIN) comparison between helix;  loop;
loop;  , sheet; and
, sheet; and  , default; and Edges are represented as follows:
, default; and Edges are represented as follows:  , hydrogen bond;
, hydrogen bond;  , peptide bond;
, peptide bond;  , salt bridge;
, salt bridge;  , ionic bond;
, ionic bond;  , Pi stacking;
, Pi stacking;  , Pi-cation; and
, Pi-cation; and  , close atom interaction.
, close atom interaction.
Although the effect of changes at individual residue site may not be prominent, collectively these changes in residue interactions could have extreme effect on protein structure, consequently altering the active site conformation. It is quite interesting to observe that in case of S83L/D87N mutant, interaction subnetwork of residues 30–34 collapses with interaction subnetworks from residues, Gln 106, Thr 123, and Glu 124, while in S83L mutant, no interactions are observed in these subnetworks. S83L and D87N mutations have apparently disrupted the RIN of WT gyrA, subsequently affecting the drug binding landscape.
The functional impact of mutations is well captured at the protein structure level, which is dependent on interactions between amino acid residues. RINs facilitate understanding the role of inter-residue interactions in the structural and functional context. Topological features derived from such networks present residue interactions quantitatively. Closeness centrality is one such measure that combines the impact of single residues on entire protein backbone. Residues involved in active site, conserved motifs/domains, or ligand binding are reported to have scored high for closeness centrality (Amitai et al., 2004). Thus, we analyzed each network corresponding to WT and MT structures to screen for the residues with high closeness values, which implies their close relationship with the protein function.
For a total of 493 nodes, WT, S83L and S83L/D87N variants formed 3905, 3893, and 4015 edges, respectively. We observed that residues in the QRDR region of WT gyrA have high closeness values ranging from 0.7 to 1.0, which is weighted as interaction score. Ser 83 and Asp 87 scored 0.92 for closeness centrality. In S83L mutant, closeness values for Leu 83 and Asp 87 were 0.23 and 0.30, respectively. In S83L/D87N variant, Leu 83 and Asn 87 scored 0.26 and 0.27, respectively, for closeness. Ser 83 and Asp87 play a crucial role in quinolone binding. Substitutions with Leu 83 and Asn 87 negatively impact gyrA structure, as revealed by reduced network closeness in inter-residue interaction graph. In S83L and S83L/D87N variants, highest closeness values were observed to be 0.34 and 0.43. For both mutant forms, closeness values for residues in the QRDR region were in the range of 0.2–0.3. Reduced values of closeness for the residues across MT structures implicate their weaker interactions with other residues in the network. Structural restraints imposed by inter-residue interactions are important from the evolutionary perspective to preserve protein functional sites.
Conclusions and Expert Outlook
Rapidly evolving computational platforms have made investigating the structural consequences of the mutations easier and feasible. To the best of our knowledge, this is the first study that reports on explicit water MD simulations of ETEC gyrA in complex with ciprofloxacin aimed at understanding the molecular mechanisms underlying quinolone resistance in gyrA variants. As evidenced by RMS deviation, RMS fluctuation, and secondary structure conservation, WT and MT trajectories explore the same conformational space and show no significant changes in backbone conformations.
Despite this, these mutations have a deleterious effect on target protein, as revealed by the disrupted residue interaction networks and altered dominant motions, which will lead to unstable active site conformation and reduced binding affinity toward quinolones. Thus, this study offers a comprehensive picture of genotype–phenotype association of quinolone resistance-associated SNPs in gyrA. The results reported in this study elucidate the effect of mutations in gyrA on quinolone binding. This study therefore provides important insights for the design of more potent antibacterial agents with high ligand efficacy for treating drug-resistant bacterial infections, including TD that continues to adversely impact global health and international travel.
Footnotes
Acknowledgment
This research received funding support from FAST TRACK Young Scientist Fellowship from DST (Department of Science and Technology), Ministry of Science and Technology, India, under grant number SB/FT/LS-278/2012. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.